

SUMMARY
Salmonella enterica serotype Typhimurium thrives in the lumen of the acutely inflamed intestine, which suggests that this pathogen is resistant to antimicrobials encountered in this environment. However, the identity of these antimicrobials and the corresponding bacterial resistance genes remains elusive. Here we show that enteric infection with S. Typhimurium evoked marked interleukin (IL)–22/IL-17 mediated induction in intestinal epithelial cells of lipocalin-2, an antimicrobial protein that prevents bacterial iron acquisition. Lipocalin-2 accumulated in the intestinal lumen of rhesus macaques during S. Typhimurium infection. Resistance to lipocalin-2, mediated by the iroBCDE iroN locus, conferred a competitive advantage upon the S. Typhimurium wild-type in colonizing the inflamed intestine of wild-type, but not of lipocalin-2 deficient mice. These data support that resistance to lipocalin-2 defines a specific adaptation to growth in the inflamed intestine.
INTRODUCTION
Salmonella enterica serotype Typhimurium (S. Typhimurium) causes a localized gastroenteritis in humans, characterized by acute intestinal inflammation and diarrhea. The pathogen encodes two type III secretion systems (T3SS-1 and T3SS-2) that are important for eliciting intestinal inflammation (Hapfelmeier et al., 2005; Tsolis et al., 1999). The initiation of inflammatory responses in tissue requires direct contact between bacteria and host cells, including epithelial cells, macrophages and dendritic cells. Macrophages and dendritic cells infected with S. Typhimurium are a source of cytokines, including interleukin [IL]-23 and IL-18, that help to amplify responses in tissue (Godinez et al., 2009; Srinivasan et al., 2007). For example, IL-23 stimulates T cells in the intestinal mucosa to produce IL-17 and IL-22 (Godinez et al., 2009), two cytokines whose expression is among the ones most prominently induced during S. Typhimurium infection (Godinez et al., 2008; Raffatellu et al., 2008). The cytokine storm ensuing from the amplification of inflammatory responses in tissue results in the activation of antimicrobial responses in the intestinal mucosa.
One important component of the antimicrobial response induced by the IL-23/IL-17 axis is the recruitment of neutrophils (Godinez et al., 2008; Godinez et al., 2009; Raffatellu et al., 2008). The extravasation of neutrophils into the mucosa provides a formidable antibacterial defense to prevent S. Typhimurium dissemination. Support for this notion comes from clinical data, showing that neutropenia is a risk factor for bacteremia with non-typhoidal Salmonella serotypes (Noriega et al., 1994). Thus S. Typhimurium appears to be susceptible to this arm of the host defense.
A second component of the antimicrobial response is the production of antimicrobial proteins in the intestinal mucosa (Godinez et al., 2009; Raffatellu et al., 2008; Zheng et al., 2008), whose release into the intestinal lumen may be responsible for the dramatic changes in the microbiota observed during inflammation (Lupp et al., 2007; Stecher et al., 2007). The cytokines IL-17 and IL-22 are required for the production in the inflamed intestine of antimicrobials (Godinez et al., 2009; Raffatellu et al., 2008), including lipocalin-2, a protein that inhibits bacterial growth by interfering with the acquisition of an essential nutrient, iron (Berger et al., 2006; Flo et al., 2004; Goetz et al., 2002). However, the cells targeted by IL-17 and IL-22 in vivo to induce production of these antimicrobials have not been identified. S. Typhimurium appears to be resistant against this arm of the host defense, because its numbers in the intestinal lumen increase dramatically during inflammation, resulting in increased fecal oral transmission (Barman et al., 2008; Lawley et al., 2008; Stecher et al., 2007). We thus hypothesized that, in addition to virulence mechanisms to induce intestinal inflammation, S. Typhimurium should possess virulence mechanisms to survive the ensuing antimicrobial responses in the intestinal lumen. While the virulence factors involved in triggering intestinal inflammation, such as T3SS-1 and T3SS-2, are well characterized, the identity of the antimicrobial responses potentially encountered in the lumen of the inflamed intestine and the identity of the corresponding bacterial resistance genes are currently unknown.
To gain further insights into this “inflammation-adapted” pathogenic lifestyle of S. Typhimurium, we set out to identify antimicrobials released by intestinal epithelial cells into the intestinal lumen during inflammation. After identifying one such antimicrobial we determined whether the corresponding S. Typhimurium resistance genes conferred an advantage during bacterial growth in the inflamed intestine.
RESULTS
Gene expression profile induced by IL–17 and IL–22 in human intestinal model epithelia
To identify antimicrobial responses elicited by IL-17 and IL-22 in the intestinal epithelium, human colonic cancer epithelial (T84) cells were polarized and basolaterally stimulated with IL–17 or IL–22. Expression of CCL20, a chemokine gene known to be induced by IL–17 in lung epithelial cells (Kao et al., 2005) was found to be induced optimally 4 hours after stimulation with IL–17 or IL–22 (data not shown). To determine the complete molecular profile of epithelial responses, RNA from four replicates was isolated 4 hours after stimulation of T84 cells with IL–22 or IL–17 and global gene expression profiles were elucidated by microarray analysis.
In general, IL–22 produced more robust responses in T84 cells than IL–17 (Supplementary Figure 1). For example, the number of genes up regulated 2-fold or more was 849 for IL–22 treatment compared to only 62 for IL–17 treatment. A table containing a complete listing of genes with significantly (P < 0.05) altered expression in T84 epithelial cells after stimulation with IL–17 or IL–22 can be accessed at the Gene Expression Omnibus database [GSE11345].
Meta analysis of in vitro and in vivo gene expression profiles suggests a contribution of epithelial cells to host defense responses during inflammation
To identify those antimicrobial responses induced in vitro in T84 cells that are also observed in the inflamed intestine in vivo, we performed meta analysis of the overlap in up regulated gene transcription (>2 fold) between T84 cells stimulated with IL–22 and the ileal mucosa of rhesus macaques infected with S. Typhimurium (Figure 1). The in vivo gene expression profile analyzed in this study had previously been generated using an adult healthy rhesus macaque that had undergone ligated ileal loop surgery (Raffatellu et al., 2008). Two ligated ileal loops had been inoculated with either sterile culture medium (mock infection) or with S. Typhimurium and 5 hours later both loops had been surgically removed to isolate RNA for gene expression profiling (Raffatellu et al., 2008). Meta analysis revealed a substantial overlap between transcripts whose levels were increased in the ileal mucosa of a rhesus macaque during S. Typhimurium infection with those whose expression was induced in human T84 epithelial cells upon stimulation with IL–22 (302 transcripts) (Figure 1A). A functional category that was statistically over-represented included genes involved in defense responses, encoding antimicrobials (LCN2, NOS2 and MUC4) and cytokines (CCL20) whose expression was increased most dramatically both in vitro in IL-22 treated T84 cells and during S. Typhimurium infection in vivo (Figure 1A). These data supported the hypothesis that IL–22 induced expression of antimicrobials in epithelial cells during S. Typhimurium infection in the intestinal mucosa.
Figure 1.
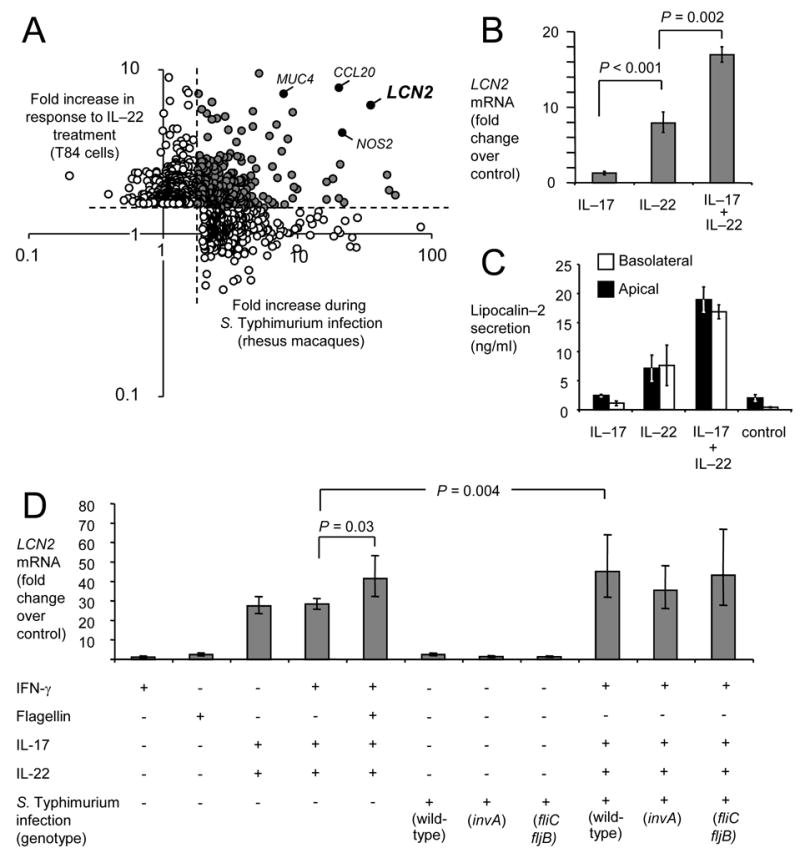
Expression of lipocalin–2 by polarized intestinal model epithelia upon stimulation with IL–17 and/or IL–22. (A) Meta analysis of the overlap (gray circles) between increases (>2 fold) in transcripts in the ileal mucosa of rhesus macaques during S. Typhimurium infection (x axis) and induction of gene expression observed in human T84 epithelial cells after treatment with IL–22 (y axis). The identity of selected genes (closed circles) is indicated. (B and D) Detection of LCN2 expression in polarized T84 cells stimulated for 4 hours with reagents listed below each graph using quantitative real-time PCR. (C) Secretion of lipocalin–2 by T84 cells into the apical (open bars) or basolateral (closed bars) compartment detected by ELISA. (B–D) Data represent means ± standard deviation from at least three different experiments.
IL–17 and IL–22 synergize to induce lipocalin-2 production in intestinal epithelial cells
Transcripts encoding the antimicrobial lipocalin-2 were among the ones most prominently increased both in vitro after IL-22 stimulation and in vivo after S. Typhimurium infection (Figure 1 A). Thus, after validating results from gene expression profiling for selected genes by real-time PCR (Supplementary Figure 2), we sought to further investigate whether intestinal epithelial cells may be a source of lipocalin-2 during S. Typhimurium infection. First, we verified that lipocalin-2 is an antimicrobial produced by intestinal epithelial cells. Quantification of changes in gene expression by real-time PCR confirmed that stimulation of T84 cells with IL–22 resulted in an induction of LCN2 expression (7.9 fold) (Figure 1B). Stimulation with IL–17 alone did not alter LCN2 transcription (1.3 fold change) but further increased LCN2 transcription when added in combination with IL–22 (17 fold). We next determined protein secretion by ELISA. Non-stimulated T84 cells secreted a small amount of lipocalin-2 apically (approximately 2ng/ml), while the amount detected in the basolateral compartment was negligible. Stimulation of T84 cells with IL–22, but not with IL–17, resulted in apical and basolateral secretion of lipocalin-2 at levels above those detected with non-stimulated controls. Stimulation with both IL–17 and IL–22 resulted in a significant (P < 0.05) further increase of lipocalin-2 secretion both apically and basolaterally, reaching substantial concentrations (approximately 20ng/ml) in both compartments (Figure 1C). In contrast, CCL20 was secreted exclusively into the basolateral compartment after cytokine stilmulation of T84 cells (Supplementary figure 2). Although IL–17 by itself did not markedly alter LCN2 transcription, our data suggested that this cytokine synergized with IL–22 in controlling production of lipocalin-2 in the intestinal epithelium.
Because lipocalin-2 was highly induced during S. Typhimurium infection in vivo, we next investigated whether expression of LCN2 was induced either by cytokines (i.e. IL-17 and IL-22) present in the inflamed intestine, by direct contact of bacteria with epithelia cells or by a combination of both mechanisms. Basolateral stimulation of T84 cells with purified flagellin or infection of T84 cells with the S. Typhimurium wild-type strain, an invasion deficient strain (invA mutant) or a non-flagellated strain (fliC fljB mutant) did not result in marked induction in LCN2 expression (Figure 1D). Treatment of T84 cells with IFN–γ did not increase LCN2 transcription. The marked increase in LCN2 transcription observed after basolateral stimulation of T84 cells with IL-17 and IL-22 was modestly increased by basolateral stimulation with purified flagellin (P = 0.03) or by S. Typhimurium infection (P = 0.004). These data suggested that induction of LCN2 transcription was mediated largely through stimulation with IL-17 and IL-22. Although direct interaction between bacteria and host cells was not sufficient to induce LCN2 transcription, these interactions could further increase LCN2 mRNA levels induced by treatment with IL-17 and IL-22.
Lipocalin–2 produced by epithelial cells after stimulation with IL–17 and IL–22 inhibits bacterial growth
To investigate the biological significance of the IL–17/IL–22–mediated lipocalin-2 production by intestinal epithelial cells, we determined whether the protein was produced at levels that exhibited an antimicrobial activity. Lipocalin–2 specifically binds enterochelin, a small molecular weight iron chelator (siderophore) produced by many members of the Enterobactericeae. As a result, lipocalin-2 exhibits a bacteriostatic effect on bacteria that depend exclusively on enterochelin to acquire iron, an essential trace element, during growth in the host (Berger et al., 2006; Flo et al., 2004; Goetz et al., 2002). The iroBCDE iroN gene cluster of S. Typhimurium encodes proteins involved in the biosynthesis and uptake of salmochelin, a glycosylated derivative of enterochelin (Baumler et al., 1998; Baumler et al., 1996; Bister et al., 2004; Hantke et al., 2003; Rabsch et al., 1999; Zhu et al., 2005). Salmochelin is not bound by lipocalin–2 and its production therefore renders S. Typhimurium lipocalin-2 resistant. However, in the absence of a functional iroBCDE iroN gene cluster, S. Typhimurium produces enterochelin as its sole siderophore and is lipocalin-2 sensitive (Crouch et al., 2008; Fischbach et al., 2006).
Growth of the S. Typhimurium wild-type (lipocalin–2 resistant) and a S. Typhimurium iroBC mutant (lipocalin–2 sensitive) were compared in medium collected either from non-stimulated T84 cells (control) or from T84 cells after stimulation with IL–17 and IL–22 (Figure 2). No differences between the S. Typhimurium wild-type and the iroBC mutant were observed in control media, however growth of the iroBC mutant was significantly (P < 0.05) reduced in media in which lipocalin-2 production had been elicited by stimulation with IL–17 and IL–22 (Figure 2A). These data suggested that the amount of lipocalin–2 produced by intestinal epithelial cells upon IL–17/IL–22 stimulation was sufficient to reduce growth of the iroBC mutant in vitro.
Figure 2.

The iroBC locus confers lipocalin-2 resistance by overcoming iron limitation. (A) Growth of the S. Typhimurium wild type or an isogenic iroBC mutant in medium from collected from polarized T84 cells that had either not been stimulated with cytokines (control, open bars) or had been stimulated with IL–17 and IL–22 for 24 hours (IL–17 + IL–22, closed bars). Bacterial numbers were determined 5 hours after inoculation with 103 bacteria. (B) Bacterial growth in rich medium (LB broth). (C) Bacterial growth in tissue culture medium (DMEM) supplemented with lipocalin-2 (LCN), ferrioxamine B (FoxB) or containing no supplements. ND, not determined. All data represent means ± standard deviation from at least three different experiments.
A mutation in iroBC did not impair bacterial growth in rich media (Luria-Bertani broth) (Figure 2B). Tissue culture medium was spiked with lipocalin-2 to confirm that the presence of this antimicrobial was responsible for the growth defect observed with the iroBC mutant. The presence of lipocalin-2 significantly (P < 0.05) reduced growth of the iroBC mutant, while growth of the S. Typhimurium wild-type was not affected (Figure 2C). Complementation of the iroBC mutant with the cloned iroBCDE iroN gene cluster restored resistance to lipocalin-2, as indicated by equal growth in tissue culture medium in the presence or absence of lipocalin-2 (Figure 2C). The lipocalin-2 mediated growth inhibition of the iroBC mutant could be prevented when iron was supplied in form of ferrioxamine B (iron-desferal), a siderophore that can be internalized by the FoxA outer membrane receptor of S. Typhimurium (Figure 2C). Collectively, these data suggested lipocalin-2 inhibited growth of the iroBC mutant through iron sequestration.
S. Typhimurium infection induces LCN2 expression in intestinal epithelial cells of rhesus macaques and secretion of lipocalin-2 into the intestinal lumen
To investigate the in vivo relevance of lipocalin-2 production by T84 cells, we examined the contribution of epithelial cells expressing this antimicrobial protein during S. Typhimurium infection in a relevant host. To this end, we first localized the LCN2 transcripts in the ileal mucosa of rhesus macaques using in situ hybridization (Figure 3). LCN2 transcripts localized to epithelial cells lining the villi and crypts, with intense staining in tissue from S. Typhimurium–infected ileal loops with a LCN2–specific cDNA probe (Figure 3A). These data suggested that LCN2 was mainly expressed by intestinal epithelial cells during S. Typhimurium infection.
Figure 3.

Spatial and quantitative analysis of lipocalin–2 expression in the ileal mucosa of rhesus macaques during S. Typhimurium infection. (A) Detection of LCN2 transcripts (brown precipitate) in representative sections from a S. Typhimurium–infected loop (left panel) and a mock–infected loop (right panel) from the same animal by in situ hybridization. Hybridization with the empty plasmid vector used for cloning the LCN2 probe was performed as a negative control (middle panel). All sections were counterstained with hematoxylin. (B) Absolute transcript levels of LCN2 and ACT1 in mock–infected (closed bars) and S. Typhimurium–infected loops (open bars) from 4 rhesus macaques. Data represent mean mRNA copy numbers per ng of RNA ± standard error. Statistically significant (P < 0.05) differences are indicated by P values. (C–E) Detection of lipocalin-2 (brown precipitate) in representative sections from a S. Typhimurium–infected loop (C and D) and a mock–infected loop (E) from the same animal using immunohistochemistry with rabbit anti–rhesus lipocalin–2 antiserum (α–lipocalin–2). Immunohistochemistry with pre–immune serum was performed as a negative control (D). (F) Lipocalin–2 secretion in mock–infected (closed bars) and S. Typhimurium-infected loops (open bars) from 4 rhesus macaques. Data represents the total amount of lipocalin–2 (ng) secreted into an approximately 5 cm long segment of the ileum. Statistically significant (P < 0.05) differences are indicated by P values.
Next, we determined the absolute number of LCN2 transcripts using quantitative real-time PCR. For each rhesus macaque (n= 4), a tissue sample from a mock–infected loop and from a S. Typhimurium infected loop were processed for mRNA isolation. Transcripts encoding β-actin (ACT1) were detected at similar levels in both treatment groups, averaging approximately 20,000 copies/ng RNA in samples from mock-infected and S. Typhimurium–infected loops (Figure 3B). Based on theoretical assumptions about the RNA content of 106 cells (5 – 8μg)(Ausubel et al., 1994), these data would predict that ACT1 was present on average in 100 – 160copies/cell. Our data were thus within the range of previous measurements by real–time PCR indicating that human leukocytes contain between 84 and 491copies of ACT1 per cell (Lupberger et al., 2002). LCN2 transcripts averaged 1,400 copies/ng RNA in samples from mock-infected loops, but the copy number was significantly (P = 0.03) increased after S. Typhimurium infection, averaging 48,000 copies/ng RNA. Collectively, these data suggested that during S. Typhimurium infection, the LCN2 gene was expressed at a very high level in the epithelium of the ileal mucosa.
We next studied the production of lipocalin-2 in situ using immunohistochemistry (Figure 3C–E). Rabbit polyclonal antibody derived from recombinant rhesus lipocalin-2 was generated. Immuno staining of formalin fixed tissue revealed that after S. Typhimurium infection, lipocalin–2 was abundant in intestinal epithelial cells and in the underlying lamina propria. These data suggested that lipocalin-2 protein is produced and secreted in tissue during the course of an infection with S. Typhimurium. Next, we wanted to detect the amount of lipocalin–2 secreted into the intestinal lumen in vivo. An ELISA was developed to detect the amount of lipocalin–2 secreted into the luminal fluid previously collected from ligated ileal loops of rhesus macaques 8 hours after inoculation with S. Typhimurium (n=4) or sterile culture medium (n=4). Luminal contents of S. Typhimurium infected loops contained on average 419ng of lipocalin–2, which was significantly higher (P = 0.007) than the 64ng lipocalin–2 detected in luminal contents of mock-infected loops (Figure 3F). Collectively, these data identified lipocalin-2 production as an antimicrobial response encountered in the lumen of the inflamed intestine.
Lipocalin-2 resistance confers an advantage during growth of S. Typhimurium in the inflamed intestine
After identifying lipocalin-2 as an antimicrobial response encountered in the intestinal lumen, we wanted to determine whether the corresponding S. Typhimurium resistance genes would confer an advantage during bacterial growth in the inflamed intestine. To establish cause and effect using lipocalin-2 deficient animals, we used the streptomycin pretreated mouse model for these studies. S. Typhimurium infection of streptomycin pretreated mice results in acute inflammation of the cecal mucosa (Barthel et al., 2003), which is accompanied by overgrowth of S. Typhimurium in the lumen of the large intestine (Que et al., 1986; Stecher et al., 2007).
We first investigated whether inactivation of iroN, encoding the IroN outer membrane salmonchelin receptor protein, would reduce the ability of S. Typhimurium to grow in the intestinal lumen of streptomycin pretreated wild-type mice (C57BL/6) (Figure 4). The iroN mutant was sensitive to lipocalin-2 mediated iron deprivation in vitro (Figure 2C). At 48 hours after infection of streptomycin pretreated mice, the iroN mutant was recovered in reduced numbers from colon contents than the S. Typhimurium wild-type strain (Figure 4A). However, there was a large animal-to-animal variation in bacterial numbers recovered from colon contents. We next investigated whether a lipocalin-2 sensitive S. Typhimurium mutant (iroN mutant) was able to compete with the S. Typhimurium wild type in vivo. Using this experimental design, the ability of the iroN mutant and the wild type to colonize the intestine is compared in each individual animal, thereby reducing the effect of animal-to-animal variation. Streptomycin pretreated wild-type mice were inoculated intragastrically with sterile LB broth (mock infection) or with a 1:1 mixture of the S. Typhimurium wild type and the iroN mutant. Compared to mock-infected mice, mice inoculated with the S. Typhimurium strain mixture exhibited markedly increased transcript levels of cytokine genes, including Il17, Il22, keratinocyte-derived cytokine (Kc, Cxcl1) and tumor necrosis factor α (Tnfa) at 48 hours after infection (Figure 5A–D). Furthermore, S. Typhimurium infection caused marked inflammatory changes in the submucosa and mucosa (Figure 6). These data confirmed that S. Typhimurium infection of streptomycin-pretreated mice was associated with acute cecal inflammation. Importantly, transcript levels of Lcn2 were markedly increased (53-fold) in S. Typhimurium-infected mice compared to mock-infected mice (Figure 7A). Expression of lipocalin-2 in the cecal mucosa was assessed at the protein levels using Western blot. While no lipocalin-2 was detectable in the cecal mucosa of mock-infected mice, the protein was highly abundant 48 hours after S. Typhimurium infection (Figure 7B). The S. Typhimurium wild type was recovered in 7-fold higher numbers from intestinal contents 48 hours after infection than the iroN mutant (Figure 4B), suggesting that IroN-mediated salmochelin uptake constituted an advantage during growth in the inflamed cecum.
Figure 4.

Growth of iroN deficient and iroN proficient S. Typhimurium strains in the murine intestine in vivo. (A) Recovery of S. Typhimurium from colon contents 48 hours after infection of mice with the S. Typhimurium wild-type or an iroN mutant. Each circle represent bacterial numbers recovered from an individual animal. Bars indicate the geometric mean. (B) Recovery of S. Typhimurium 48 hours after competitive infection with the indicated bacterial strains. Bars indicate the average competitive index (i.e. ratio of wild-type/iroN or ratio of invA spiB/invA spiB iroN) of bacteria recovered from colon contents of wild-type mice or Lcn2−/− mice 48 hours after infection with a 1:1 mixture the respective S. Typhimurium strains. Data represent geometric means ± standard error.
Figure 5.

Inflammatory responses elicited in the cecum of streptomycin-pretreated mice 48 hours after S. Typhimurium infection assessed by measuring cytokine transcription. Transcript levels of Il17a (A), Il22 (B), Kc (C) and Tnfa (D) in wild type mice (wt, gray bars) or lipocalin-2 deficient mice (Lcn2−/−, black bars) 48 hours after competitive infection (wild-type vs. iroN or invA spiB vs. invA spiB iroN) was measured by quantitative real-time PCR. Bars represent fold-changes (geometric means) in mRNA levels compared to a group of mock-infected wild type mice ± standard error.
Figure 6.

Histopathology of the cecum from mock-infected mice, or mice infected with a mixture of the indicated S. Typhimurium strains. (A) Histopathological appearance of the cecum from representative animals in each group. All images were taken from hematoxylin and eosin stained cecal sections at the same magnification (100x). Note marked edema in the submucosa (SM) of mice infected with S. Typhimurium wild-type vs. iron mutant. M, mucosa; L, lumen. (B) Pathology score determined by blinded examination of cecal sections under high power magnification. Each bar represents an individual animal.
Figure 7.
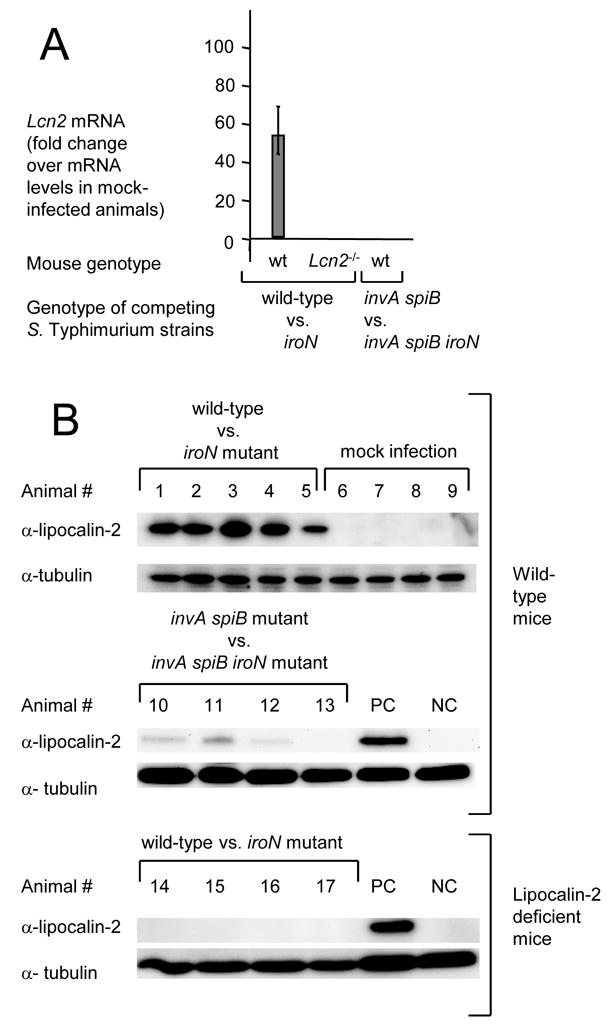
Lipocalin-2 expression detected by quantitative real-time PCR (A) or by Western blot (B) in the cecal mucosa of streptomycin-pretreated mice 48 hours after S. Typhimurium infection. (A) Bars represent fold-changes (geometric means) in mRNA levels compared a group of mock-infected wild type mice ± standard error. (B) Lipocalin-2 expression in protein extracts from the cecal mucosa of individual mice was detected with anti-mouse lipocalin-2 antibody (α-lipocalin-2). Detection of tubulin with anti-mouse tubulin antibody (α-tubulin) served as a loading control for each sample. PC, positive control sample from animal #3; NC, negative control sample from animal #8.
To investigate whether the presence of the iroN gene confers an advantage only in the presence of intestinal inflammation, we repeated the above experiments with S. Typhimurium strains that were unable to elicit this host response. The induction of cecal inflammation in streptomycin-pretreated mice requires the presence of functional T3SS-1 and T3SS-2 secretion systems (Hapfelmeier et al., 2005). We constructed a S. Typhimurium strain carrying mutations that prevent functionality of T3SS-1 (invA) and T3SS-2 (spiB). Streptomycin pretreated wild-type mice (C57BL/6) were inoculated intragastrically with sterile LB broth (mock infection) or with a 1:1 mixture of the S. Typhimurium invA spiB mutant and a S. Typhimurium invA spiB iroN mutant. Compared to mock infected mice, no marked increases in cytokine transcription (Il17, Il22, Kc, and Tnfa) were detected in the cecal mucosa of mice infected with a mixture of the S. Typhimurium invA spiB mutant and the invA spiB iroN mutant (Figure 5) and no marked inflammatory changes were detected microscopically (Figure 6). Increased expression of Lcn2 transcription in the cecal mucosa was not detected by real-time PCR and little lipocalin-2 expression was detected by Western blot (Figure 7). These data confirmed that infection with T3SS-1/T3SS-2 deficient S. Typhimurium strains does not trigger marked cecal inflammation in streptomycin-pretreated mice. Importantly, the S. Typhimurium invA spiB mutant and the invA spiB iroN mutant were recovered in equal numbers from intestinal contents 48 hours after infection (Figure 4B). These data suggested that in the absence of intestinal inflammation, the IroN-mediated salmochelin uptake did not provide a significant selective advantage during growth in the intestine.
To investigate whether intestinal inflammation provides a selective advantage for S. Typhimurium strains carrying the IroN outer membrane receptor because expression of lipocalin-2 is highly induced in the inflamed intestine, above experiments were repeated using lipocalin-2-deficient mice. Streptomycin pretreated lipocalin-2-deficient mice were inoculated intragastrically with sterile LB broth (mock infection) or with a 1:1 mixture of the S. Typhimurium wild type and the S. Typhimurium iroN mutant. Inoculation with a mixture of the S. Typhimurium wild type and the S. Typhimurium iroN mutant resulted in acute cecal inflammation, as indicated by markedly increased transcript levels of Il17, Il22, Kc, and Tnfa at 48 hours after infection and inflammatory changes in tissue (Figures 5 and 6). As expected, lipocalin-2 expression was detected neither by real-time PCR nor by Western blot in the ceca of any of the lipocalin-2-deficient mice (Figure 7). Importantly, the S. Typhimurium wild type and the iroN mutant were recovered in similar numbers from intestinal contents 48 hours after infection (Figure 4B). Furthermore, the ratio of wild type to iroN mutant (competitive index) recovered from lipocalin-2-deficient mice was significantly lower than that determined for wild type mice (P < 0.05). These data suggested that iroN confers a selective advantage during growth in the inflamed intestine because the encoded salmochelin receptor IroN mediates resistance to lipocalin-2.
DISCUSSION
One host strategy for controlling colonization of unwanted microbes is to limit the availability of iron, an essential nutrient, by sequestering this trace element both systemically by hepcidin-mediated acute phase responses (Ganz, 2006; Nemeth and Ganz, 2006), and locally by high affinity iron binding proteins, including lactoferrin secreted by neutrophils at sites of inflammation (Ong et al., 2006). To overcome this iron-withholding defense, bacteria release high-affinity iron chelators, termed siderophores, that can remove iron from host proteins and are subsequently taken up by specialized bacterial transport systems (Braun et al., 1998). The siderophore produced by most members of the Enterobacteriaceae is enterochelin, a cyclic trimer of dihydroxybenzoylserine (O’Brien and Gibson, 1970). The host can limit the ability of bacteria to use enterochelin for iron acquisition by secreting lipocalin–2, a protein whose production in tissue is induced by IL–22 (Aujla et al., 2008) and IL–17 (Conti et al., 2009; Raffatellu et al., 2008) during inflammation. Expression of LCN2 mRNA by the inflamed intestinal epithelium occurs in the human colon during inflammatory bowel disease (Nielsen et al., 1996), while murine Lcn2 mRNA is present in mouse colonic epithelium during dextran sodium sulfate-induced colitis (Playford et al., 2006). Here we show that IL–17 and IL–22 act in concert to induce LCN2 transcription in T84 colonic epithelial cells. Furthermore, LCN2 transcription was induced by S. Typhimurium infection in the intestinal epithelium of rhesus macaques, resulting in secretion of lipocalin-2 into the intestinal lumen. These data suggested that lipcalin-2 is an antimicrobial encountered in the lumen of the inflamed intestine.
Lipocalin–2 specifically binds and inactivates enterochelin, resulting in a bacteriostatic activity for some commensal bacteria, such as Escherichia coli (Berger et al., 2006; Flo et al., 2004; Goetz et al., 2002). The iroBCDE iroN gene cluster of S. Typhimurium encodes proteins involved in the biosynthesis and uptake of salmochelin, a glycosylated derivative of enterochelin (Bister et al., 2004; Hantke et al., 2003; Zhu et al., 2005). Salmochelin is not bound by lipocalin–2 and its production therefore confers lipocalin-2 resistance (Crouch et al., 2008; Fischbach et al., 2006). Here we show that lipocalin-2 resistance conferred a specific benefit during growth of S. Typhimurium in the inflamed cecum of mice. The streptomycin pretreated mouse model has limitations, because exudative intestinal inflammation is induced artificially by antibiotic treatment. However, our results show that lipocalin-2 production is also a prominent host response to S. Typhimurium infection in the intestinal mucosa of rhesus macaques, a highly relevant model for human gastroenteritis. Our results suggest that acquisition of the iroBCDE iroN gene cluster by S. Typhimurium may represent a specific adaptation to life in the inflamed intestine. A potential role of lipocalin-2 resistance in promoting colonization of the inflamed nasal mucosa has recently been proposed for Streptococcus pneumoniae and Haemophilus influenzae (Nelson et al., 2005).
The main virulence factors involved in inducing intestinal inflammation, T3SS-1 and T3SS-2 (Hapfelmeier et al., 2005; Tsolis et al., 1999), were acquired by horizontal gene transfer early after divergence of the genus Salmonella from the E. coli lineage and are present in all members of the species S. enterica (Hensel et al., 1997; Li et al., 1995; Ochman and Groisman, 1996). It seems reasonable to assume that to ensure survival, acquisition of T3SS-1 and T3SS-2 had to be accompanied by acquisition of genes that confer resistance to antimicrobials encountered in the inflamed intestine. Consistent with this idea, the iroBCDE iroN gene cluster is present in all members of the species S. enterica but is absent from the genomes of commensal E. coli strains (Baumler et al., 1997; Baumler et al., 1998). The only E. coli isolates in which the iroBCDE iroN gene cluster is present are uropathogenic E. coli (UPEC) (Bauer et al., 2002; Sorsa et al., 2003). This distribution is consistent with the concept that the iroBCDE iroN gene cluster represents an adaptation to life at inflamed mucosal surfaces, which is an environment encountered by both UPEC (cystitis) and S. Typhimurium (gastroenteritis).
In addition to overcoming lipocalin-2 mediated iron withholding, S. Typhimurium likely needs to resist many other antimicrobial defense mechanisms encountered in the inflamed intestine. Candidate antimicrobials whose transcripts are prominently induced in the intestinal mucosa during S. Typhimurium infection include inducible nitric oxide synthase (iNOS, encoded by NOS2), regenerating islet-derived 3 gamma (RegIIIγ), calprotectin A, mucin (MUC4), indoleamine 2,3-dioxygenase (IDO), dual oxidase (Duox) and defensins (Godinez et al., 2009; Raffatellu et al., 2007; Raffatellu et al., 2008; Zheng et al., 2008). Here we show that IL–22 induced expression of NOS2 and MUC4 in human intestinal epithelial cells, suggesting a possible cellular source for the high mRNA levels of these genes observed in the ileal mucosa of rhesus macaques during S. Typhimurium infection (Raffatellu et al., 2008). In mice lacking iNOS and NADPH oxidase, S. Typhimurium is recovered in reduced numbers from intestinal contents at 48 hours after infection, although this difference is not statistically significant (Ackermann et al., 2008). Research on the potential role of S. Typhimurium resistance mechanisms against the various antimicrobials encountered in the inflamed intestine represents an exciting area for future study.
MATERIALS AND METHODS
Tissue culture assays
T84 cells were grown in a 1:1 mixture of DMEM/F12 and 10% Fetal calf serum (Gibco). To achieve polarization, cells were seeded on the apical compartment in 12-mm-diameter Transwell plate (Corning Costar) and utilized when a TER of at least 1,500 Ω.cm2 was reached.
Stimulation with cytokines
Recombinant human IL-17, IL-22 and IFN-γ were obtained from R&D systems and utilized at the following final concentrations: IL-17, 100 ng/ml; IL-22, 100 ng/ml; IFN-γ, 40 ng/ml. Each stimulation experiment was performed a minimum of four times.
Gene expression profiling
Gene expression profiles (GEO accession number GSE11345) in polarized T84 cells were monitored utilizing the GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA). For each treatment group, total RNA from 4 tissue culture wells was analyzed. A minimum 2-fold difference in transcription between control and experimental samples was used as criteria for identifying a “change” in gene expression (P value<0.05; 95% confidence).
Real-time PCR
For analysis of gene expression by real-time PCR, total RNA was extracted with Tri-reagent (Molecular Research Center) and processed as described earlier (Raffatellu et al., 2007). Real-time PCR was performed using SYBR Green (Applied Biosystems) and the 7900HT Fast Real-Time PCR System. The data were analyzed using the comparative delta-Ct method (Applied Biosystems). Analysis of the copy number of rhesus LCN2 was performed by comparing crossing points of experimental cDNA to standard curves generated from a plasmid that contained a fragment of rhesus LCN2 cDNA (Hornsby et al., 2008). A list of the real-time primers used in this study is provided in Supplementary Table 1.
Bacterial strains and culture conditions
IR715 is a fully virulent, nalidixic acid resistant derivative of S. Typhimurium wild-type isolate ATCC 14028. Construction of IR715 derivatives carrying mutations in iroBC, iroN, invA, or spiB is described in the supplementary Material and Methods. A complete list of strains, plasmids and primers used for cloning and strain construction is provided in Supplementary Tables 2 and 3. All strains were grown aerobically at 37°C in Luria-Bertani (LB) broth unless otherwise noticed.
Bacterial growth in cell culture supernatant
S. Typhimurium wild-type and iroBC mutant were grown overnight in iron-limiting conditions (Nutrient Broth supplemented with 0.2 mM 2,2′-dipyridyl) at 37°C with aeration over night. Approximately 1000 CFU were inoculated into tissue culture medium (DMEM/F12), DMEM/F12 containing human lipocalin-2 (100ng/ml, R&D Systems), DMEM/F12 containing human lipocalin-2 (100ng/ml) and ferrioxamine B (1μg/ml), or the media collected from the basolateral side of transwell plate 24 hours after stimulation with IL-17 and IL-22 or mock control. CFU were enumerated by plating serial dilution 5 hours after inoculation. The experiment was performed three times.
Immunoblot
Total protein was extracted from mouse cecum using Tri-Reagent (Molecular Research Center). Total protein (15 μg) was resolved by SDS-PAGE and transferred to a PVDF membrane. Detection of mouse tubulin was performed with a primary rabbit polyclonal antibody (Cell Signaling Technology) and detection of mouse lipocalin-2 was performed with a primary rabbit polyclonal antibody (R&D systems). As secondary antibody, a secondary goat-anti-rabbit conjugate to horseradish peroxidase (HRP) (Jackson) was used.
Enzyme-linked ImmunoSorbent Assay
Supernatant from both the apical and the basolateral compartments of polarized T84 cells was collected 24 hours post-stimulation. Secretion of CCL20 and lipocalin-2 was detected by using commercially available kits (R&D systems).
In situ hybridization
Localization of rhesus mRNA transcripts in formalin fixed, paraffin embedded ileal sections was performed as described earlier (Santos et al., 2002). Probes for target genes were amplified from rhesus cDNA by using the primers reported in Supplementary table 1 and cloned into PCR2.1 (Invitrogen). Each probe (LCN2 and plasmid control) was biotin-labeled by using the NEBlot Phototype Kit according to instructions from the manufacturer (New England Biolabs).
Detection of lipocalin-2 in tissue of rhesus macaques
Generation of an anti-rhesus lipocalin–2 rabbit polyclonal antibody is described under Supplementary Material and Methods. Immunohistochemical staining of lipocalin-2 was performed on formalin fixed, paraffin embedded ileal sections by using a rabbit polyclonal antibody derived from recombinant rhesus lipocalin-2, followed by detection with a secondary anti-rabbit IgG antibody (Vector Laboratories). Staining of an adjacent section with pre-immune rabbit serum was used as a control. Detection of lipocalin–2 in the fluid collected from ligated ileal loops of rhesus macaques is described under Supplementary Material and Methods.
Mouse experiments
We used both C57BL/6 wild-type and lipocalin-2-deficient mice (Lcn2−/−). Generation of Lcn2−/− mice (Berger et al., 2006) is described in the supplementary materials and methods. Groups of 6 mice were pre-treated with streptomycin (0.1 ml of a 200 mg/ml solution in sterile water) intragastrically prior to mock-infection or inoculation with a mixture of S. Typhimurium strains (5×108 CFU/animal). At 48 hours after infection, mice were euthanized and cecum was collected for isolation of mRNA and protein and for histopathological analysis. Colon contents were collected and serial 10-fold dilutions were plated for enumerating bacterial CFUs on agar plates containing the appropriate antibiotics. Data were normalized by dividing the output ratio (CFU of the wild-type/CFU of the mutant) by the input ratio (CFU of the wild-type/CFU of the mutant).
Statistical analysis
Microarray data were analyzed using model-based algorithms (Affymetrix Gene Chip Operating System software, dChip, http://biosun1.harvard.edu/complab/dchip) and t-tests. Differences between treatment groups in fold changes in mRNA levels measured by Real-time PCR, protein levels measured by ELISA, NO production measured by Griess assays, and bacterial numbers were analyzed by ANOVA followed by Student’s t test. A P value equal or below 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service grants AI040124, AI044170 and AI079173 (A.J.B.); DK43183, DK61297 and AI43274 (S.D.); AI42081 (J.V.S.); AI50843 (C.L.B); AI050553 (R.M.T.); DE018097 (M.D.G.); National Research Service Award T32 AI60555 (M.J.H. and H.C.); Floyd and Mary Schwall Fellowship in Medical Research (S.P.N.); Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil (T.A.P. and R.L.S); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (R.L.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE
- Ackermann M, Stecher B, Freed NE, Songhet P, Hardt WD, Doebeli M. Self-destructive cooperation mediated by phenotypic noise. Nature. 2008;454:987–990. doi: 10.1038/nature07067. [DOI] [PubMed] [Google Scholar]
- Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. J. Wiley & Sons; 1994. [Google Scholar]
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer RJ, Zhang L, Foxman B, Siitonen A, Jantunen ME, Saxen H, Marrs CF. Molecular epidemiology of 3 putative virulence genes for Escherichia coli urinary tract infection-usp, iha, and iroN(E. coli) J Infect Dis. 2002;185:1521–1524. doi: 10.1086/340206. [DOI] [PubMed] [Google Scholar]
- Bäumler AJ, Heffron F, Reissbrodt R. Rapid detection of Salmonella enterica with primers specific for iroB. J Clin Microbiol. 1997;35:1224–1230. doi: 10.1128/jcm.35.5.1224-1230.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumler AJ, Norris TL, Lasco T, Voight W, Reissbrodt R, Rabsch W, Heffron F. IroN, a novel outer membrane siderophore receptor characteristic of Salmonella enterica. Journal of bacteriology. 1998;180:1446–1453. doi: 10.1128/jb.180.6.1446-1453.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumler AJ, Tsolis RM, van der Velden AW, Stojiljkovic I, Anic S, Heffron F. Identification of a new iron regulated locus of Salmonella typhi. Gene. 1996;183:207–213. doi: 10.1016/s0378-1119(96)00560-4. [DOI] [PubMed] [Google Scholar]
- Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, Fong HE, Cheung CC, Mak TW. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bister B, Bischoff D, Nicholson GJ, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Sussmuth RD. The structure of salmochelins: C-glucosylated enterobactins of Salmonella enterica. Biometals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- Braun V, Hantke K, Koster W. Bacterial iron transport: mechanisms, genetics, and regulation. Met Ions Biol Syst. 1998;35:67–145. [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. The Journal of experimental medicine. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch ML, Castor M, Karlinsey JE, Kalhorn T, Fang FC. Biosynthesis and IroC-dependent export of the siderophore salmochelin are essential for virulence of Salmonella enterica serovar Typhimurium. Mol Microbiol. 2008;67:971–983. doi: 10.1111/j.1365-2958.2007.06089.x. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, et al. The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proc Natl Acad Sci U S A. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Ganz T. Hepcidin--a peptide hormone at the interface of innate immunity and iron metabolism. Current topics in microbiology and immunology. 2006;306:183–198. doi: 10.1007/3-540-29916-5_7. [DOI] [PubMed] [Google Scholar]
- Godinez I, Haneda T, Raffatellu M, George MD, Paixão TA, Rolán HG, Santos RL, Dandekar S, Tsolis RM, Bäumler AJ. T cells help to amplify inflammatory responses induced by Salmonella enterica serotype Typhimurium in the intestinal mucosa. Infection and Immunity. 2008;76:2008–2017. doi: 10.1128/IAI.01691-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinez I, Raffatellu M, Chu H, Paixão TA, Haneda T, Santos RL, Bevins CL, Tsolis RM, Bäumler AJ. IL-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect Immun. 2009;77:387–398. doi: 10.1128/IAI.00933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Hantke K, Nicholson G, Rabsch W, Winkelmann G. Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci U S A. 2003;100:3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, Heikenwalder M, Stallmach T, Hensel M, Pfeffer K, Akira S, et al. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J Immunol. 2005;174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Bäumler AJ, Gleeson C, Blattner F, Holden DW. Analysis of the boundaries of Salmonella pathogenicity island 2 and the corresponding chromosomal region of Escherichia coli K-12. Journal of bacteriology. 1997;179:1105–1111. doi: 10.1128/jb.179.4.1105-1111.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–1057. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CY, Huang F, Chen Y, Thai P, Wachi S, Kim C, Tam L, Wu R. Up-regulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK/NF-kappaB-dependent signaling pathway. J Immunol. 2005;175:6676–6685. doi: 10.4049/jimmunol.175.10.6676. [DOI] [PubMed] [Google Scholar]
- Lawley TD, Bouley DM, Hoy YE, Gerke C, Relman DA, Monack DM. Host transmission of Salmonella enterica serovar Typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect Immun. 2008;76:403–416. doi: 10.1128/IAI.01189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ochman H, Groisman EA, Boyd EF, Solomon F, Nelson K, Selander RK. Relationship between evolutionary rate and cellular location among the Inv/Spa invasion proteins of Salmonella enterica. Proc Natl Acad Sci U S A. 1995;92:7252–7256. doi: 10.1073/pnas.92.16.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Kreuzer KA, Baskaynak G, Peters UR, le Coutre P, Schmidt CA. Quantitative analysis of beta-actin, beta-2-microglobulin and porphobilinogen deaminase mRNA and their comparison as control transcripts for RT-PCR. Mol Cell Probes. 2002;16:25–30. doi: 10.1006/mcpr.2001.0392. [DOI] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell host & microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Nelson AL, Barasch JM, Bunte RM, Weiser JN. Bacterial colonization of nasal mucosa induces expression of siderocalin, an iron-sequestering component of innate immunity. Cell Microbiol. 2005;7:1404–1417. doi: 10.1111/j.1462-5822.2005.00566.x. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annual review of nutrition. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- Nielsen BS, Borregaard N, Bundgaard JR, Timshel S, Sehested M, Kjeldsen L. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut. 1996;38:414–420. doi: 10.1136/gut.38.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega LM, Van der Auwera P, Daneau D, Meunier F, Aoun M. Salmonella infections in a cancer center. Support Care Cancer. 1994;2:116–122. doi: 10.1007/BF00572093. [DOI] [PubMed] [Google Scholar]
- O’Brien IG, Gibson F. The structure of enterochelin and related 2,3-dihydroxy-N-benzoylserine conjugates from Escherichia coli. Biochim Biophys Acta. 1970;215:393–402. doi: 10.1016/0304-4165(70)90038-3. [DOI] [PubMed] [Google Scholar]
- Ochman H, Groisman EA. Distribution of pathogenicity islands in Salmonella spp. Infect Immun. 1996;64:5410–5412. doi: 10.1128/iai.64.12.5410-5412.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong ST, Ho JZ, Ho B, Ding JL. Iron-withholding strategy in innate immunity. Immunobiology. 2006;211:295–314. doi: 10.1016/j.imbio.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Playford RJ, Belo A, Poulsom R, Fitzgerald AJ, Harris K, Pawluczyk I, Ryon J, Darby T, Nilsen-Hamilton M, Ghosh S, et al. Effects of mouse and human lipocalin homologues 24p3/lcn2 and neutrophil gelatinase-associated lipocalin on gastrointestinal mucosal integrity and repair. Gastroenterology. 2006;131:809–817. doi: 10.1053/j.gastro.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Que JU, Casey SW, Hentges DJ. Factors responsible for increased susceptibility of mice to intestinal colonization after treatment with streptomycin. Infect Immun. 1986;53:116–123. doi: 10.1128/iai.53.1.116-123.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabsch W, Voigt W, Reissbrodt R, Tsolis RM, Bäumler AJ. Salmonella typhimurium IroN and FepA proteins mediate uptake of enterobactin but differ in their specificity for other siderophores. Journal of bacteriology. 1999;181:3610–3612. doi: 10.1128/jb.181.11.3610-3612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffatellu M, Santos RL, Chessa D, Wilson RP, Winter S, Rossetti CA, Lawhon SD, Chu H, Lau T, Bevins CL, et al. The Capsule encoding viaB locus reduces IL-17 expression and mucosal innate responses in the bovine intestinal mucosa during infection with Salmonella enterica serotype Typhi. Infect Immun. 2007 doi: 10.1128/IAI.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nature Medicine. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RL, Schoffelmeer JA, Tsolis RM, Gutierrez-Pabello JA, Bäumler AJ, Adams LG. Salmonella serotype Typhimurium infection of bovine Peyer’s patches down-regulates plasma membrane calcium-transporting ATPase expression. J Infect Dis. 2002;186:372–378. doi: 10.1086/341509. [DOI] [PubMed] [Google Scholar]
- Sorsa LJ, Dufke S, Heesemann J, Schubert S. Characterization of an iroBCDEN gene cluster on a transmissible plasmid of uropathogenic Escherichia coli: evidence for horizontal transfer of a chromosomal virulence factor. Infect Immun. 2003;71:3285–3293. doi: 10.1128/IAI.71.6.3285-3293.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, Salazar-Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in salmonella-infected mice is dependent on IL-18. J Immunol. 2007;178:6342–6349. doi: 10.4049/jimmunol.178.10.6342. [DOI] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolis RM, Adams LG, Ficht TA, Bäumler AJ. Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect Immun. 1999;67:4879–4885. doi: 10.1128/iai.67.9.4879-4885.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zhu M, Valdebenito M, Winkelmann G, Hantke K. Functions of the siderophore esterases IroD and IroE in iron-salmochelin utilization. Microbiology. 2005;151:2363–2372. doi: 10.1099/mic.0.27888-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.