

Abstract
Although the acyclic nucleoside phosphonates cidofovir, adefovir and tenofovir are approved for treating human cytomegalovirus, hepatitis B and HIV infections, respectively, their utility is limited by low oral bioavailability, renal toxicity and poor cell penetration. Research over the past decade has shown that these undesirable features can be eliminated by esterifying the compounds with an alkoxyalkyl group, in effect disguising them as lysophospholipids. In this modified form, the drugs are readily taken up in the gastrointestinal tract and have a prolonged circulation time in plasma. The active metabolite also has a long half life within cells, permitting infrequent dosing. Because these modified drugs are not recognized by the transport mechanisms that cause the accumulation of acyclic nucleoside phosphonates in renal tubular cells, they lack nephrotoxicity. Alkoxyalkyl esterification also markedly increases the in vitro antiviral activity of acyclic nucleoside phosphonates by improving their delivery into cells. For example, an alkoxyalkyl ester of cyclic-cidofovir, a less soluble compound, retains antiviral activity for 3 months following a single intravitreal injection. Two of these novel compounds, hexadecyloxypropyl-cidofovir (CMX-001) and hexadecyloxypropyl-tenofovir (CMX-157) are now in clinical development. This article focuses on the hexadecyloxypropyl and octadecyloxyethyl esters of cidofovir and (S)-HPMPA, describing their synthesis and the evaluation of their in vitro and in vivo activity against a range of orthopoxviruses, herpesviruses, adenoviruses and other double-stranded DNA viruses. The extension to other nucleoside phosphonate antivirals is highlighted, demonstrating that this novel approach can markedly improve the medicinal properties of these drugs.
Keywords: lysophospholipids, ether lipids, prodrugs, alkoxyalkyl esters, hexadecyloxypropyl-cidofovir, hexadecyloxypropyl-tenfovir, hexadecyloxypropyl-cyclic-(S)-HPMPA, vaccinia virus, cowpoxvirus, ectromelia virus, cytomegalovirus, adenovirus, HIV-1, hepatitis B virus
1. Introduction
Acyclic nucleoside phosphonates were first synthesized and their antiviral activity evaluated by the groups of Antonin Holy and Erik De Clercq (De Clercq et al, 1987, Snoeck et al, 1988). There are currently three antivirals of this class approved for human use against cytomegalovirus (cidofovir, CDV, HPMPC), hepatitis B (adefovir, PMEA) and HIV (tenofovir, PMPA)(reviewed in De Clercq, 2007). These compounds are analogs of deoxycytosine monophosphate (CDV) and deoxyadenosine monophosphate (PMEA, PMPA), in which the deoxyribose has been replaced with an acyclic moiety (Figure 1). The intrinsic phosphonate of the acyclic nucleoside phosphonates is beneficial because it allows for bypass of the first phosphorylation required for activation of nucleosides during anabolic phosphorylation to the triphosphates. Therefore, compounds like cidofovir are fully active against thymidine kinase or UL-97 mutant viruses which are resistant to acyclovir or ganciclovir due to poor phosphorylation.
Figure 1.
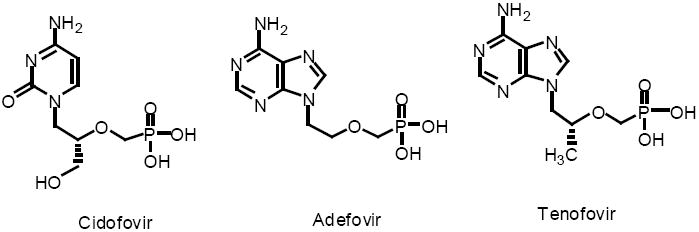
Acyclic Nucleoside Phosphonate Antivirals in Clinical Use
The principal drawbacks of acyclic nucleoside phosphonates are their poor oral bioavailability, low cellular uptake and their tendency to concentrate in the kidney proximal tubule, resulting in nephrotoxicity (Cundy, 1999). To achieve oral bioavailability, Gilead Sciences masked the dual negative charges on adefovir with dipivoxil (Noble and Goa, 1999) and on tenofovir with disoproxil fumarate (Kearney et al, 2004) leading to better oral absorption, but releasing adefovir or tenofovir in plasma where kidney proximal tubule organic anion transporters are prone to concentrate them.
This review will focus on an alternative method which improves the pharmacokinetic and antiviral performance of acyclic nucleoside phosphonates. The strategy relies on the disguise of acyclic nucleoside phosphonates as partially metabolized phospholipids (Painter and Hostetler, 2004). The rationale for these chemical modifications will be presented and the biochemistry, pharmacokinetics, in vitro and in vivo antiviral efficacy will be reviewed. The focus will be on the new alkoxyalkyl analogs of CDV (HPMPC) and (S)-HPMPA, but the generality of the approach as applied to many different types of acyclic nucleoside phosphonates will also be highlighted. The status of clinical development of two agents of this class, HDP-CDV (CMX001) and HDP-tenofovir (CMX157), will be reviewed.
2. Lysophospholipid prodrug strategy for acyclic nucleoside phosphonates
2.1 Phospholipid membrane interactions
In the fluid mosaic model, biological membranes are fluid phospholipid bilayers with embedded proteins as described by Singer and Nicolson (1972). Membrane phospholipids are comprised primarily of diacylglycerophosphocholine (phosphatidylcholine, PC) and other diacyl species such as phosphatidylserine, phosphatidylethanolamine, phosphatidylglycerol and phosphatidylinositol. Membrane phospholipids have two acyl esters and assume a cylindrical shape. They do not readily translocate from the outer to the inner leaflet of phospholipid bilayer membranes (Figure 2). In cellular membranes, the distribution of phospholipids and sphingomyelin between inner and outer membrane leaflets of plasma membranes is asymmetric implying mechanisms controlling transverse distribution (van Deenen, 1981, Zachowski, 1993). Membrane proteins have been identified (aminophosholipid translocases, flippases) which facilitate transbilayer movement of diacylphospholipids (Zachowski, 1993; Raggers et al, 2002; Kol et al, 2004). These proteins may be ATP-dependent or ATP-independent (Raggers et al, 2002). Intracellular or plasma phospholipid transfer proteins are also known which have the ability to transfer lipids between plasma lipoproteins or intracellularly between membranes (Wirtz, 2006). Finally, lipid rafts (detergent resistant microdomains) enriched in sphingomyelin, cholesterol and certain proteins, are another important membrane feature involved with cellular traffic of membranes, lipids and proteins (Simons and Vaz, 2004, Helms and Zurzolo, 2004, Hanzal-Bayer and Hancock, 2007).
Figure 2.
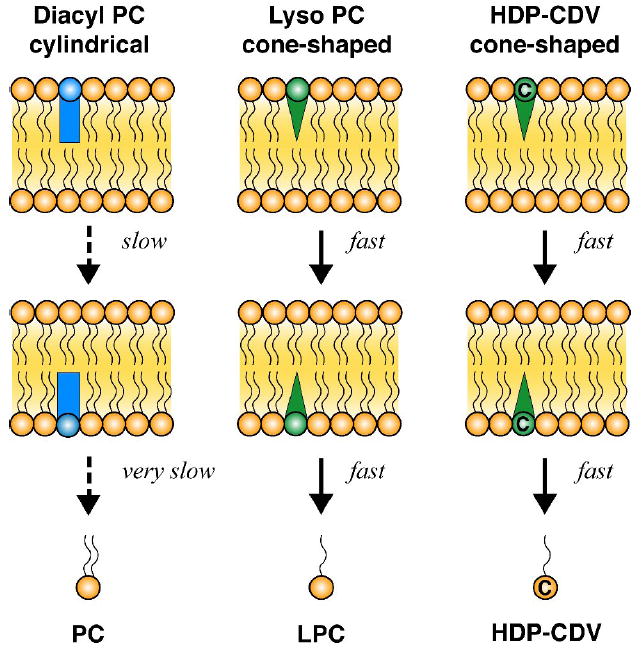
Interactions of phosphatidylcholine, lysophosphatidylcholine and HDP-CDV with phospholipid bilayer membranes
In contrast to cylinder-shaped diacylphospholipids, lysophospholipids lack the acyl ester at the sn-2 hydroxyl of glycerol, are cone-shaped and may disturb the bilayer structure which results in spontaneous transbilayer movement (Figure 2). Because lysophospholipids have a single acyl chain, their off-rate from phospholipid bilayer membranes is higher than that of diacylphospholipids which allows for rapid rates of diffusion of lysophospholipids inside the cell (Raggers et al, 2002). In principle, lysophospholipids might also undergo protein-mediated transbilayer movement but this has not been extensively studied.
Acyclic nucleoside phosphonates have a double negative charge at physiologic pH and have been found to enter cells very slowly by endocytosis (Connelly et al, 1993). In designing lipid analogs of acyclic nucleoside phosphonates to increase oral absorption in the small intestine and to facilitate cellular uptake and metabolism, we chose to focus on lysophospholipid analogs because of the ease with which they cross cellular membranes and desorb into the cytoplasm where they undergo metabolism and conversion to their diphosphates.
2.2. Design paradigm for acyclic nucleoside phosphonate analogs
Previous studies of lysophosphatidylcholine (LPC) absorption in rats or from inverted intestinal sacs indicated that about 40% of lysophosphatidylcholine is absorbed intact without further enzymatic cleavage (Scow et al, 1967, Nilsson and Borgstrom, 1967). In the small intestinal enterocyte, LPC is reacylated to PC and incorporated into chylomicrons which are secreted into intestinal lymphatics. To enhance absorption, we chose to disguise acyclic nucleoside phosphonate antivirals as LPC analogs, making some key changes in the molecule to optimize stability and oral bioavailability. The structure of LPC is shown in Figure 3 (left panel) and the arrows indicate the enzymatic cleavage points of phospholipases A1, C, D and lysophospholipase (van den Bosch, 1982). There are several key features of the structure of HDP-CDV (right panel, Figure 3) that make it more stable and more prone to good absorption from the intestine. First, the acyl ester bond at the sn-1 position of LPC has been replaced with an ether linkage, preventing hydrolysis of the acyl group by lysophospholipase during absorption. The alkyl ether also increases compound stability. Second, the hydroxyl at the sn-2 position of glycerol in LPC has been replaced with a hydrogen atom to prevent reacylation by lysophosphatidylcholine acyltransferases present in small intestinal enterocytes and other tissues. Finally, it should be noted that compounds like CDV and (S)-HPMPA have a phosphonate (-P-CH2-) linkage to the acyclic nucleoside which is not subject to cleavage by a phospholipase D or a phosphodiesterase. This means that the only metabolic cleavage which can readily occur is that catalyzed by phospholipase C (Figure 3), an enzyme which we first demonstrated in mammalian tissues in 1980 (Matsuzawa and Hostetler, 1980; Hostetler and Hall, 1980). Phospholipase C is not present in plasma or pancreatic secretions, providing stability for HDP-CDV and other compounds of this type during oral absorption and transport in plasma to tissues. This general approach is applicable to all nucleoside phosphonates.
Figure 3.
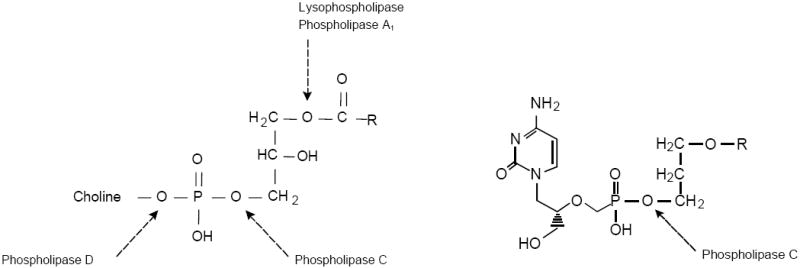
Predicted major metabolic cleavage sites for lysophosphatidylcholine and HDP-CDV
3. Antiviral evaluation of hexadecyloxypropyl and octadecyloxyethyl esters of CDV and (S)-HPMPA
3.1. Antiviral Effects in vitro
Cidofovir (CDV) and (S)-HPMPA were converted to their hexadecyloxypropyl and octadecyloxyethyl esters (Figure 4) and compared their antiviral activity with CDV and (S)-HPMPA (Beadle et al, 2002; Beadle et al, 2006; Beadle, 2007).
Figure 4.
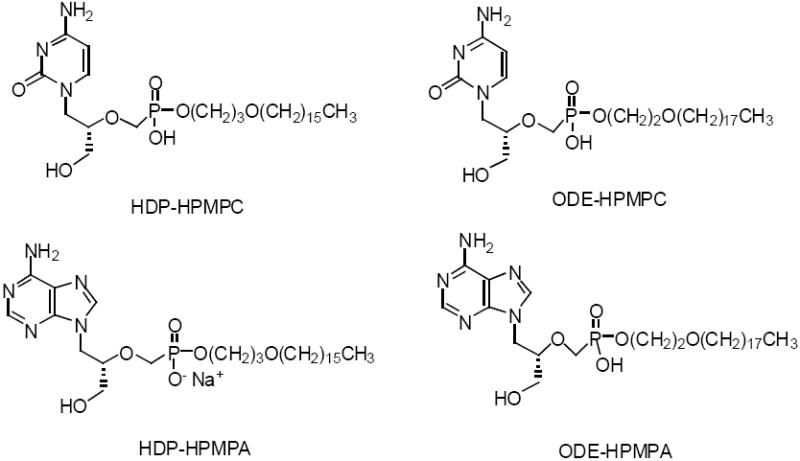
Structure of Key Alkoxyalkyl Esters of CDV and (S)-HPMPA
CDV Analogs
HDP-CDV and ODE-CDV showed large increases in antiviral activity against four poxviruses compared with CDV (Table 1). Unmodified CDV has EC50 values ranging from 12 to 46 μM while HDP-CDV and ODE-CDV had EC50s ranging from 0.003 to 0.9 (Kern et al, 2002; Buller et al, 2004). Variola appeared to be more sensitive to inhibition by HDP-CDV and ODE-CDV than other poxviruses with EC50 values of 0.10 and 0.03 μM, respectively (Huggins et al, 2002). Against herpesviruses, HDP and ODE esters of CDV showed remarkable activity in the low nanomolar EC50 range (0.9 nM for HCMV and 8 to 60 nM for HSV-1,-2 and HHV-8), versus unmodified CDV which ranged from 0.38 (HCMV) to 65 μM (EBV) (Williams-Aziz et al, 2005). Increases in antiviral activity of several logs were routinely noted versus unmodified CDV. HDP and ODE esters were generally similar in activity against herpesviruses. Against five strains of adenovirus, the order of activity was ODE-CDV > HDP-CDV ≫ CDV, and increases of activity of 2 to 3 logs were commonly seen versus unmodified CDV (Hartline et al, 2005). HDP-CDV and ODE-CDV also showed increased activity against BK virus (Randhawa et al, 2006) and orf viruses (Dal Pozzo et al, 2007).
Table 1.
Antiviral Effects of CDV, HDP-CDV and ODE-CDV on Replication of Poxviruses, Herpesviruses, Adenoviruses and Polyoma viruses, in vitro
| EC50, μM | |||
|---|---|---|---|
| Poxviruses | CDV | HDP-CDV | ODE-CDV |
| Cowpox Brighton | 44.7a | 0.9a | 0.3a |
| Vaccinia Copenhagen | 46.2a | 0.8a | 0.2a |
| Variola Bangladesh | 27.3b | 0.10b | 0.03b |
| Ectromelia virus | 12.0c | 0.50c | 0.20c |
| Rabbitpox virus | 39.0d | 0.504 | - |
| Herpes viruses | |||
| HCMV | 0.38e | 0.0009e | 0.0009e |
| HSV-1 | 5.5f | 0.06f | 0.02f |
| HSV-2 | 5.1f | 0.01f | 0.008f |
| HHV-8 | 2.6f | 0.02f | 0.03f |
| EBV | 65.6f | 0.03f | 0.10f |
| VZV | 0.5f | 0.0004f | 0.0001f |
| Adenoviruses | |||
| AdV-3 | 2.0g | 0.01g | <0.006g |
| AdV-5 | 0.5g | <0.009g | <0.008g |
| AdV-7 | 1.3g | 0.02g | <0.009g |
| AdV-8 | 1.0g | 0.03g | <0.008g |
| AdV-31 | 1.4g | 0.28g | 0.090g |
| Polyoma virus | |||
| BK virus | 115h | 0.13h | 0.083h |
| ORF virus | |||
| IT-C2 | 0.24i | 0.037i | 0.0032i |
| NZ2 | 1.26i | 0.18i | 0.03i |
References:
Buller et al, 2003;
(S)-HPMPA Analogs
(S)-HPMPA is the adenine analog of CDV (HPMPC) and is known to be more active against poxviruses than CDV. HDP and ODE esters of (S)-HPMPA were tested against cowpox, vaccinia, variola and ectromelia viruses in vitro (Beadle et al., 2006; Huggins, personal communication, 2006). (S)-HPMPA itself was 3 to 10 times more active against poxvirus replication than CDV. Esterification to HDP-(S)-HPMPA or ODE-(S)-HPMPA resulted in large increases in antipoxvirus activity ranging from 160 to 270 fold (Beadle et al, 2006). In HCMV infected cells, the antiviral activity of HDP-(S)-HPMPA and ODE-(S)-HPMPA was increased by 270 fold versus (S)-HPMPA (Beadle et al 2006) (Table 2). Studies with other herpesviruses have not yet been reported. In adenovirus infected cells, ODE-(S)-HPMPA was the most active compound with EC50 values of 0.04 to 0.16 μM compared with 0.19 to 1.1 μM with HDP-(S)-HPMPA (Hartline et al, 2005). (S)-HPMPA was previously shown to be inactive against HIV-1 (Balzarini et al, 1993). However, when HDP and ODE esters were prepared and tested against HIV-1, EC50 values in the 0.4 to 7 nanomolar range were found (Hostetler et al, 2006). HDP-(S)-HPMPA and ODE-(S)-HPMPA are also active in the low nanomolar range against BK virus (Randhawa et al, 2006, Randhawa et al, 2008) and orf viruses (Dal Pozzo et al, 2007).
Table 2.
Antiviral Effects of (S)-HPMPA, HDP-(S)-HPMPA and ODE-(S)-HPMPA on Replication of Poxviruses, Herpesviruses, Adenoviruses, HIV, Polyoma, ORF and Hepatitis B viruses, in vitro
| EC50, μM | |||
|---|---|---|---|
| Poxviruses | (S)-HPMPA | HDP-(S)-HPMPA | ODE-(S)-HPMPA |
| Cowpox | 4.0a | 0.02a | 0.02a |
| Vaccinia | 2.7a | 0.01a | 0.01a |
| Variola | 7.9b | <0.05b | <0.05b |
| Ectromelia | 0.24c | 0.049c | 0.032c |
| Herpes viruses | |||
| HCMV | 0.82a | 0.003a | 0.003a |
| Adenoviruses | |||
| AdV-3 | 1.9d | 0.43d | 0.14d |
| AdV-5 | 0.50d | 0.19d | 0.04d |
| AdV-7 | 2.2d | 1.0d | 0.06d |
| AdV-8 | 1.1d | 1.1d | 0.07d |
| AdV-31 | 2.5d | 0.29d | 0.16d |
| HIV-1 | |||
| HIV-1lai | 78e | 0.007e | 0.0004e |
| Polyoma virus | |||
| BK virus | n.d. | 0.02f | 0.03f |
| ORF virus | |||
| IT-C2 | 0.18g | 0.011g | 0.0026g |
| NZ2 | 0.61g | 0.036g | 0.006g |
| Hepatitis B virus | |||
| HBV wt | 0.99h | 0.25h | 0.11h |
Huggins, J. personal communication 2006;
Buller and Hostetler, unpublished, 2008;
Dal Pozzo et al, 2006;
Esterification of CDV and (S)-HPMPA with HDP and ODE was designed to increase oral bioavailability based on the resemblance to lysophosphatidylcholine (Figure 3). However, the large increases in antiviral activity in vitro were unexpected. Both CDV and (S)-HPMPA have been reported to inhibit replication of all double stranded DNA viruses and the increased antiviral activity observed after alkoxyalkyl esterification does not appear to be specific for a particular type of virus (Tables 1,2). Although (S)-HPMPA was reported to be inactive against HIV (Balzarini et al, 1993), HDP-(S)-HPMPA and ODE-(S)-HPMPA are highly active with low nanomolar EC50 values in vitro (Hostetler et al, 2006), suggesting that the alkoxyalkyl ester strategy can extend a compound’s range of antiviral activity in some cases (Table 2).
3.2. Effect of alkyl chain length and linker moiety
To determine the optimal length for the alkoxyalkyl chain, a series of analogs of CDV were synthesized with alkoxyalkyl chains ranging from 12 to 24 atoms. Their activity against HCMV and ganciclovir-resistant HCMV isolates is shown in Figure 5. The optimal chain length is 20 atoms, representing a 16 carbon alkyl group (hexadecyl) and an oxypropyl linker. HDP-CDV represents the optimal structure for both wild type HCMV (left panel) and three drug resistant isolates of HCMV (right panel) having UL97 and DNA polymerase mutations (Wan et al, 2005, Williams-Aziz et al, 2005). Alkoxyalkyl esters with shorter chains (12 to 16 atoms) or long chains (24 atoms) exhibit less activity. This is probably due to increased water solubility of the short chains, while the 24 atom compound has a lower fluidity and may be hindered in its ability to traverse membranes or by slow hydrolysis by phospholipase C. HDP-CDV retains substantial antiviral activity against the CMV strains resistant to ganciclovir, foscarnet and CDV, even those having DNA polymerase mutations or double mutations in UL97 and DNA polymerase retaining excellent EC50 values from 0.8 to 7 nanomolar (Figure 5). CDV compounds lacking a linker are generally less active and those having the oxyethyl linker are usually the most active at any given chain length compared with the oxypropyl moiety (Wan et al, 2005; Williams-Aziz et al, 2005).
Figure 5.
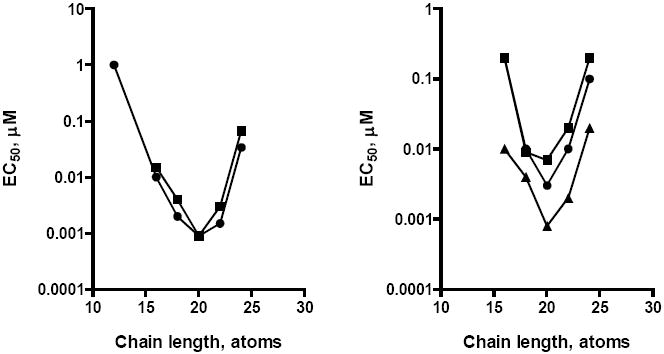
Effect of alkoxypropyl-cidofovir chain length on antiviral activity against HCMV, MCMV and drug-resistant HCMV isolates
Left Panel: Wild type CMVs; HCMV (AD169), circles; MCMV, squares; Right panel: Drug resistant CMV isolates: VR4760 (GCV/PFA resistant, UL97/DNA pol mutations), triangles; C8914-6 (GCV resistant, UL 97 mutation), circles; 759 D100 (GCV/CDV resistant, DNA pol mutation), squares. Adapted from Wan et al, 2005 and Williams-Aziz et al, 2005
4. Mechanism of increased antiviral activity of HDP-CDV and HDP-(S)-HPMPA
4.1. Cell uptake and anabolic phosphorylation
Antiviral studies with CDV, HDP-CDV and (S)-HPMPA and HDP-(S)-HPMPA showed dramatic increases in antiviral activity against many dsDNA viruses as discussed in the prior section. In some cases, compounds previously reported to be inactive against HIV-1 ((S)-HPMPA) were shown to have low nanomolar EC50 values when converted to HDP-(S)-HPMPA or ODE-(S)-HPMPA (Hostetler et al, 2006). To evaluate the mechanisms responsible for the increased antiviral activity, 14C-labeled CDV, HDP-CDV, (S)-HPMPA, and HDP-(S)-HPMPA were incubated with MRC-5 human lung fibroblasts and cell uptake and anabolic phosphorylation of the compounds to the active metabolites (CDV and (S)-HPMPA diphosphates) was determined (Aldern et al, 2003; Magee et al, 2008). CDV and (S)-HPMPA uptake increased very slowly over 24 hrs reaching levels of 3.6 and 2.8 picomoles/well (Figure 6), consistent with their reported uptake by fluid phase endocytosis (Connelly, 1999; Palu et al, 1991). In contrast, HDP-CDV and HDP-(S)-HPMPA uptake increased rapidly over 6 hrs and progressively to 24 hrs achieving cellular levels of 187 and 155 picomoles/well, respectively (Figure 6), representing an increase of 52-55 fold versus unmodified CDV and (S)-HPMPA. The area under curve for HDP-CDV and HDP-(S)-HPMPA was 41 and 42 fold greater than that of CDV and (S)-HPMPA. Interestingly, the uptake profile for the two HDP esters in MRC-5 cells is very similar, suggesting that the hexadecyloxypropyl lipid moiety (not the nucleoside polar head group) is driving cellular uptake because of the propensity of these compounds to insert spontaneously into the phospholipid bilayer membrane of cells (Figure 2).
Figure 6.
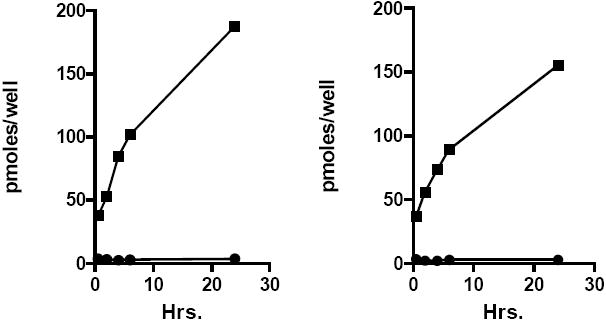
Cellular uptake of CDV, (S)-HPMPA and their HDP esters in MRC-5 cells
Left panel: CDV, circles; HDP-CDV, squares; Right panel: (S)-HPMPA, circles; HDP-(S)-HPMPA, squares. Adapted from Aldern et al, 2003, and Magee et al, 2008.
Increased entry of HDP-CDV and HDP-(S)-HPMPA into the cell is not sufficient to explain the marked increases in antiviral activity noted above. To have antiviral activity, HDP-CDV and HDP-(S)-HPMPA must be hydrolyzed to CDV and (S)-HPMPA by a cellular phospholipase C followed by anabolic phosphorylation to their diphosphates (CDVpp and (S)-HPMPApp) (Hostetler, 2007). In the MRC-5 uptake studies, we examined the metabolites in the cells and measured levels of CDV, CDVp and CDVpp and (S)-HPMPA, (S)-HPMPAp and (S)-HPMPApp by Partisil SAX HPLC (Figure 7). With exposure to 10 μM CDV, cell levels of CDVpp at 24 hr were 1.3 picomoles/well versus 132 picomoles/well with HDP-CDV, an increase of 100 fold (Aldern et al, 2003). When exposed to 10 μM (S)-HPMPA, (S)-HPMPApp levels at 24 hr were 21 picomoles/well compared with 451 picomoles/well with HDP-(S)-HPMPA, and increase of 21 fold (Magee et al, 2008). A timed study showed that HDP-(S)-HPMPA conversion to (S)-HPMPApp is much more rapid than of HDP-CDV (Aldern et al, 2006). When the cells were exposed to HDP-CDV or HDP-(S)-HPMPA for 24 hrs followed by washout of the compounds, the disappearance of the respective diphosphates in the cell was slow with a T½ of about 7 days which suggests that less frequent dosing might be possible in vivo (Aldern et al, 2003; Aldern et al, 2006).
Figure 7.
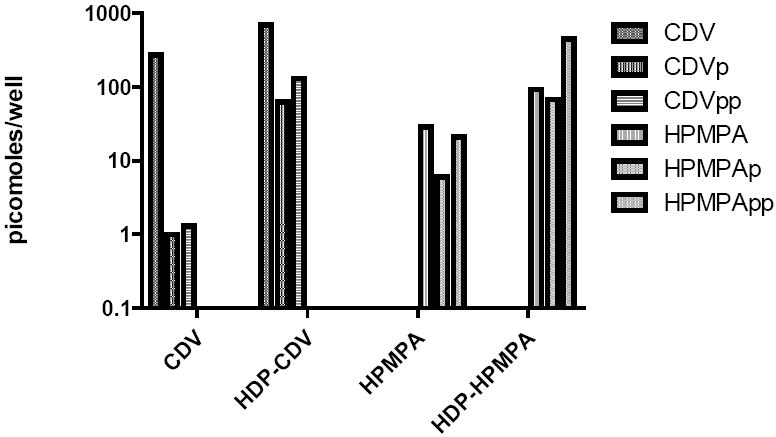
Metabolism and anabolic phosphorylation of CDV, (S)-HPMPA and their HDP esters in MRC-5 cells in vitro
Legend: 10 μM 14C-labeled CDV, (S)-HPMPA, HDP-CDV and HDP-(S)-HPMPA were applied to wells containing confluent MRC-5 cells and incubated for 24 hrs. Anabolic phosphorylation to their monophosphates (CDVp and (S)-HPMPAp) and diphosphates (CDVpp and (S)-HPMPApp) was measured by Partisil SAX HPLC. Adapted from Aldern et al, 2003, and Magee et al, 2008.
The cellular metabolism studies comparing CDV with HDP-CDV and (S)-HPMPA with HDP-(S)-HPMPA indicate that the cell uptake and conversion of the HDP analogs to their diphosphates is 0.5 to 1 log greater than the unmodified compounds. This, in turn, probably accounts for the greater antiviral activity of the alkoxyalkyl esters of the acyclic nucleoside phosphonates. Slow uptake of CDV and (S)-HPMPA is due to poor penetration of the cell membranes because of the dual negative charge at physiologic pH and the slow nature of fluid phase endocytosis, their common mechanism of uptake (Connelly et al, 1993, Palu et al, 1991). In contrast, HDP-CDV and HDP-(S)-HPMPA uptake is very rapid because these compounds spontaneously insert into cell membranes, translocate to the inner membrane leaflet and desorb readily to be metabolized by phospholipase C and anabolic phosphorylation to the antiviral diphosphates (Figures 2, 6, 7).
4.2. Mechanism of action of cidofovir and (S)-HPMPA diphosphates on the vaccinia E9L DNA polymerase
Cidofovir inhibition of the CMV and Vaccinia polymerases
CDV undergoes anabolic phosphorylation in the cell to CDV diphosphate (CDVpp), the active antiviral. Prior studies with the DNA polymerase of CMV showed that CDVpp, an analog of dCTP, inhibits the polymerase by incorporation into the growing DNA chain opposite template Gs. Incorporation of a single CDV causes a 31% decrease in the rate of elongation but when two successive CDVs are added, chain termination results and 3’-to-5’ exonuclease activity is blocked (Xiong et al, 1997).
However, with the E9L DNA polymerase of vaccinia virus, the mechanism of inhibition is somewhat different. CDVpp is a poor substrate for DNA synthesis relative to dCTP but when incorporated into the growing DNA strand, CDV terminated primers are good substrates for the next dNTP addition. However, after the addition of CDV + 1 base by the E9L polymerase, the rate of primer extension is greatly slowed and the 3’-to-5’ exonuclease activity is completely blocked (Magee et al 2005). Recently, an additional mechanism of action was discovered; it was shown that templates containing a CDV residue cannot be extended beyond the CDV base by the E9L polymerase, effectively blocking further rounds of replication (Table 3) (Magee et al 2008).
Table 3.
Mechanism of Action of CDV and (S)-HPMPA diphosphates versus CMV and Vaccinia DNA polymerases
| Viral Polymerase | Compound | Action | Result | 3’-to-5’ Exo-nuclease | Template strand extension | Ref. |
|---|---|---|---|---|---|---|
| CMV DNA polymerase | CDVpp | Incorporation of 1 CDV base + any base | Slows chain extension | Inhibited | ? | Xiong et al, 1997 |
| Incorporation of 2 consecutive CDV bases | Chain termination | |||||
| Vaccinia E9L DNA polymerase | CDVpp | Incorporation of CDV + any base | Primer extension greatly slowed | Inhibited | Blocked | Magee et al, 2005, 2008 |
| Vaccinia E9L DNA polymerase | (S)-HPMPApp | Incorporation of one or more (S)-HPMPAs | No effect on primer extension | Inhibited | Blocked | Magee et al, 2008 |
(S)-HPMPA inhibition of the Vaccinia DNA polymerase
(S)-HPMPA was found to block adenovirus DNA polymerase at the level of chain elongation although the detailed mechanism is not clear (Mul et al 1989). Against vaccinia virus replication, (S)-HPMPA is 17 times more active than CDV (Tables 1 and 2). Surprisingly, unlike CDVpp, (S)-HPMPApp is an excellent substrate for the E9L polymerase with a Km and Vmax similar to that of dATP. It is readily incorporated into the growing DNA strand and, unlike CDVpp, it does not slow chain extension but blocks 3’-to-5’ exonuclease activity when in the penultimate position. At the primer terminus HPMPA can still be excised. Why then is (S)-HPMPA 17 times more inhibitory to vaccinia replication than CDV? Because (S)-HPMPApp is readily incorporated into DNA and does not slow chain extension, many (S)-HPMPApp residues may be incorporated into the template strand and templates containing (S)-HPMPA cannot be extended, blocking further rounds of replication leading to template strand inhibition (Magee et al 2008).
5. Pharmacokinetics and tissue distribution of HDP-CDV and HDP-(S)-HPMPA in mice
5.1 Oral pharmacokinetics of HDP-CDV
HDP-CDV was designed to resemble lysophosphatidylcholine, a natural phospholipid metabolite which is substantially absorbed without further hydrolysis (Scow et al 1967). We replaced the phosphocholine moiety of LPC with CDV, eliminated the sn-2 hydroxyl of glycerol to prevent reacylation by acylCoA:1-acyl-sn-glycero-3-phosphocholine-O-acyltransferase [EC 2.3.1.62]. The sn-1 acyl ester bond was changed to an ether linkage to prevent cleavage by pancreatic or cellular lysophospholipases. To see if this approach would increase oral bioavailability, radioactive CDV and HDP-CDV were prepared. Oral administration of equimolar doses (17.8 μmoles/kg) of [2-14C]-CDV or HDP-[2-14C]-CDV to mice produced very low plasma levels with CDV and substantially greater levels with HDP-CDV (Figure 8). The Cmax for CDV was 0.8 μM versus 2.3 μM for HDP-CDV and the area under curve for HDP-CDV was 12 fold greater than that of CDV. The relative oral bioavailability was estimated to be 88% for HDP-CDV and the T½ for HDP-CDV was 14.9 hrs (Ciesla et al 2003). Analysis of the plasma indicated that about 50% of the radioactivity was intact HDP-CDV, the remainder being CDV and a metabolite. Similar results were obtained with HDP-[8-14C]-HPMPA which was estimated to have a Cmax of 1.1 μM, a relative oral bioavailability of 73% and a T½ of 11 hrs (Hostetler et al, 2005 and Magee et al, 2008).
Figure 8.
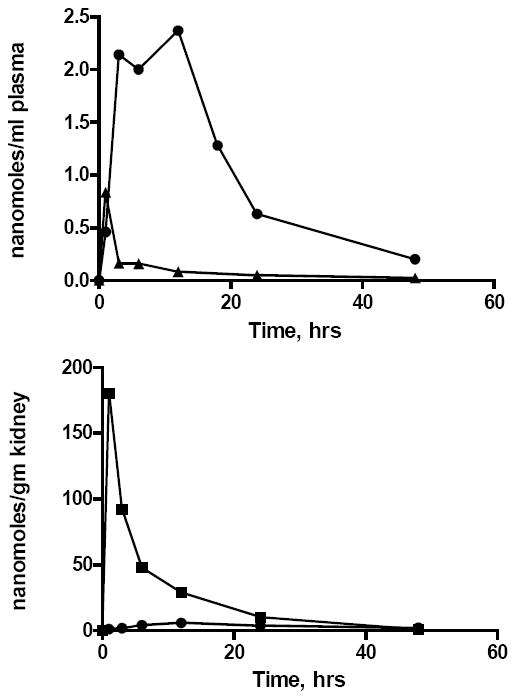
Key pharmacokinetic features of [2-14C]-CDV and HDP-[2-14C]-CDV after administration to mice: plasma and kidney levels of drug and metabolites.
Legend. Upper panel: Plasma levels of [14C]-CDV (triangles) or HDP-[14C]-CDV (circles) after oral administration to mice at 17.8 μmoles/kg. Lower panel: Kidney levels of [2-14C]-CDV (squares) after intraperitoneal administration (17.8 μmoles/kg) or HDP-[2-14C]-CDV (circles) after oral administration (17.8 μmoles/kg) to mice. Adapted from Ciesla et al, 2003.
Nephrotoxicity is the dose limiting toxicity of intravenous CDV in humans because the compound is concentrated by the organic anion transporter in renal proximal tubule cells (Izzedine et al 2005). With HDP-CDV, another key pharmacokinetic finding was that 17.8 micromoles/kg of HDP-CDV given orally results in very low kidney uptake, Cmax 5.9 nanomole/gm. However, in mice, when an equimolar dose of CDV was given intraperitoneally, the Cmax in kidney was 180 nanomole/gm, a 30 fold increase (Figure 8). The area under curve in kidney was 170 nanomole hrs/gm for HDP-CDV versus 1170 nanomole hrs/gm for CDV, an increase in AUC of 6.9 fold (Ciesla et al 2003). Thus, masking of one of the negative charges on the phosphonate of CDV by esterification with HDP greatly reduces kidney uptake, the major site of dose-limiting toxicity with CDV.
A scheme showing the proposed pathways of intestinal absorption of HDP-CDV is shown in Figure 9. Like lysophosphatidylcholine (LPC), HDP-CDV is thought to be absorbed at the apical brush border in mixed micelles of bile salts, monoglycerides and lysophospholipids. Following absorption, HDP-CDV enter the portal vein directly leading to high levels of drug and metabolites in liver (Ciesla et al, 2003). LPC is reacylated in the enterocyte endoplasmic reticulum to phosphatidylcholine (PC) and PC and HDP-CDV (red symbols) are incorporated in to lipid-rich chylomicrons. Assembled chylomicrons contain triglyceride and cholesterol esters in the core while the surface coat consists of apolipoprotein B48, PC and HDP-CDV (Figure 9). Chylomicrons are secreted into the small intestinal lymphatic system which leads successively to the thoracic duct, left subclavian vein, innominate vein and ultimately to the superior vena cava. After returning to the right side of the heart, chylomicrons pass through the lung and then, via the left side of the heart, into the arterial circulation. Interestingly, it appears to be possible to manipulate the fraction of the acyclic nucleoside phosphonate lipid prodrug passing through the chylomicron pathway (lung) versus the portal vein (liver) by making the lipid ester more hydrophobic. For example, 1-O-octadecyl-2-O-benzyl-sn-glycero-CDV, a more hydrophobic prodrug than HDP-CDV because of the 2-O-benzyl group (instead of hydrogen), has been shown to give rise to higher lung levels than HDP-CDV after oral administration due to greater partitioning into the chylomicron pathway (Hostetler et al, 2007).
Figure 9.
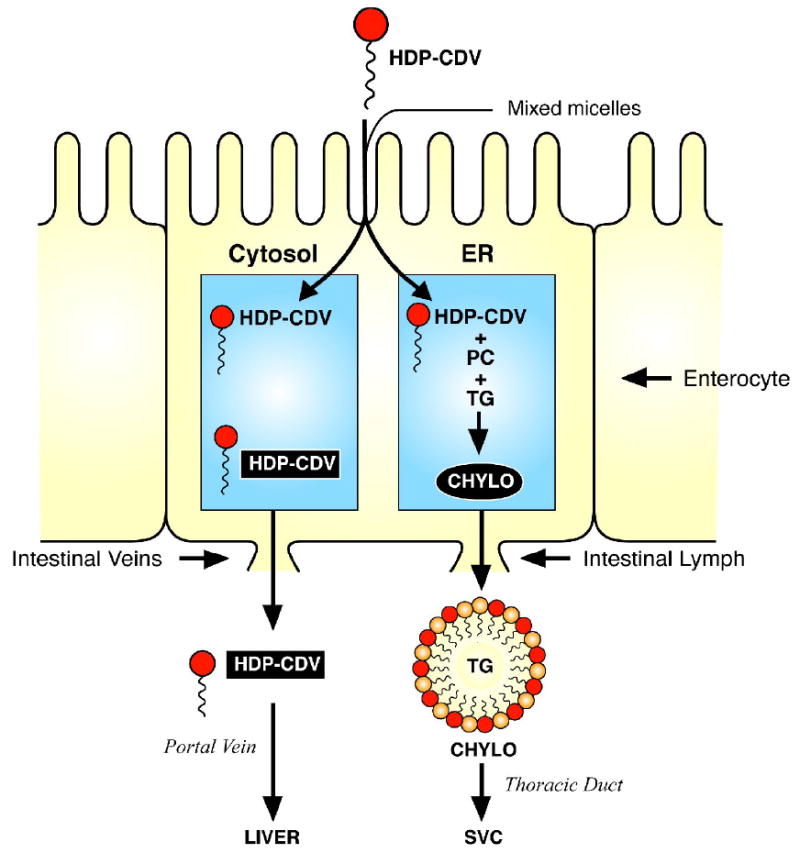
Proposed pathways of intestinal absorption of HDP-CDV
Legend: HDP-CDV (dark symbols) in mixed micelles of bile salts, monoglycerides, and lysophospholipids is absorbed at the brush border of the enterocyte. HDP-CDV enters the portal vein directly. Chylomicrons are formed in the endoplasmic reticulum with a core consisting of triglycerides (TG) and cholesterol esters and a surface coat consisting of apoprotein B-48 (not shown in the diagram), phosphatidylcholine (light symbols) and HDP-CDV (dark symbols) and secreted into the small intestinal lymphatics.
5.2. Oral pharmacokinetics of HDP-(S)-HPMPA
Oral administration of HDP-(S)-[8-14C]-HPMPA to mice as a single dose of 10 mg/kg gives a Cmax for HDP-(S)-HPMPA and metabolites of 1.1 μM at 1 hr. The plasma AUC0-24 hr was 12.1 nanomole hrs/ml versus 16.4 nanomole.hr/ml for intraperitoneal administration indicating a relative oral bioavailability of 74% compared with 24% for (S)-HPMPA (Quenelle et al, 2007). The dose-limiting toxicity of HDP-(S)-HPMPA in rodents was gastrointestinal and no evidence of nephrotoxicity was apparent (Hostetler, unpublished data, 2007). Thus, the esterification of CDV and HPMPA with HDP leads to marked increases in oral bioavailability and is probably a general finding with compounds of this class.
In summary, conversion of CDV and (S)-HPMPA to HDP-CDV and HDP-(S)-HPMPA results in the following:
Increased cellular uptake and conversion to CDVpp and (S)-HPMPApp, the active antiviral metabolites.
Multiple log increases antiviral activity against double stranded DNA viruses compared with the unmodified nucleobases.
Bypass of the initial phosphorylation required with conventional nucleoside analogs.
Oral bioavailability
Full activity against ganciclovir and acyclovir resistant herpes group viruses
Elimination of nephrotoxicity
Oral activity in animal models of vaccinia, cowpox, mousepox, rabbitpox, human CMV, murine CMV, adenovirus infections and viral retinitis.
6. Efficacy of HDP-CDV and related compounds in animal models of disease
6.1. Orthopoxvirus infections
Treatment of cowpox and vaccinia infection
HDP-CDV was originally designed to address the need for oral prophylaxis or treatment of smallpox because of concerns about possible bioterrorism with this agent. HDP-CDV was first tested orally in lethal models of cowpox (CPX) and vaccinia virus (VV) infection in mice. Both HDP-CDV and ODE-CDV were effective in reducing mortality when given orally prior to or 1, 2 or 3 days following intranasal infection with CPX or VV in BALB/c mice (Quenelle et al, 2004). Smee et al (2004) treated mice 24 hr after infection with VV-IHD strain with a single oral dose of 100, 50 or 25 mg/kg of HDP-CDV and found highly significant protection with zero to 20% mortality versus 100% mortality in untreated mice.
Treatment of ectromelia virus infection
In lethal aerosol challenge model with ectromelia virus (ECTV) infection, HDP-CDV and ODE-CDV given orally at 5 and 10 mg/kg prevented mortality from a high dose challenge in A/Ncr mice. Viral titers in liver and spleen were reduced below the limit of detection at 7 days while lung titers were reduced only slightly at 10 mg/kg (Buller et al, 2004).
In more detailed studies, high dose (10,000 × LD50) ECTV infection by the aerosol or intranasal route was treated with HDP-CDV (CMX001) 10 mg/kg on the first day followed by 2.5 mg/kg on day 3. Untreated A/Ncr mice had 100% mortality by day 12 versus 25 % mortality in HDP-CDV treated animals by day 20 (Parker et al, 2008). With high dose intranasal infection, animals treated with 10 mg/kg on day 1 and 2.5 mg/kg of HDP-CDV on day 3 were completely protected versus 100% mortality by day 7 in untreated mice. The dose of HDP-CDV required to produce 100% survival varies with the infectious dose of ECTV in the intranasal model. At high viral input doses (500-5,000 pfu), 8 mg/kg HDP-CDV provided complete protection while 4 mg/kg × 5 days gave 62 to 75% protection. With 50 pfu of ECTV, 2 mg/kg of HDP-CDV for 5 days provided 87% protection. However, with low dose infections with less than 5 pfu, 2 mg/kg of HDP-CDV for 5 days provided 100% protection (Parker et al, 2008). Since the upper limit of natural infections with variola may approach 50 pfu, attempts were made to determine the dose of HDP-CDV required providing full protection. Infected A/Ncr mice were treated with doses of HDP-CDV ranging from 0.3 to 5 mg/kg daily for 14 days. Daily treatment with doses of 1.25 mg/kg or greater for 14 days provided complete protection from mortality but 2.5 mg/kg or greater was required to minimize infection-related weight loss in this model (Parker et al, 2008).
Synergistic efficacy of HDP-CDV and ST-246 treatment of cowpox infection
ST-246 is an orally bioavailable drug which inhibits poxvirus infection by interfering with intracellular maturation of the virus (Yang et al, 2005), while HDP-CDV inhibits the viral DNA polymerase by inhibiting chain elongation (Magee et al, 2005) and produces template strand inhibition when the templates contain CDV residues (Magee et al, 2008). Quenelle at al (2007) found that in mice infected with CPX where treatment was delayed until 6 days post-infection, various doses of ST-246 (1 to 10 mg/kg) or HDP-CDV (0.3 to 3 mg/kg) alone did not separately provide survival benefits. However, when several ineffective doses of ST-246 and HDP-CDV were used in combination starting 6 days after infection and continuing for 5 days, nearly full protection could be achieved (Quenelle et al, 2007b).
Targeting CDV analogs to lung in ectromelia virus infection
HDP-CDV and ODE-CDV protect from poxvirus mortality and lower viral titers in liver and spleen but do not greatly reduce viral titers in lung (Quenelle et al 2004, Buller et al, 2004). By making the lipid ester more hydrophobic by adding an O-benzyl group (1-O-octadecyl-2-O-banzyl-sn-glycero-3-CDV, ODBG-CDV) (Hostetler et al, 2007), it is possible to increase the incorporation of the compound into the chylomicron surface coat during assembly in the enterocyte (see Figure 9). This leads to a higher proportion of the absorbed compound passing through the intestinal lymphatics to the superior vena cava and eventually to the right side of the heart followed by passage through the lungs (instead of passing to the liver first via the portal vein). Although the relative oral bioavailability of ODBG-[2-14C]-CDV was 32% compared with 88% for HDP-[2-14C]-CDV, the peak level in lung for ODBG-CDV was 100 nanomoles/gm compared with 1 nanomole/mg for HDP-CDV. Oral administration of 2 and 8 mg/kg of HDP-CDV and ODBG-CDV to animals given a high dose intranasal challenge with ECTV resulted in 80% survival at 2 mg/kg and 100% survival at 8 mg/kg versus 100% mortality in untreated A/Ncr mice (Hostetler et al, 2007). While this approach does not appear to confer any survival benefits in poxvirus infections, it may represent a useful way to design other drugs to target lung infections and neoplasia.
HDP-CDV treatment of rabbitpox virus infection
Infection of rabbits with rabbitpox virus (RPV) has been reported to exhibit all aspects of human infection with variola virus (Adams et al, 2007). This is important because approval of an anti-smallpox drugs by the FDA requires demonstration of activity in two or more poxvirus infection models having a well defined pathogenesis resembling that of human variola virus infection (Roberts et al, 2008). Rabbits injected intradermally with RPV develop a systemic disease with fever, viremia, secondary skin and mucocutaneous lesions, severe respiratory disease, natural aerosol transmission between animals and death in a high percentage of infected animals by nine days. In this system, HDP-CDV was given at doses of either 1 mg/kg or 5 mg/kg twice a day for 5 days beginning 24 hrs before challenge with 500 pfu of RPV. Untreated animals exhibited typical symptoms of RPV infection including fever, weight loss, skin and mucocutaneous lesions and 100% mortality by day 8. The group treated with 1mg/kg of HDP-CDV exhibited signs of disease and 50% mortality at day 8 while animals receiving 5 mg/kg showed little sign of disease except for a transient fever and <5 secondary skin lesions. The authors concluded that RPV infection of rabbits reprises most of the typical features of human smallpox, providing an excellent model for testing of drugs and vaccines (Adams et al, 2007).
Treatment of CDV-resistant vaccinia virus infection
Serial passage of vaccinia virus (VV) in the presence of increasing concentrations of CDV gives rise to CDV-resistant virus (Smee et al, 2002; Andrei et al, 2006; Kornbluth et al, 2006). The CDV-resistant VV-WR was found to have 5 mutations, 4 in the exonuclease domain (H296Y, A314V, H319W, S338F) of the vaccinia E9L polymerase and one in the polymerase domain (R604S). The CDV resistant VV-WR had an EC50 value of 29 μM representing 11-14 fold resistance (Table 4). The CDV-resistant VV-WR was found to be 10 to 30 times less virulent in mice. However, a single dose of HDP-CDV of 100 or 50 mg/kg given once 24 hrs after exposure provided 80 to 90% survival versus 20% in untreated animals. CDV was also effective; a single intraperitoneal dose of CDV 50 or 100 mg/kg gave 60 to 80% survival (Table 4) (Kornbluth et al, 2006). Andrei et al (2006) did similar studies with vaccinia Lederle (VV-LED) and found two mutations, A314T and A684V, which led to 9 fold resistance to CDV in vitro (Table 4). The CDV resistance mutations, A314T and A684V in a vaccinia WR background, were nonlethal in mice with doses of 4,000 pfu/mouse. Nevertheless, it was shown that 10 or 50 mg/kg of subcutaneous CDV daily for 5 days reduced weight loss in mice infected with a high dose of CDV resistant VV-WR (Andrei et al, 2006). Investigators agree that CDV resistant vaccinia is less virulent and in spite of the 9 to 14 fold in vitro resistance, the disease in mice can be treated effectively with parenteral CDV (Smee et al, 2005, Andrei et al, 2006) or oral HDP-CDV (Kornbluth et al, 2006).
Table 4.
Properties of CDV-resistant vaccinia virus
| EC50, μM | Mutations: E9L DNA Polymerase | ||||||
|---|---|---|---|---|---|---|---|
| Virus | Wt | CDV-R | Fold Res. | Exonuclease domain | Polymerase domain | CDV-R Virulence | Treatment of CDV-R vaccinia infections in mice |
| Vaccinia WR a | 2.1 | 29 | 13.8 | H296Y A314V H319W S338F | R604S | Decreased 10 to 30 fold | Single oral dose of 50 or 100 mg/kg HDP-CDV 10-20% mortality vs 100% mortality in untreated |
| Vaccinia Lederle b | 2 | 18 | 9 | A314T | A684V | Nonlethal in vaccinia-WR background | 10 or 50 mg/kg CDV s.c. for 5 days reduced weight loss compared with the untreated controls. All animals survived. |
Abbreviations : Wt, wild type; WR, Western Reserve, CDV-R, CDV-resistant
HDP-(S)-HPMPA and ODE-(S)-HPMPA treatment of cowpox and vaccinia infections
The in vitro antiviral activity of HDP- and ODE-(S)-HPMPA EC50s against vaccinia Copenhagen are 0.01 μM versus 0.8 μM for HDP-CDV, representing an 80 fold increase in activity (Beadle et al, 2006). These two compounds are somewhat less active against vaccinia WR in vitro with EC50s ranging from 0.1 to 0.3 μM versus 1.1 μM for HDP-CDV (Quenelle et al, 2007). When BALB/c mice were infected intranasally with lethal doses of vaccinia WR and oral treatment delayed for 24, 48 or 72 hrs, 10 mg/kg and 30 mg/kg HDP-(S)-HPMPA and ODE-(S)-HPMPA daily for 5 days both provided significant protection (87 to 100% survival). Similar results were observed in animals infected with cowpox Brighton. When treatment was delayed for 72 hrs, 30 mg/kg treatment with both compounds provided lesser, but statistically significant protection (Quenelle et al, 2007). In spite of their greater in vitro antiviral activity, HDP-(S)-HPMPA and ODE-(S)-HPMPA do not appear to offer significant benefits over HDP-CDV (CMX001).
6.2. HCMV infection
HDP-CDV and ODE-CDV are highly active in vitro against human cytomegalovirus (HCMV) with EC50s of 1 nanomolar (Beadle et al, 2002, Wan et al, 2005). The antiviral properties of oral HDP-CDV and ODE-CDV were tested in SCID mice having either human fetal retinal tissue or human fetal thymus/liver implants which were later infected with HCMV. Implants were infected with HCMV and 24 hrs later oral treatment was started with HDP-CDV or ODE-CDV at 5 and 10 mg/kg daily for 28 days. In fetal retinal implants at 28 days, HDP-CDV at 5 and10 mg/kg reduced HCMV infection in the implants with 7% positive for HCMV versus 71% positive in untreated implants. In thy/liv implants, 5 mg/kg HDP-CDV and ODE-CDV both eliminated HCMV infection in 100% of implants at 28 days (Bidanset et al, 2004). In SCID implanted thy/liv mice, HDP-(S)-HPMPA and ODE-(S)-HPMPA at 10 mg/kg for 28 days were also effective reducing the percentage of HCMV infected implants from 45% in untreated to 9% and 11%, respectively. Viral load in the treated implants remaining infected was reduced by 1.4 to 2.7 logs (Quenelle et al, 2008). While effective, HDP-(S)-HPMPA and ODE-(S)-HPMPA do not appear to offer any advantages over HDP-CDV (CMX001) in the animal models of HCMV infection.
6.3. Murine CMV infection
The Smith strain of murine CMV produces lethal model of disseminated disease after intraperitoneal injection and has been used to evaluate potential antiviral therapies (Kern, 1991). HDP-CDV and ODE-CDV have in vitro EC50s of 1 nanomolar against MCMV in vitro (Kern et al 2004). When treatment was delayed for 24 or 48 hrs after intraperitoneal injection of MCMV, both HDP-CDV and ODE-CDV given daily for five days produced statistically significant survival benefits, 67% to 100%, compared with no survival in untreated animals. Surprisingly, with a treatment delay of 24 hrs, a single oral dose of 3, 10 or 30 mg/kg HDP-CDV or ODE-CDV resulted in 87 to 100% survival compared with 7 to 10% survival in untreated animals. Survival benefits were also noted at 48 hr treatment delays (Kern et al, 2004). HDP-(S)-HPMPA was also active in the lethal MCMV model at 10 mg/kg and 30 mg/kg following a 24 hr treatment delay. However, the compound does not show activity superior to HDP-CDV in the lethal MCMV model (Quenelle et al, 2008).
6.4. Adenovirus infection
HDP-CDV is highly active in vitro against adenovirus serotypes 3,5,7,8 and 31 (Table 1 and Hartline et al, 2005). Toth et al (2008) recently found that HDP-CDV (CMX001) given orally at 2.5 mg/kg 1 day prior to infection with adenovirus type 5 (Ad5) and daily for 18 days thereafter prevented Ad5-induced mortality in immunosupressed Syrian hamsters. Untreated animals had 50% mortality by day 6 post infection. Treated animals also had significantly lower levels of serum ALT (a liver enzyme) and lower serum viral titers. In animals treated with CMX-001, serum and liver viral titers were undetectable at 15 days post infection versus serum and liver viral titers of 104 TCID50 /ml and 106 TCID50/gm in surviving untreated animals. CMX001 also reduced Ad5 titers in salivary gland and pancreas and prevented extensive liver damage, a major target of adenovirus infection (Toth et al, 2008).
CMX001 was also effective when given 6 hrs to 2 days after infection with Ad5. Treatment with 2.5 mg/kg of CMX001 given 6 hrs or 1 day after infection provided complete protection while one animal of eight in the 2 day postinfection group died versus three of eight in the untreated group. Animals with a 2 day treatment delay had a lower virus burden in the liver at day 7 and no infectious virus was found in liver at day 16. Animals with a 6 hr or 1 day treatment delay had liver enzyme and viral titer parameters indistinguishable from animals treated prophylactically (Toth et al, 2008).
6.5. Prevention of viral retinitis by pretreatment with intraocular HDP-cyclic-CDV
The animal studies discussed above utilized the HDP or ODE esters of CDV or (S)-HPMPA because of their tendency to insert into biological membranes and their oral bioavailability. However, less soluble forms of these compounds can be prepared by utilizing HDP esters of cyclic-CDV. Because both negative charges of CDV are masked, HDP-cyclic-CDV is sparingly soluble and has poor oral pharmacokinetics (Hostetler, unpublished data). To obtain long acting antiviral preparations for intraocular use, HDP-cyclic-CDV and HDP-cyclic-HPMPA have been evaluated (Cheng et al 2004; Tammewar et al 2007).
Intraocular CDV is effective in treating human CMV retinitis but causes a decrease in intraocular pressure, an unacceptable side effect. In fact a large number of acyclic nucleoside phosphonates, including (S)-HPMPA, had this property in animal testing (Banker et al 1998). However, HDP-cyclic-CDV, a poorly soluble crystalline analog of CDV, given by intravitreal injection in rabbits was nontoxic and doses of 100 μg/eye did not cause decreased intraocular pressure. The depot of HDP-cyclic-CDV cleared slowly from the vitreous with a half life of 6.3 days. Control and pretreated rabbits were then challenged with intraretinal HSV-1 at varying times after pretreatment. A single intravitreal dose of HDP-cyclic-CDV prevented HSV retinitis for 68 days versus pretreatment with vehicle or CDV, and statistically significant reductions in HSV retinitis were observed 100 days (Cheng et al 2004). HDP-cyclic-CDV is expected to be even more active against HCMV retinitis because its EC50 value is 42 nanomolar, lower than the EC50 for HSV-1,100-600 nanomolar (Wan et al 2005; Williams-Aziz et al, 2005).
HDP-cyclic-(S)-HPMPA is also highly active against HCMV with an EC50 of 20 nanomolar (Tammewar et al 2007). Intravitreal injection of the highest nontoxic dose of the sparingly soluble compound in the rabbit eye (55 mg/eye) gives an estimated intravitreal “concentration” of 65 μM, more than 3,000 times the EC50 against HCMV. HDP-cyclic-(S)-HPMPA did not cause decreased intraocular pressure in the guinea pig eye and has a residence time in vitreous of over 4 months (Tammewar et al 2007). Both HDP-cyclic-CDV and HDP-cyclic-(S)-HPMPA are worthy of further evaluation as possible long acting intraocular treatments for CMV retinitis.
7. Application of the same strategy to other acyclic nucleoside phosphonates
In the foregoing sections, the effect of esterification of HPMPC (CDV) and HPMPA with alkoxyalkyl groups was reviewed including their effects to enhance antiviral activity and oral bioavailability and to reduce nephrotoxicity. In this section, the effect of alkoxyalkyl esterification of compounds with other acyclic nucleoside phosphonate side chains and other nucleobases will be discussed and the most interesting results highlighted.
There are numerous alternative acyclic structures. Structures of the side chains of the ones in current clinical use, such as those of adefovir (PME) and tenofovir (PMP), are shown in Figure 10. In this figure, B denotes a nucleobase (A,C,T,G or substituted nucleobases) and R represents an alkoxyalkyl group such as hexadecyloxypropyl or octadecyloxyethyl. The antiviral activity of alkoxyalkyl esterified acyclic nucleoside phosphates of interest is shown grouped by the respective acyclic structures and compared with the unmodified compounds (Table 5).
Figure 10.
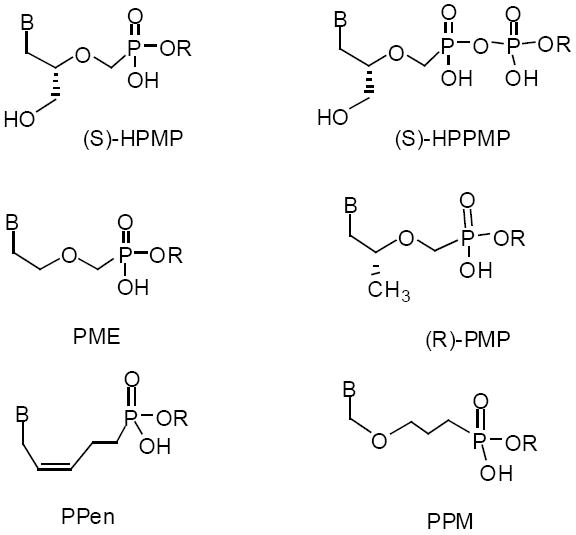
Structure of alkoxyallkyl esters of various antiviral acyclic nucleoside phosphonates
Abbreviations: B, nucleobase; R, alkoxyalkyl; HPMP, 3-hydroxy-2-(phosphono-methoxy)propyl; PME, 2-(phosphonomethoxy)ethyl; PPen, 5-phosphono-pent-2-en-1-yl; HPPMP, 3-hydroxy-2-(phosphophosphomethoxy)propyl; PMP, 3-hydroxy-2-(phosphono-methoxy)propyl; PPM, phosphonopropoxymethyl.
Table 5.
Antiviral activity of alkoxyalkyl esters of various types of acyclic nucleoside phosphonates
| Compound |
EC50, μM |
||
|---|---|---|---|
| HPMP nucleosides | Vaccinia | HCMV | HIV-1 |
| (S)-HPMPA | 2.7a | 0.82a | 77.5b |
| HDP-(S)-HPMPA | 0.01 | 0.003 | 0.007 |
| (S)-HPMPC (Cidofovir, CDV) | 46.2c | 0.38d | - |
| HDP-CDV (CMX001) | 0.8 | 0.009 | - |
| cHPMP-5-aza-C | 2.12 e | 0.070 e | - |
| HDE-cHPMP-5-aza-C | 0.037 | 0.00038 | >0.80 |
| PME nucleosides | Vaccinia | HCMV | HIV-1 |
| PMEA (Adefovir) | - | - | 1.1f |
| HDP-PMEA | - | - | 0.000015 |
| PMEDAP | >10g | 13g | 0.08f |
| HDP-PMEDAP | 0.6 | 0.5 | 0.000016 |
| PME-N6-cyPr-DAP | 4.0g | 0.4g | 0.07f |
| HDP-PME- N6-cyPr-DAP | 0.10 | 0.02 | <0.00001 |
| PPen nucleosides | HIV-1 | HBV (2.2.15) | HBV (AD38) |
| PPen A,C,G,T | >100h | >100h | >30h |
| HDP-PPen-A | >30 | 15.7 | 1.5 |
| HDP-PPen-C | >20 | 2.9 | 3.9 |
| HDP-PPen-G | 7.5 | 1.8 | 2.4 |
| HDP-PPen-T | >20 | 0.66 | 2.0 |
| PMP nucleosides | HIV-1 | HBV (2.2.15) | HBV (AD38) |
| PMPA (Tenofovir) | 3.20i | 7.2i | 2.25i |
| HDP-PMPA (CMX157) | 0.012 | 1.6 | 0.49 |
| PPM nucleosides |
HCMV |
||
| PPMG (ACV methylene phosphonate) | 5.4j | ||
| HDP-PPMG | 0.24 | ||
| PPMDAP | >100j | ||
| HDP-PPMDAP | 2.85 | ||
| PPM-N6-cyPr-DAP | >90j | ||
| HDP-PPM- N6-cyPr-DAP | 1.6 | ||
| HPPMP nucleoside | Vaccinia | HCMV | HSV-1 |
| HPMPC (CDV) | 31 | 1.2 | 3.3 |
| HDP-P-HPMPC | 3.2k | 0.26k | 0.06k |
References:
Ruiz et al, 2005;
Abbreviations: HPMPA, 9-(S)-(3-hydroxy-2-phosphonomethoxy-propyl)adenine; PMEA, 9-(2-phosphonomethoxyethyl)adenine; PMEDAP, 9-2-(phosphonomethoxyethyl)-2,6-diaminopurine; PME-N6-cyPr-DAP, 9-(2-phosphonomethoxyethyl) -2-amino-6-cyclopropylaminopurine; PPen-A, 9-(5-phosphono-pent-2-en-1-yl)adenine; PMPA, 9-(R)-(2-phosphonomethoxypropyl)adenine; PPMG, 9-(phosphonopropoxymethyl)-guanine; HDP-P-HPMPC, hexadecyloxypropyl-phospho-cidofovir; HDP, hexadecyloxypropyl; HDE, hexadecyloxyethyl.
7.1. HPMP nucleosides
Cidofovir (HPMPC, Vistide®) is a representative of his class, having the phosphonomethoxypropyl acyclic side chain (Figure 10). Cidofovir is in clinical use for cytomegalovirus retinitis and was the first to be synthesized and evaluated as its hexadecyloxypropyl ester (HDP-CDV, CMX001). HDP esters of both (S)-HPMPA and CDV show multiple log increases in antiviral activity when tested in vitro against vaccinia (50-270x) or HCMV (40-270x) (Table 5). (S)-HPMPA had been reported previously to be inactive against HIV (Balzarini et al, 1993) but the HDP ester was found to have an EC50 value of 7 nanomolar (Table 5; Hostetler et al, 2006). This appears to be due to extraordinarily poor cellular lymphocyte uptake and conversion of (S)-HPMPA to HPMPA diphosphate versus that observed with HDP-(S)-HPMPA (Aldern and Hostetler, unpublished 2007). Esterification of HPMP-cyclic-5-aza-cytosine with hexadecyloxyethyl results in a multiple log increase in the activity of this compound against vaccinia and HCMV (Krečmerová et al, 2007).
7.2. PME nucleosides
Adefovir (PMEA, Hepsera®) is a drug used for treating hepatitis B (as the dipivoxil ester). It is also active against HIV infection (Kahn et al, 1999). The acyclic side chain of PMEA has the phosphonomethoxyethyl structure (Figure 10). In contrast to HPMPA and CDV, PMEA lacks a secondary hydroxylmethyl group in its acyclic side chain and is an obligatory chain terminator. Like CDV, nephrotoxicity is the most significant toxicity in humans chronically treated with adefovir (Kahn et al, 1999). HDP-PMEA is strikingly active against HIV-1 with an EC50 of 15 picomolar (Table 5) and selectivity index of 4,000 (Valiaeva et al, 2006). The increased activity against HIV-1 of HDP-PMEA versus PMEA is the largest increase in activity noted to date with HDP esters of acyclic nucleoside phosphonates. The PME analogs of diaminopurine (PMEDAP) and N6-cyclopropyl-diaminopurine (PME-N6-cPr-DAP) also show dramatic increases in antiviral activity against HIV-1 when converted to HDP-PMEDAP and HDP-PME-N6-cPr-DAP with EC50 values of 16 and <10 picomolar. Increased activity is also noted for both compounds against vaccinia and HCMV (Prichard et al, 2008). It is worth noting that HDP-PME-N6-cPr-DAP has an excellent EC50 against HCMV of 20 nanomolar and 100 nanomolar against vaccinia virus suggesting possible preclinical evaluation for use against poxvirus and herpesvirus diseases (Table 5).
7.3. PMP nucleosides
The PMP nucleosides have a side chain with the structure phosphonomethoxypropyl (Figure 10). The acyclic side chain has a methyl group in place of the hydroxymethyl group in the HPMP nucleoside phosphonates making PMPA an obligatory chain terminator. PMPA is an example of a drug in clinical use for treatment of HIV infection as tenofovir disoproxil fumarate (Viread®). Esterification of PMPA with HDP increases antiviral activity from EC50 values of 3.2 μM for PMPA to 0.012 μM for HDP-PMPA in HIV-1 infected PBMCs (Painter et al, 2007), representing 260 fold increase (Table 5). HDP-PMPA also exhibits increased activity versus PMPA in HBV although the increased activity is less, 4-5 fold (Table 5). HDP-PMPA (CMX157) is in preclinical development for HIV infection and will be discussed in more detail in section 8.
7.4. PPen nucleosides
A new class of acyclic nucleoside phosphonate was recently reported having a 5-phosphono-pent-2-en-1-yl acyclic side chain loosely based on the structure of d4T (Choo et al 2007a). Unmodified acyclic nucleoside phosphonates of this class are inactive against HSV-1. However, when esterified with HDP, the HDP-PPen-T compound exhibits antiviral activity against HSV-1 in vitro (Choo et al, 2007a). None of the unmodified PPen nucleosides are active against HIV or HBV (Choo et al, 2007b). However, when converted to their HDP esters (Table 5), HDP-PPen-G is active against HIV-1 and HBV with EC50 values of 7.5 μM (HIV) and 1.8-2.4 μM (HBV). Against HBV, HDP-PPen-T was the most active compound against HBV with an EC50 of 0.66 μM. All HDP-PPen nucleosides (A,C,G,and T) were active against HBV, but only HDP-PPen-G had significant activity against HIV (Table 5). It is presumed that the lack of activity of unmodified PPen nucleosides is due to poor cellular uptake as reported previously for CDV (Aldern et al, 2003) and (S)-HPMPA (Magee et al, 2008).
7.5. PPM nucleosides
PPM nucleosides have a phosphonopropoxymethyl acyclic side chain (Figure 10); an example is phosphonopropoxymethylguanine (PPMG) which is also known as acyclovir methylene phosphonate. PPMG is a weak inhibitor of HCMV with an EC50 of 5.4 μM. HDP-PPMG has an EC50 value of 0.24 μM, an increase of 22 fold (Ruiz et al, 2006). PPMDAP and PPM-N6-cyPr-DAP are essentially inactive against HCMV in vitro, but their HDP esters have EC50s of 2.8 and 1.6 μM respectively (Table 5).
7.6. HPPMP nucleoside
When HPMPC (CDV) is esterified with HDP-phosphate, by forming a pyrophosphate link between the phosphonate and the phosphate, one obtains HDP-P-CDV (Ruiz et al, 2007). This compound shows increased antiviral activity against vaccinia, HCMV and HSV-1 compared with unmodified CDV (Table 5).
7.7. Summary: alkoxyalkyl prodrugs of other nucleoside phosphonates
From the results summarized in this section and in Table 5, it is clear that the enhancement of antiviral activity resulting from esterification of an acylic nucleoside phosphonate with an alkoxyalkyl residue like hexadecyloxypropyl is not confined to one particular type of acyclic phosphonate side chain or to one nucleoside base. All types of acyclic nucleoside phosphonates show enhancement of antiviral activity, presumably due to increased cellular penetration of the alkoxyalkyl esterified compounds and increased metabolic conversion to their diphosphates (triphosphate equivalents) (Aldern et al 2003, Magee et al 2008). In a number of instances, acyclic nucleoside phosphonates which were reported to be inactive showed antiviral activity as their HDP esters (Hostetler et al, 2006, Choo et al, 2007a, Choo et al, 2007b, Prichard et al, 2008). This emphasizes the probability that earlier in vitro antiviral screening with unmodified acyclic nucleoside phosphonates in virus-infected cells has systematically underestimated the antiviral activity of most members of this class.
8. Status of clinical development of HDP-CDV (CMX001) and HDP-tenofovir (CMX157)
HDP-CDV was originally synthesized and evaluated as an oral treatment for smallpox under biodefense initiatives of the National Institutes of Allergy and Infectious Disease (NIAID) and the Department of Defense. However, as the antiviral evaluations proceeded, it became apparent that HDP-CDV was a broad spectrum antiviral with potent activity against orthopoxviruses (including variola), cytomegalovirus (wild type and drug resistant), herpes simplex virus, adenovirus, Epstein Barr virus, VZV, HHV-6, HHV-8, BK virus, human papilloma virus and orf virus. Under an NIAID grant to Chimerix Inc. of Durham, NC, HDP-CDV (CMX001) is currently in Phase I/II human clinical trials (www.ClinicalTrials.gov, NCT00780182 and NCT00793598).
8.1. HDP-CDV
Preclinical studies
HDP-CDV (CMX001) is being developed for prevention and treatment of smallpox and for various double stranded DNA virus infections. HDP-CDV antiviral activity against orthopoxviruses, various herpes viruses, adenoviruses, BK virus and orf virus has been described (Table 1). CMX001 is active against HCMV strains and drug resistant HCMV (Figure 3), all herpes group viruses (Williams-Aziz et al 2006) and human papilloma virus (HPV 18) (Painter et al, 2008a). CMX001 is orally bioavailable and results in substantial systemic exposure in rabbits, mice and monkeys. Doses of 4 and 10 mg/kg were effective in models of orthopoxvirus, CMV and adenovirus infections as discussed above. Safety studies in mice, rats and monkeys showed dose-dependent enteritis which was reversible after discontinuance of the drug. It is expected that reversible gastrointestinal toxicity will be the dose limiting toxicity of CMX001 (Painter et al, 2008a). Animals maintained normal clinical pathology values and, in contrast to CDV, there was no evidence of nephrotoxicity. CMX001 undergoes omega oxidation to a short chain carboxylic acid metabolite (CMX064) due to small intestine and liver cytochrome P450 enzymes. CMX064 is devoid of antiviral activity. Formation of CMX064 is greatest in the monkey and much lower in humans and mice (Painter et al, 2008a).
Clinical Studies
A placebo-controlled, dose-escalating Phase I safety pharmacokinetic study in healthy volunteers showed dose-proportional Cmax and AUC with single doses ranging from 0.1 to 1 mg/kg (Painter et al, 2008a; Painter et al, 2008b). Oxidative metabolism of CMX001 to CMX064 when matched by AUC was much lower in humans than in monkeys, rabbits or mice and multiple dose studies in man are currently in process (NCT00780182). It was concluded that plasma concentrations of CMX001 in normal human volunteers are as much as 20 fold greater than those observed in animals at comparable doses per kg of body weight (Painter et al, 2008b). BK virus infection is an important cause of renal allograft rejection (Hirsch et al, 2002) and there is no approved treatment. A Phase Ib/IIa study is currently in progress for a post-organ transplant study of CMX001 in patients exhibiting viruria with BK virus (NCT00793598).
8.2. HDP-tenofovir (CMX157)
Preclinical studies
The HDP ester of PMPA (tenofovir) was synthesized and evaluated in vitro against HIV-1 in MT-2 cells where it was found to have an EC50 of <0.00001 μM versus 0.65 μM for tenofovir. In PBMCs the EC50 was 0.012 μM compared with 3.20 μM for tenofovir (Painter et al, 2007). Selectivity indexes for CMX157 in MT-2 cells and PBMCs were >5 × 106 and 12,000, respectively. Oral administration of single or multiple doses of 10, 30 or 100 mg/kg to rats showed dose proportional plasma levels of CMX157. On day one the Tmax was noted at 0.5 to 2 hrs and with doses of 10, 30 and 100 mg/kg, the Cmax and AUC0-24hrs values were 85, 379 and 905 ng/ml and 442, 1830 and 5410 ng.hrs/ml. Similar results were noted at day seven. Tenofovir was the other significant metabolite in plasma. Toxicity studies in rats given 10, 30 and 100 mg/kg for 7 days showed no signs of toxicity, no alterations in body weight, no changes in clinical chemistry parameters and no mortality. On necropsy, there were no changes in organ weights or abnormalities of histopathology. CMX157 was tested against a panel of thirty NRTI resistant HIV variants and found to be substantially more active (295-472 fold) than tenofovir with low nanomolar EC50 values. This appears to be due in part to higher levels of tenofovir diphosphate (TFV-PP) in cells exposed to CMX157 compared to the levels observed with tenofovir exposure; in PBMCs incubated with 1μM CMX157, TFV-PP was 33 fold higher than in cells incubated with the same amount of tenofovir (Lanier et al, 2008). The increased activity may also be attributed, in part, to CMX157 binding directly to HIV virions and subsequently inhibiting viral replication in “untreated” cells. Neither direct binding to HIV nor subsequent inhibition of viral replication was observed with tenofovir (Lanier et al, 2009). CMX157 is currently in preclinical development for treatment of HIV infection.
9. Conclusions
HDP-CDV and HDP-(S)-HPMPA are alkoxyalkyl prodrugs of CDV and (S)-HPMPA which exhibit a several log increase in antiviral activity against orthopoxviruses and herpes group viruses because of their ability to traverse biological membranes more rapidly than CDV and (S)-HPMPA, resulting in much higher levels of their diphosphates in the cells. Alkoxyalkyl esters with 18-22 atoms are optimally active. These compounds are orally bioavailable and are highly efficacious in animal models of orthopoxvirus, cytomegalovirus and adenovirus infections. Esterification of acyclic nucleoside phosphonates with alkoxyalkyl groups produces increases their antiviral activity and oral bioavailability, and is not specific for any particular nucleobase or acyclic phosphonate side chain. Alkoxyalkyl ester prodrugs of a wide variety of acyclic nucleoside phosphonates exhibit multiple-log increases in antiviral activity. Certain acyclic nucleoside phosphonates which were reported to be inactive in vitro, were found to have substantial antiviral activity after conversion to their hexadecyloxypropyl or octadecyloxyethyl esters. Preclinical and clinical studies of HDP-CDV reveal no nephrotoxicity, in contrast to CDV and adefovir, where nephrotoxicity is dose-limiting.
HDP-CDV (CMX001) is in clinical development for orthopoxvirus and dsDNA virus infections, and studies to date have revealed no problematic adverse effects. Another compound of this class, HDP-tenofovir (CMX157), is also orally bioavailable, highly active against NRTI resistant variants of HIV-1 and has the unique attribute of inserting directly into the phospholipid bilayer of the virus and inhibiting subsequent replication of HIV virions. Animal safety studies have been benign. CMX157 is now in preclinical development for treatment of HIV infection. The alkoxyalkyl ester strategy for acyclic nucleoside phosphonates reviewed here is a general approach which provides many benefits for developing new orally available drugs for a range of viral infections.
Acknowledgments
Drs. Karel Wirtz, David Evans, Randall Lanier and James Beadle kindly reviewed portions of the manuscript. This work was supported by NIH/NIAID grants AI074057, AI076558, AI066499 and AI064615. Dr. Hostetler is a consultant and equity holder in Chimerix Inc. The terms of this relationship have been reviewed and approved by the University of California, San Diego in accordance with their conflict of interest policies. Ms. Stephanie McKinney assisted in preparation of the manuscript.
ABBREVIATIONS
- Ad5
adenovirus type 5
- ALT
alanine aminotransferase
- AUC
area under curve
- CDV
cidofovir
- CDVp
cidofovir monophosphate
- CDVpp
cidofovir diphosphate
- CMV
cytomegalovirus
- cPr-DAP
N6-cyclopropyl-2,6-diaminopurine
- CPX
cowpox
- dATP
deoxyadenosine triphosphate
- dCTP
deoxycytosine triphosphate
- ECTV
ectromelia virus
- GCV
ganciclovir
- HBV
hepatitis B virus
- HCMV
human cytomegalovirus
- HD
hexadecyl
- HDP
hexadecyloxypropyl
- HDP-CDV
hexadecyloxypropyl-cidofovir
- HDP-PMEDAP
hexadecyloxypropyl-9-(2-phosphono-methoxyethyl)-2,6-diaminopurine
- HDP-PMPA
hexadecyloxypropyl-(R)-9-(2-phosphono-methoxypropyl)adenine
- HHV
human herpes virus
- HIV
human immunodeficiency virus
- HPMP
3-hydroxy-2-(phosphonomethoxy)propyl
- HPMPA
(S)-9-(3-hydroxy-2-phosphono-methoxypropyl)adenine
- HPMPAp
HPMPA monophosphate
- HPMPApp
HPMPA diphosphate
- HPPMP
3-hydroxy-2-(phosphophosphonomethoxy)propyl
- HPMPC
(S)-1-(3-hydroxy-2-phosphonomethoxypropyl)cytosine (cidofovir)
- HPMPDAP
(S)-9-(3-hydroxyl-2-phosphono-methoxypropyl)-2,6-diaminopurine
- HPV
human papilloma virus
- IHD
the IHD strain of vaccinia virus
- LPC
lysophosphatidylcholine
- NRTI
nucleoside reverse transcriptase inhibitor
- ODBG
1-O-octadecyl-2-O-benzyl-sn-glycerol
- ODE-CDV
octadecyloxyethyl-cidofovir
- PBMC
peripheral blood mononuclear cells
- PC
phosphatidylcholine
- Pfu
plaque forming unit
- PME
2-(phosphonomethoxy)ethyl
- PMEA
9-(2-phosphonylmethoxyethyl)adenine
- PMEDAP
9-(2-phosphonomethoxyethyl)-2,6-diaminopurine
- PMP
3-hydroxy-2-(phosphonomethoxy)propyl
- PMPA
(R)-9-(2-phosphonomethoxypropyl)adenine
- PPen
5-phosphono-pent-2-en-1-yl
- PPM
phosphonopropoxymethyl
- PPMDAP
phosphonopropoxymethyl-2,6,-diaminopurine
- RPV
rabbitpox virus
- SCID
severe combined immunodeficiency
- TCID
tissue culture infectious dose
- VV
vaccinia virus
- VV-LED
vaccinia virus-Lederle
- VV-WR
vaccinia virus Western Reserve
- VZV
varicella zoster virus
- Wt
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MM, Rice AD, Moyer RW. Rabbitpox virus and vaccinia virus infection of rabbits as a model for human smallpox. J Virol. 2007;81:11084–11095. doi: 10.1128/JVI.00423-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. The increased antiviral activity of 1-O-hexadecyloxypropyl-cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol Pharmacol. 2003;63:678–681. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- Aldern KA, Beadle JR, Hostetler KY. Comparison of the Intracellular Metabolism of Cidofovir and (S)-HPMPA and Their Hexadecyloxypropyl Esters. Antiviral Res. 2006;70:126, A57. [Google Scholar]
- Andrei G, Gammon DB, Fiten P, De Clercq E, Opdenakker G, Snoeck R, Evans DH. Cidofovir resistance in vaccinia is linked to diminished virulence in mice. J Virol. 2006;80:9391–9401. doi: 10.1128/JVI.00605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J, Holy A, Jindrich J, Naesens L, Snoeck R, Schols D, De Clercq E. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethyoxypropyl)-2,6-diaminopurine. Antimicrob Agents Chemother. 1993;37:332–338. doi: 10.1128/aac.37.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker AS, De Clercq E, Taskintuna I, Keefe KS, Bergeron-Lynn G, Freeman WR. Influence of intravitreal injections of HPMPC and related nucleoside analogs on intraocular pressure in guinea pig eyes. Invest Ophthalmol Vis Sci. 1998;39:1233–1242. [PubMed] [Google Scholar]
- Beadle JR. Current Protocols in Nucleic Acid Chemistry. Unit 152 Supplement 29. Chapter 15. John Wiley & Sons, Inc; 2007. Synthesis of Cidofovir and (S)-HPMPA Ether Lipid Prodrugs. [DOI] [PubMed] [Google Scholar]
- Beadle JR, Rodriquez N, Aldern KA, Hartline C, Harden E, Kern ER, Hostetler KY. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication, in vitro. Antimicrob Agents Chemother. 2002;46:2381–2386. doi: 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle JR, Wan WB, Ciesla SL, Keith KA, Hartline C, Kern ER, Hostetler KY. Synthesis And Antiviral Evaluation of Alkoxyalkyl Derivatives of 9-(S)-(3-Hydroxy-2-phosphono-methoxypropyl)adenine Against Cytomegalovirus and Orthpoxviruses, in vitro. J Med Chem. 2006;49:2010–2015. doi: 10.1021/jm050473m. [DOI] [PubMed] [Google Scholar]
- Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. Oral activity of ether lipid prodrugs of cidofovir against experimental human cytomegalovirus infections. J Virol. 2004;190:499–503. doi: 10.1086/421912. [DOI] [PubMed] [Google Scholar]
- Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318:474–81. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Cheng L, Hostetler KY, Lee J, Koh HJ, Beadle JR, Bessho K, Toyoguchi M, Aldern K, Bovet JM, Freeman WR. Characterization of a Novel Intraocular Drug-Delivery System Using Crystalline Lipid Antiviral Prodrugs of Ganciclovir and Cyclic Cidofovir. Invest Ophthalmol Vis Sci. 2004;45:4138–4144. doi: 10.1167/iovs.04-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo H, Beadle JR, Chong Y, Trahan J, Hostetler KY. Synthesis of the 5-phosphono-pent-2-en-1-yl nucleosides: A new class of antiviral acyclic nucleoside phosphonates. Bioorg Med Chem. 2007a;15:1771–1779. doi: 10.1016/j.bmc.2006.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo H, Beadle JR, Kern ER, Prichard MN, Keith KA, Hartline CB, Trahan J, Aldern KA, Korba BE, Hostetler KY. Antiviral activities of novel 5-phosphono-pent-2-en-1-yl nucleosides and their alkoxyalkyl phosphonoesters. Antimicrob Agents Chemother. 2007b;51:611–615. doi: 10.1128/AAC.00444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesla SL, Trahan J, Winegarden KL, Aldern KA, Painter GR, Hostetler KY. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–171. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- Connelly MC, Robbins BL, Fridland A. Mechanism of uptake of the phosphonate analog (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC) in Vero cells. Biochem Pharmacol. 1993;46:1053–1057. doi: 10.1016/0006-2952(93)90670-r. [DOI] [PubMed] [Google Scholar]
- Cundy KC. Clinical pharmacokinetics of the antiviral nucleotide analogs cidofovir and adefovir. Clin Pharmacokinet. 1999;36:127–143. doi: 10.2165/00003088-199936020-00004. [DOI] [PubMed] [Google Scholar]
- Dal Pozzo F, Andrei G, Lebeau I, Beadle JR, Hostetler KY, De Clercq E, Snoeck R. In Vitro Evaluation Of The Anti-Orf Virus Activity Of Alkoxyalkyl Esters Of CDV, cCDV And (S)-HPMPA. Antiviral Res. 2007;75:52–57. doi: 10.1016/j.antiviral.2006.11.010. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem Pharmacol. 2007;73:911–922. doi: 10.1016/j.bcp.2006.09.014. [DOI] [PubMed] [Google Scholar]
- De Clercq E, Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov. 2005;4:28–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- De Clercq E, Sakuma T, Baba M, Pauwels R, Balzarini J, Rosenberg I, Holy A. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antiviral Res. 1987;8:261–272. doi: 10.1016/s0166-3542(87)80004-9. [DOI] [PubMed] [Google Scholar]
- Hanzal-Bayer MF, Hancock JF. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–2104. doi: 10.1016/j.febslet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Hartline CB, Gustin KM, Wan WB, Ciesla SL, Beadle JR, Hostetler KY, Kern ER. Activity of Ether Lipid Ester Prodrugs of Acyclic Nucleoside Phosphonates Against Adenovirus Replication In Vitro. J Infect Dis. 2005;191:396–9. doi: 10.1086/426831. [DOI] [PubMed] [Google Scholar]
- Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- Hirsch HH, Knowles W, Dickenmann M, Passweg J, Klimkait T, Mihatsch MJ, Steiger J. A prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N Engl J Med. 2002;347:488–496. doi: 10.1056/NEJMoa020439. [DOI] [PubMed] [Google Scholar]
- Hostetler KY. Synthesis and antiviral evaluation of broad spectrum, orally active analogs of cidofovir and other acyclic nucleoside phosphonates. In: De Clercq E, editor. Advances Antiviral Drug Design. Vol. 5. 2007. pp. 169–186. [Google Scholar]
- Hostetler KY, Aldern KA, Wan WB, Ciesla SL, Beadle JR. Alkoxyalkyl esters of (S)-HPMPA are potent inhibitors of HIV-1 replication, in vitro. Antimicrob Agents Chemother. 2006;50:2857–2859. doi: 10.1128/AAC.01223-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler KY, Beadle JR, Trahan J, Aldern KA, Owens G, Schriewer J, Melman L, Buller RM. Oral 1-O-octadecyl-2-O-benzyl-sn-glycero-3-cidofovir targets the lung and is effective against a lethal respiratory challenge with ectromelia virus in mice. Antiviral Res. 2007;73:212–218. doi: 10.1016/j.antiviral.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler KY, Hall LB. Phospholipase C activity of rat tissues. Biochim Biophys Res Comm. 1980;96:388–393. doi: 10.1016/0006-291x(80)91227-9. [DOI] [PubMed] [Google Scholar]
- Hostetler KY, Korba BE, Beadle JR. Synthesis and antiviral evaluation of hexadecyloxy-propyl-(S)-HPMPA against HBV and drug resistant HBV variants in vitro. Global Antiviral J. 2005;1(Suppl 2):31. Abst 29. [Google Scholar]
- Huggins JW, Baker RO, Beadle JR, Hostetler KY. Orally active ether lipid prodrugs of cidofovir for the treatment of smallpox. Antiviral Res. 2002;53:A66, 104. [Google Scholar]
- Izzedine H, Launay-Vacher V, Deray G. Antiviral drug-induced nephrotoxicity. Am J Kidney Dis. 2005;45:804–817. doi: 10.1053/j.ajkd.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kahn J, Lagakos S, Wulfsohn M, Cherng D, Miller M, Cherrington J, Hardy D, Beall G, Cooper R, Murphy R, Basgoz N, Ng E, Deeks S, Winslow D, Toole JJ, Coakley D. Efficacy and safety of adefovir dipivoxil with antiretroviral therapy: a randomized controlled trial. JAMA. 1999;282:2305–12. doi: 10.1001/jama.282.24.2305. [DOI] [PubMed] [Google Scholar]
- Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- Kern ER. Value of animal models to evaluate agents with potential activity against human cytomegalovirus. Transplant Proc. 1991;23(3 Suppl 3):152–5. [PubMed] [Google Scholar]
- Kern ER, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Quenelle DC. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir. Antimicrob Agents Chemother. 2004;48:3516–22. doi: 10.1128/AAC.48.9.3516-3522.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother. 2002;46:991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol MA, de Kroon AIPM, Killian JA, de Kruijff B. Transbilayer movement of phospholipids in biogenic membranes. Biochemistry. 2004;43:2673–2681. doi: 10.1021/bi036200f. [DOI] [PubMed] [Google Scholar]
- Kornbluth RS, Smee DF, Sidwell RW, Snarsky V, Evans DH, Hostetler KY. Mutations in the E9L Polymerase Gene of Cidofovir Resistant Vaccine WR are Responsible for the Drug-Resistant Phenotype. Antimicrob Agents Chemother. 2006;50:4038–4043. doi: 10.1128/AAC.00380-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krečmerová M, Holý A, Pohl R, Masojídková M, Andrei G, Naesens L, Neyts J, Balzarini J, De Clercq E, Snoeck R. Ester prodrugs of cyclic 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: synthesis and antiviral activity. J Med Chem. 2007;50:5765–5772. doi: 10.1021/jm0707166. [DOI] [PubMed] [Google Scholar]
- Lanier ER, Lampert B, Trost L, Painter GR, Almond M. Hexadecyloxypropyl-tenofovir (CMX157) has enhanced potency in vitro against NRTI resistant HIV relative to tenofovir and a favorable prelinical profile. Antiviral Ther. 2008;13(suppl 3):A6. Abst 4. [Google Scholar]
- Lanier ER, Lampert B, Robertson A, Almond M, Painter G. Hexadecyloxypropyl Tenofovir (HDP-TFV, CMX157) Associates Directly with HIV and Subsequently Inhibits Viral Replication in “Untreated” Cells. Proceedings of the 16th Conference on Reteroviruses and Opportunistic Infections; February 8-11, 2009; Montreal, Canada. 2009. Abstract J-117. [Google Scholar]
- Magee WC, Aldern KA, Hostetler KY, Evans DH. Cidofovir and (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenine are highly effective inhibitors of vaccinia virus DNA polymerase when incorporated into the template strand. Antimicrob Agents Chemother. 2008;52:586–597. doi: 10.1128/AAC.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee WC, Hostetler KY, Evans DH. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate. Antmicrob Agents Chemother. 2005;49:3153–3162. doi: 10.1128/AAC.49.8.3153-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa Y, Hostetler KY. Properties of phospholipase C isolated from rat liver lysosomes. J Biol Chem. 1980;255:646–652. [PubMed] [Google Scholar]
- Mul YM, van Miltenburg RT, De Clercq E, van der Vliet PC. Mechanism of inhibition of adenovirus DNA replication by the acyclic nucleoside triphosphate analogue (S)-HPMPApp: influence of the adenovirus DNA binding protein. Nucleic Acids Res. 1989;17:8917–29. doi: 10.1093/nar/17.22.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble S, Goa KL. Adefovir dipivoxil. Drugs. 1999;58:479–487. doi: 10.2165/00003495-199958030-00010. [DOI] [PubMed] [Google Scholar]
- Nilsson A, Borgstrom B. Absorption and metabolism of lecithin and lysolecithin by intestinal slices. Biochim Biophys Acta. 1967;137:240–254. doi: 10.1016/0005-2760(67)90100-2. [DOI] [PubMed] [Google Scholar]
- Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, De Clercq E, Korba BE, Aldern KA, Beadle JR, Hostetler KY. Evaluation of hexadecyloxypropyl-9-R-[2-(phosphonomethoxy)propyl]adenine, CMX157, a potential treatment for human imunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob Agents Chemother. 2007;51:3505–3509. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter GR, Hostetler KY. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol. 2004;22:423–427. doi: 10.1016/j.tibtech.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Painter GR, Lampert B, Trost LC, Frazer N, Robertson A. Development of HDP-cidofovir (CMX001) for double-stranded (dsDNA) virus infections. 21st International Conference Antiviral Research; Montreal. 2008b. Late breaker abstract. [Google Scholar]
- Painter GR, Trost LC, Lampert BM, Almond MR, Buller RM, Kern ER, Painter GP, Robertson AT, O’Mahony R. CMX001: Anti-smallpox agent, Anti-cytomegalovirus agent, viral polymerase inhibitor. Drugs of the Future. 2008a;33:655–661. [Google Scholar]
- Palu G, Stefanelli S, Rassu M, Parolin C, Balzarini J, De Clercq E. Cellular uptake of phosphonylmethoxyalkylpurine derivatives. Antivir Res. 1991;16:115–119. doi: 10.1016/0166-3542(91)90063-w. [DOI] [PubMed] [Google Scholar]
- Parker S, Touchette E, Oberle C, Almond M, Robertson A, Trost LC, Lampert B, Painter GR, Buller RM. Efficacy of therapeutic intervention with an oral ether-lipid analog of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res. 2008;77:39–49. doi: 10.1016/j.antiviral.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Hartline CB, Harden EA, Daily SL, Beadle JR, Valiaeva N, Kern ER, Hostetler KY. Inhibition of herpesvirus replication by hexadecyloxypropyl esters of purine and pyrimidine based phosphonomethoxyethyl nucleoside phosphonates. Antimicrob Agents Chemother. 2008 doi: 10.1128/AAC.00918-08. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, Collins DJ, Herrod BP, Keith KA, Trahan J, Beadle JR, Hostetler KY, Kern ER. Effect of oral treatment with hexadecyloxypropyl-[(S)-9-(3-hydroxy-2-phosphonylmethoxy-propyl)adenine] [(S)-HPMPA] or octadecyloxyethyl-(S)-HPMPA on cowpox or vaccinia virus infections in mice. Antimicrob Agents Chemother. 2007;51:3940–3947. doi: 10.1128/AAC.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, Collins DJ, Hostetler KY, Beadle JR, Wan WB, Kern ER. Oral treatment of cowpox and vaccinia infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother. 2004;48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, Collins DJ, Pettway LR, Hartline CB, Beadle JR, Wan WB, Hostetler KY, Kern ER. Effect of oral treatment with (S)-HPMPA, HDP-(S)-HPMPA or ODE-(S)-HPMPA on replication of murine cytomegalovirus or human cytomegalovirus in animal models. Antiviral Res. 2008;79:133–135. doi: 10.1016/j.antiviral.2008.01.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, Prichard MN, Keith KA, Hruby DE, Jordan R, Painter GR, Robertson A, Kern ER. Synergistic efficacy of the combination of ST-246 with CMX001 against orthopoxviruses. Antimicrob Agents Chemother. 2007b;51:4118–4124. doi: 10.1128/AAC.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggers RJ, Pomorski T, Holthuis JCM, Kalin N, van Meer G. Lipid traffic: the ABC of transbilayer movement. Traffic. 2002;1:226–234. doi: 10.1034/j.1600-0854.2000.010305.x. [DOI] [PubMed] [Google Scholar]
- Randhawa P, Farasati NA, Shapiro R, Hostetler KY. Ether lipid derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob Agents Chemother. 2006;50:1564–1566. doi: 10.1128/AAC.50.4.1564-1566.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa P, Zemlicka J, Sauerbrei A, Meier C, Hostetler KY, Beadle JR, Farasati NA, Huang Y, Bradley M. Anti-BK virus activity of nucleoside analogs. Antimicrob Agents Chemother. 2008;52:1519–1521. doi: 10.1128/AAC.01241-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Styrt B, McCune S. FDA perspective on antivirals against biothreats: communicate early and often. Antiviral Res. 2008;78:60–63. doi: 10.1016/j.antiviral.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Ruiz JC, Aldern KA, Beadle JR, Hartline CB, Kern ER, Hostetler KY. Synthesis and antiviral evaluation of alkoxyalkyl esters of phosphonopropoxymethyl-guanine and phosphonopropoxymethyl-diaminopurine. Antiviral Chem Chemother. 2006;17:89–95. doi: 10.1177/095632020601700204. [DOI] [PubMed] [Google Scholar]
- Ruiz JC, Beadle JR, Aldern KA, Keith KA, Hartline CB, Kern ER, Hostetler KY. Synthesis and antiviral evaluation of alkoxyalkyl phosphate conjugates of cidofovir and adefovir. Antiviral Res. 2007;75:87–90. doi: 10.1016/j.antiviral.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scow RO, Stein Y, Stein O. Incorporation of dietary lecithin and lysolecithin into lymph chylomicrons in the rat. J Biol Chem. 1967;242:4919–4924. [PubMed] [Google Scholar]
- Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–95. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GN. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Smee DF, Sidwell RW, Kefauver D, Bray M, Huggins JW. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrob Agents Chemother. 2002;46:1329–133. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Wong M-H, Bailey KW, Beadle JR, Hostetler KY, Sidwell RW. Effects of highly active antiviral substances on lethal vaccinia virus (IDH strain) respiratory infections in mice. Int J Antimicrob Agents. 2004;23:430–437. doi: 10.1016/j.ijantimicag.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Smee DF, Wandersee MK, Bailey KW, Hostetler KY, Holy A, Sidwell RW. Characterization and treatment of cidofovir-resistant vaccinia (WR strain) virus infections in cell culture and in mice. Antiviral Chem Chemother. 2005;16:203–211. doi: 10.1177/095632020501600306. [DOI] [PubMed] [Google Scholar]
- Snoeck R, Sakuma T, De Clercq E, Rosenberg I, Holy A. (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob Agents Chemother. 1988;32:1839–1844. doi: 10.1128/aac.32.12.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammewar AM, Cheng L, Hostetler KY, Falkenstein I, Beadle JR, Barron EC, Kozak I, Freeman WR. Intraocular properties of an alkoxyalkyl derative of cyclic 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine, an intravitreally injectable anti-HCMV drug in rabbit and guinea pig. J Ocular Pharmacol Therap. 2007;23:433–444. doi: 10.1089/jop.2007.0018. [DOI] [PubMed] [Google Scholar]
- Toth K, Spencer JF, Dhar D, Sagartz JE, Buller RML, Painter GR. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in permissive immunosuppressed animal models. Proc Nat Acad Sci. 2008;105:7293–7297. doi: 10.1073/pnas.0800200105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiaeva N, Beadle JR, Aldern KA, Trahan J, Hostetler KY. Synthesis and antiviral evaluation of alkoxyalkyl esters of acyclic purine and pyrimidine nucleoside phosphonates against HIV-1 in vitro. Antiviral Res. 2006;72:10–19. doi: 10.1016/j.antiviral.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Van Deenen LLM. Topology and dynamics of phospholipids in membranes. FEBS letters. 1981;123:3–15. doi: 10.1016/0014-5793(81)80007-5. [DOI] [PubMed] [Google Scholar]
- Van den Bosch H. Phospholipases. In: Hawthorne JN, Ansell GB, editors. “Phospholipids,” New Comprehensive Biochemistry. Vol. 4. Elsevier Biomedical; Amsterdam: 1982. pp. 313–357. [Google Scholar]
- Wan WB, Beadle JR, Hartline CB, Kern ER, Ciesla SL, Valiaeva N, Hostetler KY. Comparison of the antiviral activity of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro. Antimicrob Agents Chemother. 2005;49:656–662. doi: 10.1128/AAC.49.2.656-662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Aziz SL, Hartline CB, Harden EA, Daily SM, Prichard MN, Kushner NL, Beadle JR, Wan WB, Hostetler KY, Kern ER. Comparative Activity Of Lipid Esters Of Cidofovir And Cyclic Cidofovir Against Replication Of Herpesviruses In Vitro. Antimicrob Agents Chemother. 2005;49:3724–3733. doi: 10.1128/AAC.49.9.3724-3733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz KWA. Phospholipid transfer proteins in perspective. FEBS letters. 2006;580:5436–5441. doi: 10.1016/j.febslet.2006.06.065. [DOI] [PubMed] [Google Scholar]
- Xiong X, Smith JL, Chen MS. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob Agents Chemother. 1997;41:594–599. doi: 10.1128/aac.41.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J Virol. 2005;79:13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachowski A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J. 1993;294:1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]