

SUMMARY
αβ and γδ T-cells arise from a common thymocyte progenitor during development in the thymus. Emerging evidence suggests that the pre-T cell receptor (pre-TCR) and γδ T-cell receptor (γδTCR) play instructional roles in specifying the αβ and γδ T-lineage fates, respectively. Nevertheless, the signaling pathways differentially engaged to specify the fate and promote the development of these lineages remain poorly understood. Here we show that differential activation of the ERK - early growth response gene (Egr) - inhibitor of DNA binding 3 (Id3) pathway plays a defining role in this process. In particular, Id3 expression serves to regulate adoption of the γδ fate. Moreover, Id3 is both necessary and sufficient to enable γδ-lineage cells to differentiate independently of Notch signaling and become competent IFNγ-producing effectors. Taken together, these findings identify Id3 as a central player that controls both adoption of the γδ fate and their maturation in the thymus.
INTRODUCTION
T cells comprise two major lineages that arise from a common progenitor and are identified by the T cell receptor (TCR) complex they express, αβ and γδ (Ciofani et al., 2006; Dudley et al., 1995). Nevertheless, the molecular processes that underlie specification of the αβ and γδ fates remain poorly understood. Two models have been advanced to explain the role of the TCR in this fate decision: stochastic and instructional (Hayday and Pennington, 2007). The stochastic model stipulates that fate adoption is independent of TCR signals, which serve only to rescue survival of the appropriately matched, pre-determined fate. Conversely, the instructional model posits that TCR signals direct adoption of the fate that matches the TCR from which the signal is derived (i.e., pre-TCR for the αβ fate and γδTCR for the γδ fate). While existing data do not easily fit either model, as originally proposed, emerging evidence supports an instructional model in which TCR signal strength dictates fate adoption (Ciofani et al., 2006; Haks et al., 2005; Hayes et al., 2005). Indeed, recent single cell progenitor analyses defined the CD4-CD8- CD44-CD25+ (DN3) stage as the developmental step at which T-lineage commitment is completed (Ciofani et al., 2006). These studies further demonstrated that ectopic expression of a pre-TCR or γδTCR in Rag2-deficient DN3 thymocytes was capable of instructing commitment to the αβ and γδ lineages, respectively (Ciofani et al., 2006). Moreover, it was recently shown that the linkage between specific TCR expression and fate selection can be severed genetically, in that a single TCR complex could induce the differentiation into either fate when the resulting signals were modulated (Haks et al., 2005; Hayes et al., 2005). These findings support the hypothesis that the strength of the TCR signal is a critical, lineage-determining factor, and that this determination can be made irrespective of the TCR complex from which the signals arise.
Our previous analysis suggested that the stronger signals that promote adoption of the γδ-fate involve pronounced activation of the ERK-Egr-Id3 pathway (Haks et al., 2005; Hayes et al., 2005). Nevertheless, it remained unclear how differential ERK-Egr-Id3 signaling might function in regulating fate choice or how it might be integrated with signaling input from other pathways, such as Notch. One attractive target of strong TCR signals is E proteins, which are basic helix-loop-helix transcription factors that bind DNA at E-box motifs (CANNTG) (Murre, 2005). Mice in which E protein genes are ablated (e.g., mice lacking E2A, HEB, or both) exhibit severely perturbed development of αβ T cells, but show relatively mild effects on γδ T cell differentiation (Bain et al., 1999; Barndt et al., 2000; Wojciechowski et al., 2007). Accordingly, strong TCR signals might selectively impair αβ T cell development by phenocopying E protein deficiency, either through marked induction of Id3, which can heterodimerize with E proteins and block their function, or by increased ERK activation, which can induce E protein degradation through a phosphorylation-dependent mechanism (Nie et al., 2003; Ordentlich et al., 1998).
E proteins may also serve as a common downstream focal point for TCR and Notch signaling. Notch molecules are surface receptors involved in a wide variety of cell fate decisions, including αβ/γδ lineage commitment (reviewed by (Hernandez-Hoyos and Alberola-Ila, 2003; Radtke et al., 2004). Recently, it was determined that αβ-lineage precursors are dependent upon Notch signaling throughout their pre-TCR-induced differentiation program to the CD4+ CD8+ DP stage; however, γδ-lineage thymocytes become Notch-independent upon expression of the γδTCR complex (Ciofani et al., 2006). Like TCR signaling, Notch signaling can suppress E protein function through ERK activation or Id3 induction (Nie et al., 2003; Ordentlich et al., 1998; Reynaud-Deonauth et al., 2002). We suggest a model in which the lineage-specific requirement for Notch signaling is determined by the relative strength of TCR signals, through effects on E protein activity. Accordingly, we hypothesize that Notch independent-differentiation is conferred on γδTCR-expressing cells through strong TCR signals, which are capable of repressing E protein activity below the threshold required for γδ-differentiation without Notch signals. In contrast, the pre-TCR signals that promote the αβ-fate are too weak without the assistance of Notch to suppress E protein activity sufficiently to surmount the E protein mediated checkpoint at the DN3 stage (Engel et al., 2001; Engel and Murre, 2004). Because of the ability of Id3 to regulate of E protein activity, our model suggests that Id3 would be a key molecular mediator that facilitates γδ-T cell commitment and differentiation.
Here we show that Id3 is required for strong TCR signals to both promote adoption of the γδ-fate and oppose the αβ-fate outcome. Moreover, while in most cases Id3 is required for the differentiation of the broader repertoire of non-autoreactive γδ-T cells, in certain cases when γδ-TCR affinity for ligand is very high, Id3 appears to restrict the differentiation potential of that particular TCR-Vγ subset. Id3 also plays a critical role in conferring Notch-independence to developing γδ-lineage cells and in arming them to proliferate and produce IFNγ in response to stimulation. This function of Id3 requires its ability to interact with and suppress E protein targets. Taken together, these findings implicate Id3 as an important molecular determining factor in enforcing T cell fate decisions, integrating Notch and TCR signals, and enabling functional maturation.
RESULTS
Id3 is required for Egr1-mediated regulation of fate adoption
We have previously shown that the Erk-Egr1-Id3 pathway plays an important role in the specification of T-lineage fates (Haks et al., 2005). Expression of Egr1, Egr3 and Id3 mRNA levels are higher in thymic γδ cells than in DN3 and DN4 precursors, representing a mixture of αβ and γδ lineage progenitors (Figure 1A) (Ciofani et al., 2006; Dudley et al., 1995). Moreover, as reported previously, retroviral transduction of Egr1 into C57BL/6 E13.5 fetal thymocytes markedly increased the frequency and number of mature γδ T cells (i.e., CD24lo TCRγδ+) at the expense of αβ-lineage-committed DP thymocytes (Figure 1B,C) (Haks et al., 2005). The ability of ectopically expressed Egr1 to simultaneously promote γδ development and oppose the development of αβ lineage cells is dependent on the Egr target, Id3, as these effects are markedly diminished in Id3-deficient cells (Figure 1B,C). Id3-deficiency did not completely eliminate the ability of Egr1 transduction to alter cell fate, perhaps because of partial compensation by other Egr targets, such as Id2 (Figure S1, in Supplemental Data available on line). Taken together, these data implicate the Erk-Egr-Id3 pathway as a key component of the strong signals that modulate αβ/γδ lineage fate in the thymus.
Figure 1. Id3 is required for Egr1-mediated promotion of γδ development.

(A) Egr1, Egr3, and Id3 mRNA are more highly expressed in developing γδ lineage cells. Egr and Id mRNA levels were quantified in triplicate in the indicated cell populations by Real time PCR, standardized using β-actin, and normalized to levels in DN3. Results ± standard deviation are depicted graphically.
(B-C) Induction of γδ development by enforced expression of Egr1 is dependent on Id3. E13.5 Id3+/- and Id3-/- fetal thymocytes were transduced with empty vector (LZRS) or Egr1 (LZRS-Egr1), seeded into thymic lobes and placed in fetal thymic organ culture for 2 days. Developmental progression was assessed by flow cytometry on electronically gated transduced (GFP+) cells. The absolute number of γδ lineage cells (γδTCR+ or γδTCR+CD24lo) and αβ lineage DP thymocytes per lobe is depicted graphically in (C). Results are representative of 3 experiments performed.
Id3 is required for development of the normal repertoire of γδ T cells
Since Id3-deficiency impaired the Egr-induced development of fetal γδ T cells, we asked whether γδ cell development were similarly perturbed in adult mice. Surprisingly, we found that Id3-deficiency resulted in an increased number of CD24lo γδ-lineage cells in the thymus (Figure 2A). Consistent with a recent report, the expansion appeared to be restricted to a subpopulation of Vγ1.1+ γδ T cells (Ueda-Hayakawa et al., 2009), as other γδ cells including the Vγ2 and Vγ3 dendritic epidermal T cell (DETC) subsets were reduced (Figure 2A,B). Unlike the conventional Vγ2 and Vγ3 subsets, Vγ1.1+ γδ cells have been suggested to represent so called NK γδ T cells thought to be autoreactive and selected on high affinity ligands (Felices et al., 2009; Lees et al., 2001; Qi et al., 2009). We reasoned that such cells might normally be deleted by excessively strong signals, but are allowed to survive and expand in the absence of Id3. Since the putative selecting ligand(s) for the Vγ1.1+ subset have not been identified, we sought to test this possibility by using the KN6 γδ TCR transgenic (Tg) mice in which KN6 γδTCR Tg thymocytes are positively selected in the thymus on the non-classical MHC class Ib molecule T22d ligand, but are deleted by the 10-fold higher affinity T-10/22b ligand (Figure 2C) (Adams et al., 2008; Pereira et al., 1992). Our hypothesis predicts that deletion of KN6 Tg thymocytes by T-10b ligand should be prevented by Id3-deficiency. Indeed, Id3-deficiency blocks deletion of KN6 Tg thymocytes by T-10/22b ligand, resulting in a marked expansion of KN6 Tg thymocytes with a mature CD24lo phenotype. These data suggest an apparent dichotomy of Id3 function, with Id3 restricting the development of TCR-Vγ subsets bearing high affinity γδTCRs, while at the same time being required for development of the broader repertoire of non-autoreactive γδ-T cells.
Figure 2. Id3-deficiency disrupts the development of γδ T cells.

(A-B) Id3-deficiency results in expansion of the Vγ1.1+ subset. Id3-/- mice were backcrossed to the C57BL/6 background for 3 generations before tissues from mice of the indicated genotypes were analyzed by flow cytometry. Results are depicted as histograms and summarized graphically on the right.
(C-D) Id3-deficiency rescues deletion of autoreactive γδ precursors. Thymocyte preparations were produced from Rag2+/- KN6 Tg mice expressing positively selecting ligand (T-22d) or higher affinity negatively selecting ligand (T-10/22d/b) and from KN6 Tg mice expressing high affinity ligand but lacking Id3. Preparations were analyzed by flow cytometry and gated DN cells were displayed as histograms as above. Differences in absolute number of the indicated subsets that are marked by asterisks are statistically significant (p<0.05). Results are representative of 3 experiments performed with a minimum of 2 mice per genotype in each experiment.
Regulation of lineage fate by the Egr-Id3 pathway is not dependent upon alterations in TCR repertoire
The selective inhibition of particular Vγ subsets by Id3-deficiency could be effected at the level of lineage commitment, selection and maturation, or at the level of gene-rearrangement (Ueda-Hayakawa et al., 2009). Consistent with the latter possibility, the E protein targets, whose functions are antagonized by Id3, have been shown to be involved in regulating TCR gene rearrangement (Agata et al., 2007; Bain et al., 1999). Accordingly, it was possible that ectopic expression of Egr1 and Id3-deficiency were modulating lineage fate indirectly by altering the TCR repertoire. To test this possibility, we revisited the role of superinduction of Egr1 and Id3 on T cell fate in a model with a fixed TCR repertoire. For this purpose, we employed KN6 γδTCR Tg Rag2-deficient mice in which lineage choice can be manipulated by modulating access to ligand or signaling output from a single TCR complex (Haks et al., 2005; Pereira et al., 1992). Thymocytes from KN6 Tg mice expressing T22d ligand adopt the γδ fate (CD4- CD8- γδTCR+ CD24lo; Figure 3A) whereas those lacking ligand (i.e., β2M-deficient) commit to the αβ-lineage, undergo proliferative expansion, and develop into DP thymocytes (Figure 3A). Accordingly, we employed the KN6 Tg model to assess the ability of Egr1 and Id3 to modulate lineage fate independently of alterations in the TCR repertoire. To determine whether ectopic expression of Egr1 would divert cells from the αβ lineage to the γδ-fate, we ectopically expressed Egr1 in KN6 Tg mice lacking ligand (i.e., committing to the αβ lineage; Figure 3B, left panel) (Miyazaki, 1997). Indeed, we found that the Egr1 Tg both antagonized αβ-lineage development, as indicated by the reduced number of DP thymocytes and promoted development of mature CD24lo γδ-lineage cells (Figure 3B). We next investigated whether the Egr1 target, Id3, was required for strong signals (generated by γδTCR-ligand interactions) to promote γδ development and simultaneously oppose development of αβ-lineage cells. In agreement, Id3-deficiency interfered with the ligand-induced development of mature CD24lo γδ-lineage cells, while increasing the number of αβ-lineage DP thymocytes (Figure 3C). Importantly, Id3-deficiency caused proportional changes in αβ-lineage DP and CD24lo γδ-lineage cells, consistent with an effect on lineage commitment. These data suggest that the Egr-Id3 pathway plays a critical role in lineage assignment that does not require alterations in the TCR repertoire. Id1-deficiency had no affect on lineage assignment in this model (data not shown). Notably, the ability of strong signals to promote the γδ-lineage was associated with markedly reduced expression of E proteins (E47) relative to that observed in cells committing to the αβ fate in the absence of ligand (Figure S2).
Figure 3. Egr1 and Id3 can modulate lineage choice without affecting the TCR repertoire.

(A) Schematic representation of lineage assignment in the KN6 γδTCR Tg model.
(B) Enforced expression of Egr1 diverts KN6 Tg thymocytes to the γδ fate. Thymocytes from mice of the indicated genotypes were analyzed by flow cytometry: 1) Lig-, KN6 Tg Rag2-/-β2M-/-; 2) Lig-Egr1Tg, KN6 Tg Rag2-/-β2M-/- Egr1Tg. Results are depicted as histograms and summarized graphically on the right with each symbol corresponding to an individual mouse.
(C) The strong TCR signals that promote adoption of the γδ fate require Id3. Mice of the indicated genotypes were analyzed by flow cytometry: 1) Lig+, KN6 Tg Rag2-/-; 2) Lig+Id3-/-, KN6 Tg Rag2-/-Id3-/-. (B-C) Differences between the absolute number of DP and CD24lo DN thymocytes between these mice are statistically significant (asterisk indicates p<0.05).
Effects of Id3-deficiency on fate adoption are associated with changes in survival and proliferation
We next wished to determine how strong signals employ Id3 to both promote development of γδ-lineage cells and oppose αβ-lineage fate. Previous studies indicated that the E proteins can control cell growth and survival (Engel et al., 2001; Engel and Murre, 2004; Xi et al., 2006). Accordingly, we reasoned that repression of E proteins by Id3 might differentially influence growth and survival of developing αβ and γδ T-lineage cells. Interestingly, the impaired generation of mature CD24lo γδ-lineage cells in ligand-expressing Id3-/- KN6 Tg mice is associated with increased Annexin V staining, suggesting that Id3 is required to promote the survival of these cells (Figure 4A). The CD24lo population also exhibited increased BrdU incorporation. Perhaps this contributes to the increased apoptosis among these cells, since cycling cells are more sensitive to apoptotic stimuli (Komarova et al., 1997). Effects on survival and proliferation also appear to be associated with the ability of Id3 to oppose αβ-lineage development (Figure 4B). Indeed, Id3-deficiency caused an increase in survival amongst cells that differentiate to the αβ-lineage DP stage (i.e., through a CD8 immature single positive or ISP intermediate) and accumulate in Id3-/- mice expressing ligand (Figure 4B). Moreover, Id3-deficiency increased the fraction of BrdU+ ISP cells, suggesting that Id3, in the context of strong signals, normally suppresses their growth. Taken together, these findings indicate that following strong TCR signals, Id3 functions to promote the production of CD24lo γδ-lineage cells and opposes the development of αβ-lineage and this is associated with effects on growth and survival.
Figure 4. Id3 promotes the γδ fate and opposes the αβ fate and these effects are associated with alternations in growth and survival.

(A-B) Id3 promotes survival of mature CD24lo γδ lineage cells and impairs growth and survival of CD8ISP and αβ lineage DP. Apoptosis and proliferation of the indicated populations were evaluated flow cytometrically by Annexin staining and BrdU incorporation, respectively. Each symbol represents an individual experiment involving at least 3 animals, with a line connecting the animals with a different genotype contained within a single experiment (upper panels). The difference in % apoptotic cells between Lig+ and Lig+Id3-/- populations was significant (p<0.05) for the following populations: 1) γδTCR+CD24lo; 2) ISP; and 3) DP. The mean percent BrdU incorporation ± standard deviation is presented graphically (bottom panels) with a minimum of 10 mice per condition. *, p<0.05; **, p<0.01.
(C) Effects of Id3 on growth and survival are associated with differential regulation of Bcl2 family members. Expression of the indicated genes was measured by real time PCR on thymocyte subsets purified by flow cytometry. Expression levels were standardized using β-actin, and normalized to levels in Rag2-/- DN3 thymocytes. Data are representative of two different experiments.
To gain insight into the molecular basis for the differences in survival and proliferation observed in Id3-deficient mice, we examined the expression levels of molecular effectors of these pathways. An attractive set of candidates was the RORγt-BclXL axis, shown recently to require the E protein targets of Id3 for expression (Xi et al., 2006). In accord with this model, expression of RORγt and the pro-survival factor BclXL was elevated in Id3-/- ISP cells relative to that in Id3+/+ ISP cells, consistent with their increased survival (Figure 4C). Interestingly, expression of RORγt and BclXL was not decreased in Id3-/- CD24lo γδ-lineage and so did not correlate with the increased apoptosis of these cells (Figure 4C). However, Bcl-2 expression was significantly reduced in Id3-/- CD24lo γδ-lineage cells, consistent with their increased apoptosis (Figure 4C). These data suggest that Id3 induction in the context of a strong signal is associated with the increased survival of γδ-lineage cells and decreased survival of αβ-lineage cells. Further, these fates correlate with differential, cell-context-dependent effects on the expression of the pro-survival factors, Bcl-2 and BclXL, respectively. Expression patterns of other the E protein targets, CDK6 and Bim, were not consistent with alterations in growth and survival (Figure S3)
Because Id3-deficiency is associated with changes in growth and survival of αβ and γδ lineage cells occurring under the influence of strong TCR signals, we thought it possible that Id3 may regulate lineage selection prior to TCR signaling by acting on pre-committed progenitors. Lineage fate has been shown to be complete in DN3 progenitors and can be controlled by ectopic expression of TCR complexes in Rag2-/- DN3 thymocytes (Ciofani et al., 2006). To determine whether Id3 induction might be functioning by selectively inducing apoptosis among pre-committed αβ-lineage precursors, we co-expressed Id3 (to mimic a strong signal) with TCRβ (pre-TCR-induced αβ-fate) in Rag2-/- DN3 thymocytes. Our results showed that Id3-transduction did not result in selective loss of αβ-lineage precursors, suggesting that strong signals through Id3 are acting on the specification or commitment process to promote γδ-lineage differentiation and oppose the αβ fate (Figure S4). Conversely, to determine whether strong signals promote the generation of CD24lo γδ-lineage cells by preferentially expanding a small pre-committed γδ-lineage population, we ectopically expressed the KN6 γδTCR in Rag2-/- thymocytes, which lack pre-committed γδ-lineage cells, and assessed effects on fate adoption in the presence and absence of ligand (Figure S5). Importantly, KN6 γδTCR-expressing precursors adopted the γδ-fate in the presence of ligand (at the expense of the αβ-fate) and, conversely, were diverted to the αβ-fate (at the expense of the γδ-fate) when ligand expression was knocked down by shRNA (Figure S5). Altogether, these data suggest that strong signals and Id3 are able to influence lineage commitment per se. The effects on ISP/DP survival that we observed in vivo likely occur in a minority of cells that escape ligand-engagement early in development. Recent observations suggest that ligand engagement of uncommitted DN thymocytes induces commitment to the γδ-lineage; however, cells which escape ligand development and commit to the αβ-fate appear to become susceptible to deletion by ligand as they differentiate to the ISP or DP stages (Kreslavsky et al., 2008).
γδ-biased profile-gene Rgs1 is closely linked with γδ T cell function
Since Id3-deficiency interfered with γδ cell maturation in response to strong TCR signals, we investigated whether effector function was similarly compromised. Id3-deficiency impaired both TCR-induced proliferation and IFNγ production (Figure 5). Additionally, this effect was largely restricted to γδ-lineage cells, as the TCR-induced proliferation of αβ-lineage CD4 T cells was not affected by Id3-deficiency, although proliferation of Id3-/- CD8 T cells was somewhat delayed following TCR activation (Figure S6) (Pan et al., 1999; Rivera et al., 2000). Of note, both proliferation and IFNγ production occur only in mature γδ-lineage cells that are CD24lo (Figure S7) (Pereira et al., 1992).
Figure 5. Induction of Id3 by strong signals during γδ development is required for functional competence.

(A-C) Id3-deficiency impairs TCR-dependent proliferation and production of IFNγ. DN thymocytes from KN6+Lig+ and KN6+Lig+Id3-/- mice were labeled with CFSE, stimulated for 48h on plate-bound anti-CD3 Ab (10μg/ml), and analyzed by FACS (A). DN thymocytes from the same mice as in (A) were stimulated as above and cellular expansion was evaluated on triplicate wells by MTT assay. (B). DN thymocytes were stimulated with plate-bound anti-CD3 Ab for 24 hours, following which the proportion of IFNγ producing cells was determined by intracellular staining and flow cytometry. (C) Mean ± standard deviation is presented graphically.
(D) Expression of the γδ biased gene profile and functional competence can be separated. Expression of the indicated genes was measured on flow cytometrically isolated cell populations by real time PCR as in Figure 4. Mean ± standard deviation is presented graphically.
Recent reports have suggested that full functional maturation of γδ-lineage cells requires trans-presentation of lymphotoxin-β (LTβ) to developing γδ-lineage cells by DP thymocytes (Pennington et al., 2003; Silva-Santos et al., 2005). This induces a set of genes termed the “γδ-biased gene profile” whose expression is enriched in γδ cells and linked to function: ICER, Nurr1, Nor1, Nurr77, and Rgs1 (Pennington et al., 2003; Silva-Santos et al., 2005). Since ligand-selected KN6-Tg γδ-lineage cells are functional (Figure 5A-C), yet develop in the near absence of DP thymocytes (Figure 3C), we examined whether these cells express the γδ-biased gene profile. Surprisingly, KN6 Tg γδ cells from Id3-sufficient, ligand-expressing mice exhibited increased expression for all of the γδ-biased genes examined (Figure 5D), indicating that DP-mediated trans-conditioning may not be absolutely required for their induction. These findings support the notion that elaboration of the γδ-biased gene profile is associated with γδ function (Pennington et al., 2003; Silva-Santos et al., 2005); however, Id3-deficiency appeared to sever this linkage. Indeed, despite the fact that proliferation and IFNγ production in response to TCR engagement are impaired in γδTCR+ cells from Id3-deficient mice, these cells displayed elevated expression of a subset of the γδ-biased profile-genes (Figure 5D), with the notable exception of Rgs1 expression, which was reduced in these cells. One potential explanation for this observation is that particular members of the γδ-biased profile may be more closely linked to γδ-function than others. Specifically, Rgs1 expression correlated well with γδ cell function in that it was highly-induced in functional KN6 Lig+ γδ cells and reduced in those lacking Id3 (Figure 5D); expression of the Nr4a family members (Nurr1, Nor1, and Nur77) was not. The elevated expression of the Nr4a genes in the nonfunctional, Id3-deficient γδTCR+ CD24lo cells may their role in regulating cell death in this misdirected population exhibiting increased apoptosis (Figure 4) (Cheng et al., 1997; Liu et al., 1994). Our data indicate that induction of some genes within the γδ-biased profile, such as Rgs1, is tightly linked to γδ T cell function, whereas others appear to be separable.
Id3 induction by strong signals is necessary and sufficient to promote Notch-independent maturation
The above findings provide support for Id3 as an important molecular effector of the strong signals that dictate γδ fate choice and maturation. However, it remained unclear whether other aspects of γδ-lineage development might rely on Id3. We wished to determine whether Id3 induction in the context of strong signals was required to confer Notch-independent maturation upon γδTCR-expressing cells (Ciofani et al., 2006). To test this possibility, we ectopically expressed either a pre-TCR or γδTCR in Rag2-/- DN3 cells and then assessed the role of Id3 in determining the Notch-dependence of their development in vitro (Figure 6A). As reported previously (Ciofani et al., 2006), pre-TCR (TCRβ-transduced) expressing Rag2-deficient DN3 cells proliferated robustly and developed to the DP stage (Figure 6A). This differentiation was independent of Id3, as it was not impaired by Id3-deficiency, but was dependent upon Notch signaling (i.e., required a Delta-Notch ligand, DL1; Figure 6A). Even the αβ-lineage DP cells that developed in the γδTCR-expressing cultures were dependent upon Notch-signals (Figure 6A)(Ciofani et al., 2006; Garbe et al., 2006). In contrast, γδ-lineage commitment and development into mature DN CD24lo γδ cells was not dependent upon Notch signals, but was dependent upon Id3. Indeed, Id3-deficiency markedly reduced the numbers of CD24lo γδ-lineage cells, instead diverting the γδTCR-expressing cells to the αβ-lineage DP stage (Figure 6A), as we observed in vivo (Figure 3). These data indicate that Id3-function is particularly important for elaboration of the γδ-lineage, but is dispensable for adoption of the αβ-fate. Interestingly, expression of the E protein target of Id3, E47, was reduced by Notch signaling, even in the absence of TCR (preTCR or γδTCR) expression (Figure 6B). However, consistent with the requirement of Notch signals for differentiation of preTCR-expressing Rag2-deficient DN3 cells, we observed that E47 levels were dramatically reduced only when both preTCR and Notch signals are present (Figure 6B, left side), indicating that the ERK-Notch-mediated degradation of E proteins may be critically important for reducing E protein activity at the β-selection checkpoint (Engel et al., 2001; Engel and Murre, 2004; Huang et al., 2004). This finding provides a potential explanation for why the generation of αβ-lineage DP cells is clearly dependent upon Notch, yet independent from Id3 (Figure 6A,B) (Huang et al., 2004; Nie et al., 2003; Ordentlich et al., 1998). On the other hand, Rag2-deficient DN3 cells transduced to express the KN6 γδTCR showed a strong reduction in E47 levels irrespective of Notch signals (Figure 6B, right side).
Figure 6. Id3 induction by strong signals is necessary and sufficient to confer Notch-independence on γδ lineage cells.

(A) Notch independence of γδ lineage cells is impaired by Id3-deficiency. Rag2-/- or Rag2-/-Id3-/- DN3 cells were transduced as indicated, cultured on OP9-DL1 or OP9-Cntl cells for 5 d, and the expression of CD4 and CD8 was analyzed by flow cytometry. Bar graphs on the right show the fold increases (relative to control transduced cultures) in absolute numbers of DP or γδ+ CD24lo DN cells from the indicated transductions, culture conditions and genotype.
(B) Notch signals reduce E protein expression. Equal quantities of protein from detergent extracts of cells cultured as in (A) were immunoblotted with the indicated Ab.
(C-D) Ectopic expression of Id3 is sufficient to confer Notch independence on TCR- thymocytes. Rag2-/-Id3-/- DN3 cells were transduced as indicated, cultured on OP9-DL1 or OP9-Cntl cells for 6 d, with the fold increases in cellularity over input shown in (C), and the expression of CD24 and forward-light scatter (FSC) analysis by flow cytometry are shown in (D). These data are representative of at least 3-independent experiments.
Since these data suggest that Id3 plays an important role in conferring Notch-independence to γδ-lineage cells, we next examined whether Id3-induction was sufficient. In fact, ectopic expression of Id3 in Rag2/Id3-deficient DN3 cells led to increased cellular expansion (Figure 6C) and maturation (e.g., CD24 down-modulation; Figure 6D and Table S1), which were both independent of Notch signaling. Moreover, the ability of Id3 to confer these capabilities on Rag2/Id3-deficient DN3 cells was abrogated by a mutation that eliminated the ability of Id3 to interact with and suppress E protein function (S->P; Figure 6C,D) (Deed et al., 1997). Id3 expression following transduction was approximately 10-fold higher than is expressed endogenously by CD24lo γδ cells (Figure S8).
Id3 induction by strong signals is necessary and sufficient to promote functional competence of γδ-lineage cells
A recent report implicated ligand “experience” in the thymus as an important determinant of effector function, arming such γδ cells as IFNγ-producing effectors (Jensen et al., 2008). Since we have found that ligand-mediated induction of Id3 was necessary to arm KN6 Tg γδ cells for TCR-induced proliferation and IFNγ production (Figure 5C), we asked whether Id3 was sufficient to promote the functional maturation of Rag2-deficient DN3 cells. Indeed, ectopic expression of Id3 in Rag2/Id3-deficient DN3 cells enabled these cells to become responsive to stimulation. In response to mitogenic stimulation, Id3-transduced cells both increased in size (Figure 7A) and produced IFNγ at levels comparable to mature CD24lo γδ cells (Figure 7B). Furthermore, Id3-transduction conferred these capabilities to Rag2/Id3-deficient precursors in the absence of Notch receptor-ligand interactions (Figure 7B). As above, the ability of Id3 to promote Notch-independent acquisition of effector function was lost when the Id3 mutant construct was used (Figure 7A,B). The Id3-induced acquisition of effector function was accompanied by the induction of ICER and Rgs1, a γδ-biased gene profile member that we have found to be closely linked to γδ-function (Figure 7C), but not induction of Nurr1, whose expression appears to be separable from γδ cell function (Figure 5). Sox13 is another recently described marker of γδ lineage cells (Melichar et al., 2007); however, while Sox13 expression was not detected in DP cells, it was also not found to be elevated in CD24lo γδ-lineage cells, in agreement with recent evidence suggesting it may not reliably mark all γδ cells (Kreslavsky et al., 2008). Taken together, these data suggest that Id3 is both necessary and sufficient for adopting the γδ-lineage fate, and for arming γδ T cells to produce IFNγ in response to ligand-experience.
Figure 7. Ectopic expression of Id3 is able to confer functional competence on TCR-thymocytes.
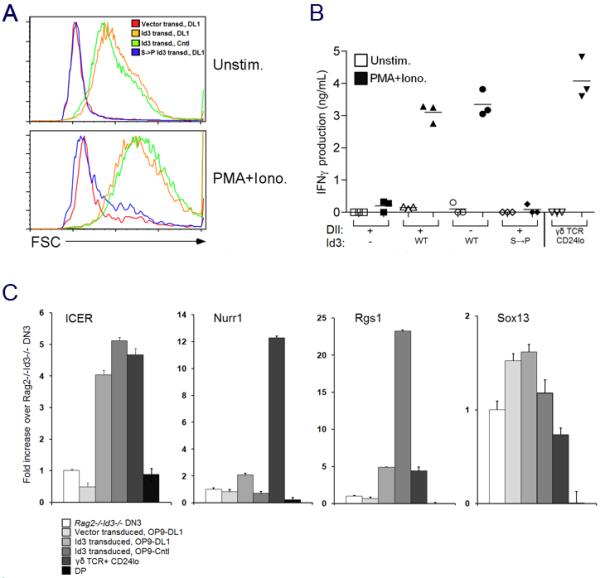
(A) Id3 transduced cells increase in size upon PMA + ionomycin stimulation.
(B) Id3 transduction confers competence to produce IFNγ on Rag2/Id3-deficient DN3 cells. (A-B) Sorted CD45+ GFP+ cells from transduced DN3 cells (as indicated) were stimulated for 36 h with PMA/Ionomycin (PMA/Iono) or placed in culture without stimulation (Unstim) and (A) analyzed by flow cytometry to determine cell size by forward-light scatter (FSC). (B) Levels of IFNγ in culture supernatants were quantified by an antibody-capture ELISA.
(C) Id3 transduction confers expression of some γδ-biased genes. RNA transcript levels of Rgs1, ICER, Nurr1, and Sox13 were measured by real time PCR as in Fig. 5 from sorted CD45+ GFP+ cells from transduced DN3 cells cultured on OP9-Cntl or OP9-DL1 cells as indicated.
DISCUSSION
The present report not only provides new insights into the role of Id3 in regulating T lineage fate assignment in response to strong signals, but also into the role of Id3 in rendering γδ-lineage cells independent of Notch signals and functionally competent. Id3 is required for strong signals to promote adoption of the γδ-lineage and oppose adoption of the αβ-fate. We recently reported that γδ-lineage cells are far less dependent on Notch signaling than αβ-lineage cells (Ciofani et al., 2006). Based on our present findings, it is now clear that Id3 is both necessary and sufficient for Notch independence of γδ-lineage cells; and that this independence requires a functional helix through which Id3 heterodimerizes with, and suppresses the function of, E proteins. Finally, Id3 is also necessary and sufficient to arm γδ-lineage cells for effector function defined by TCR- or mitogen-induced proliferation and IFNγ production. These findings place Id3 as a central molecular mediator of the strong signals that influence T cell fate, as well as their developmental and functional characteristics.
It has been suggested for some time that αβ- and γδ-lineage precursors are differentially dependent upon Notch signaling, though this has been controversial until recently (Garcia-Peydro et al., 2003; Hernandez-Hoyos and Alberola-Ila, 2003; Radtke et al., 2004; Washburn et al., 1997). Indeed, we showed that while αβ-lineage precursors remain Notch-dependent during their pre-TCR induced transition to the DP stage, γδ-precursors become independent of Notch signaling upon expression of the γδTCR (Ciofani et al., 2006). Nevertheless, it was unclear how γδTCR signaling permitted Notch-independent differentiation of γδTCR-expressing DN cells. We now show that Id3 is an important molecular effector of this process. The molecular basis for this effect has not been established; however, recent evidence from the Murre lab (as well as genetic analysis from Drosophila) suggests interplay between E proteins and the Notch pathway (Ikawa et al., 2006; Schwartz et al., 2006). Moreover, numerous reports support the notion that suppression of E protein function (at least transiently) is required for early thymocyte differentiation (Engel and Murre, 2004; Koltsova et al., 2007; Murre, 2005). We now propose a model whereby lineage commitment and Notch-dependence are determined by graded reductions in E protein activity mediated by differences in TCR signal strength (Figure S9). DN3 thymocytes are prevented from further development by E protein-mediated enforcement of the β-selection checkpoint, as evidenced by the ability of E2A-deficiency to enable pre-TCR deficient thymocytes to traverse the β-selection checkpoint and differentiate to the DP stage (Engel and Murre, 2004; Wojciechowski et al., 2007). Paradoxically, E protein deficiency blocks the development of pre-TCR expressing cells beyond the β-selection checkpoint to the DP stage, suggesting that the induction of αβ-lineage development by pre-TCR signals is dependent upon partial or temporally-restricted suppression of E protein activity (Engel et al., 2001). Accordingly, pre-TCR signals may suppress E protein activity, in part by induction of Id3 but are unable by themselves to suppress E protein activity beyond the threshold required for αβ-lineage development and thus require Notch-ligand interactions to do so. Notch signaling has been reported to suppress E protein function both by ERK-dependent induction of E protein degradation (Nie et al., 2003; Ordentlich et al., 1998) and through induction of Id3, which can directly repress E protein function (Reynaud-Deonauth et al., 2002). Unlike the weak signals that promote αβ-lineage development, the strong signals that confer Notch-independent differentiation upon γδ-lineage cells are dependent upon Id3 induction and are sufficient to suppress E protein activity beyond the threshold required for γδ-lineage commitment and development without assistance from Notch activity.
Because effects on growth and survival are associated with the regulation of lineage fate by Id3 in the context of strong TCR signals, it is possible that Id3 is not affecting lineage fate per se, but rather is affecting the growth and survival of pre-committed αβ or γδ lineage progenitors. Nevertheless, a number of lines of evidence argue against this perspective and for a role of Id3 in influencing lineage commitment. If strong signals were simply eliminating αβ-lineage cells, then Id3 transduction would be predicted to preferentially eliminate pre-TCR expressing αβ-lineage progenitors; however, Id3-transduction does not do so (Figure S4). Regarding the promotion of the γδ fate by strong signals (or Egr-transduction), the converse argument could be made that strong signals promote the generation of CD24lo γδ-lineage cells by preferentially expanding a small pre-committed population. However, we demonstrated that ectopic expression of the KN6 γδTCR in Rag2-/- thymocytes (which lack pre-committed γδ-lineage cells) promoted the γδ fate and opposed the αβ fate in the presence of ligand and produced the converse effect in the absence of ligand (Figure S5). It should be noted that this represents the first example where the expression of a specific γδTCR ligand was demonstrated to be required for selection and development of γδ-lineage precursors. Altogether, these data suggest that strong signals and Id3 drive lineage commitment.
While we had previously reported impaired development of γδ T cells in fetal Id3-deficient mice, the role of Id3 as a critical molecular effector in γδ cell specification and development is also revealed by the perturbation of γδ development in adult Id3-deficient mice. A recent report suggested that Id3 functions to restrain the development of γδ-lineage cells, as γδ T cell numbers were increased in adult Id3-deficient mice (Ueda-Hayakawa et al., 2009). The expansion was accompanied by enhanced V(D)J recombination at the TCR loci (γ,δ, and β) and an outgrowth of the Vγ1.1+ γδ cell subset. We observed similar effects of Id3-deficiency, but propose an alternative interpretation. We suggest that Id3-serves to terminate V(D)J recombination following TCR expression (either pre-TCR or γδTCR expression), thereby limiting the developmental time window during which TCR rearrangement can occur and explaining why TCR rearrangements were more extensive in Id3-deficient mice. We further demonstrated that while γδ T cell numbers were increased in Id3-defcient mice, the expansion appeared to be restricted to the Vγ1.1+ subset, as the Vγ2 and Vγ3 γδ subsets were severely reduced. We think these differential effects on particular Vγ subsets relate to their respective affinities for selecting ligands, with Id3-deficiency impairing selection of those with intermediate affinity, while allowing those of high affinity to escape deletion. This interpretation is supported by our demonstration that Id3-deficiency impairs selection of KN6 γδTCR Tg thymocytes on the intermediate affinity T-22d ligand, while rescuing them from deletion by T-10/22b, for which their affinity is 10-fold higher (Adams et al., 2008). Indeed, at least some Vγ1.1+ γδ cells, which are expanded in Id3-/- mice, are thought to belong to an NK γδ T cell lineage selected on high affinity ligand (Felices et al., 2009; Lees et al., 2001; Qi et al., 2009). Taken together, these findings suggest an apparent dichotomy of Id3 function, restricting production of γδ cells whose TCR affinity for ligand is very high, while promoting differentiation of the broader repertoire of non-autoreactive γδ-T cells.
Along with its role in promoting the Notch independence of developing γδ T cells, Id3 plays an important role in the acquisition of effector function by γδ-lineage cells. We found that Id3-deficiency impairs the ability of KN6 γδTCR Tg+ cells to proliferate and produce IFNγ in response to TCR ligation. Conversely, ectopic expression of Id3 in the absence of TCR expression, conferred the ability to proliferate and produce IFNγ in response to mitogenic signals. The Hayday lab has reported that developing γδ T cells require presentation of LTβ in trans by DP thymocytes for both functional competence and the expression of a number of genes termed the “γδ-biased gene profile” (Pennington et al., 2003; Silva-Santos et al., 2005). DP thymocytes are not present in appreciable numbers in either the ligand-expressing KN6 γδTCR Tg mice or among Id3-transduced DN3 cells cultured on OP9-monolayers in vitro. Nevertheless, despite the absence of DP thymocytes, the γδ T cells that developed in these models were functional and expressed several of the genes comprising the “γδ-biased gene profile” (Pennington et al., 2003; Silva-Santos et al., 2005). Interestingly, we found that Id3-deficiency separated γδ-function from expression of the γδ-biased gene profile in that the Id3-deficient γδ cells expressed a subset of the profile genes, with the notable exception of Rgs1, and yet were not functional (Figures 5 and 7). One possible explanation is that only a subset of the profile genes is uniquely associated with γδ-function. For example, although the function of Rgs1 in T cells is unknown, our data suggest that Rgs1 expression is tightly correlated with γδ T cell function, raising the interesting possibility that Rgs1 plays an active role in promoting the functional competence of γδ T cells.
The role of Id3 in promoting functional competence also provides an important mechanistic insight into a recent report regarding the role of ligand in shaping γδ T cell effector function. The report contends that ligand-mediated selection in the thymus does not play a significant role in shaping the γδ TCR repertoire but does influence the nature of effector function (Jensen et al., 2008). That is, γδ T cells that purportedly did not encounter a selecting ligand during development (antigen-naïve) are programmed to produce IL-17, whereas antigen-experienced γδ T cells produce IFNγ. While ruling out the possibility of ligand-mediated selection in the thymus is extremely difficult, we would agree that ligand-mediated selection arms γδ T cells to produce IFNγ (Figures 5 and 7). Indeed, our data supports the notion that the induction of Id3 following TCR-ligand engagement plays a critical role.
Taken together, our findings place Id3 as a central molecular mediator of the strong signals that influence T cell fate, enable developing γδ T cells to gain Notch-independent maturation, and shape their functional attributes. While it is likely that Id3 (perhaps with assistance from Id2) mediates these effects by suppressing the function of E proteins, this interpretation remains to be fully characterized. Future efforts will be directed at determining how the ERK-Egr-Id3 pathway, as well as other mediators of strong signals, is differentially employed to specify T-lineage fates in the thymus.
EXPERIMENTAL PROCEDURES
Mice
All mice were maintained in the fully accredited animal facilities at either the Fox Chase Cancer Center or the Sunnybrook Research Institute. All mouse strains used were described previously (Haks et al., 2005). Id3-/- mice were kindly provided by Drs. R. Benezra (Memorial Sloan-Kettering Cancer Center) and Y. Zhuang (Duke University) (Pan et al., 1999) and used both on the 129 background and after backcrossing to C57BL/6 for 3 generations. Egr1Tg mice were provided by Toru Miyazaki (Univ. Tokyo) (Miyazaki, 1997).
Retroviral transduction and culture
LZRS-pBMN-linker-IRES-eGFP (LZRS) and LZRS-Egr1 vectors have been previously described (Carleton et al., 2002). Murine Id3 was cloned by PCR using primers which spanned the start and stop codons. Serine 49 of Id3 was mutated to P (S49->P) by standard methodology and following sequence verification was subcloned into pMiY using standard methodology. Viral supernatants were produced as described and used to spin infect progenitors prior to seeding fetal thymic lobes (IL2Rγ-/-Rag2-/-) or plating on OP9 stromal layers as described (Ciofani et al., 2006; Haks et al., 2003). The effect of ectopically expressed genes on thymocyte development was assessed flow cytometrically by gating on cells positive for the fluorescent indicator protein. DN3 cells (d7 HSC OP9-DL1 cultured) were transduced by overnight co-culture using stable retrovirus-producing GP+E.86 packaging cells as previously described followed by coculture on OP9 monolayers in OP9-medium (α-mem supplemented with 1 ng/mL mouse IL-7 (Peprotech) and 5 ng/mL human recombinant Flt-3 ligand (Peprotech)) (Ciofani et al., 2006).
Flow cytometry
Skin preparations were performed as described (Havran et al., 1991). All single cell suspensions were stained with commercially available Ab (BD PharMingen, eBiosciences, and Caltag) and analyzed on either a FACSVantageSE (Becton Dickinson) or BDLSR using Flowjo software (Treestar, Inc.). Anti-Vγ Ab were generously provided by Dr. P. Pareira (Pasteur Institute). BrdU staining was performed according to manufacturers specifications following a 4h pulse with BrdU (i.p. injection of 100μg/mouse). Where applicable, dead cells were excluded from the analyses using propidium iodide (PI) or DAPI gating.
Real time PCR
Thymocyte populations were purified by flow cytometry, where necessary, following depletion using magnetic anti-CD8 beads (Miltenyi Biotec). RNA was extracted using the Rneasy kit according to manufacturer’s specifications (Qiagen) and converted to cDNA using the SuperscriptII kit (Invitrogen). Expression of the indicated genes was measured by real time PCR using stock Taqman probes purchased from Applied Biosystems, Inc., as described, on a ABI Prism 7500 Real Time PCR machine (Koltsova et al., 2007). Some of the quantitative PCR analyses were performed using SYBR GreenER (Invitrogen). Primer/probe set order numbers will be supplied upon request. β-Actin expression was used to normalize cycles thresholds, which were then converted into fold difference. All samples were analyzed in triplicates or quadruplicates for +RT, duplicates for -RT, and a nontemplate control was added to each PCR reaction.
Immunoblotting
Cells lysates and nuclear extracts were prepared as previously described (Ciofani et al., 2004; Engel et al., 2001), resolved by SDS-PAGE through 10% acrylamide gels, and transferred onto PVDF membranes (Amersham Biosciences, Baie d’Urfé, QC). Immunoblotting was performed with antibodies specific for E47 (N-649), HEB (A-20), Tata Binding Protein (ITBP-18), and GAPDH, all from Santa Cruz.
Proliferation and cytokine production
DN thymocytes prepared by anti-CD8 bead depletion were stimulated on plate bound anti-CD3 Ab and subjected to MTT assay as described (Haks et al., 2005). DN thymocytes were labeled with CFSE according to manufacturer’s directions (Molecular Probes, Invitrogen) and stimulated for 48 hours with plate-bound anti-CD3 Ab (5μg/ml). CFSE labeling was detected by flow cytometry. For detection of IFNγ producing cells, DN thymocytes were stimulated for 24 hours with anti-CD3 Ab. For the last 4 hours of stimulation GolgiPlug™( Benton Dickinson) was added to block protein secretion. The cells were permabilized and stained for intracellular IFNγ using the BD Cytofix/Cytoperm™ Solution Kit and the percentage of IFNγ producing cells determined by flow cytometry.
PMA/Ionomycin stimulation
CD45+ GFP+ cells were sorted from transduced DN3 cultured for 6d on either OP9-Cntl or OP9-DL1 cells. Sorted cells (5 × 104 cells/well of a 96-well plate) were subsequently cultured with 50 ng/mL PMA (Sigma) and 500 ng/mL Ionomycin (Sigma) in OP9 medium supplemented with 50 U/mL IL-2 (Peprotech). Supernatants were harvested after 36 h and IFNγ levels were quantified using the DuoSet ELISA Development System (R&D systems) according to the manufacturer’s protocol.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. A. Singer and M.K. Anderson for critically reviewing the manuscript. We acknowledge the following core facilities at Fox Chase Cancer Center: Cell Culture, DNA Sequencing, DNA Synthesis, Flow Cytometry, and Laboratory Animal; the Sunnybrook Research Institute Centre for Cytometry and Scanning Microscopy, and the Division of Comparative Research. This work was supported by NIH grants (DLW) AI081314, NIH core grant P01CA06927, Center grant #P30-DK-50306, and an appropriation from the Commonwealth of Pennsylvania; and by a grant (JCZP) from the Canadian Institute of Health Research (CIHR-MOP42387). JCZP is supported by a Canada Research Chair in Developmental Immunology. GWW and MC were supported by a CIHR Scholarship. JPL and SYL were supported by the Fox Chase Cancer Plain & Fancy and Greenwald Postdoctoral Fellowships, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no competing financial interests.
REFERENCES
- Adams EJ, Strop P, Shin S, Chien YH, Garcia KC. An autonomous CDR3delta is sufficient for recognition of the nonclassical MHC class I molecules T10 and T22 by gammadelta T cells. Nature Immunology. 2008;9:777–784. doi: 10.1038/ni.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agata Y, Tamaki N, Sakamoto S, Ikawa T, Masuda K, Kawamoto H, Murre C. Regulation of T cell receptor beta gene rearrangements and allelic exclusion by the helix-loop-helix protein, E47. Immunity. 2007;27:871–884. doi: 10.1016/j.immuni.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Bain G, Romanow WJ, Albers K, Havran WL, Murre C. Positive and negative regulation of V(D)J recombination by the E2A proteins. Journal of Experimental Medicine. 1999;189:289–300. doi: 10.1084/jem.189.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barndt RJ, Dai M, Zhuang Y. Functions of E2A-HEB heterodimers in T-cell development revealed by a dominant negative mutation of HEB. Mol Cell Biol. 2000;20:6677–6685. doi: 10.1128/mcb.20.18.6677-6685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton M, Haks MC, Smeele SA, Jones A, Belkowski SM, Berger MA, Linsley P, Kruisbeek AM, Wiest DL. Early growth response transcription factors are required for development of CD4(-)CD8(-) thymocytes to the CD4(+)CD8(+) stage. Journal of Immunology. 2002;168:1649–1658. doi: 10.4049/jimmunol.168.4.1649. [DOI] [PubMed] [Google Scholar]
- Cheng LE, Chan FK, Cado D, Winoto A. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO Journal. 1997;16:1865–1875. doi: 10.1093/emboj/16.8.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Knowles GC, Wiest DL, von Boehmer H, Zuniga-Pflucker JC. Stage-specific and differential notch dependency at the alphabeta and gammadelta T lineage bifurcation. Immunity. 2006;25:105–116. doi: 10.1016/j.immuni.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, Maryanski JL, Zuniga-Pflucker JC. Obligatory role for cooperative signaling by pre-TCR and Notch during thymocyte differentiation. J Immunol. 2004;172:5230–5239. doi: 10.4049/jimmunol.172.9.5230. [DOI] [PubMed] [Google Scholar]
- Deed RW, Hara D, Atherton GT, Peters G, Norton JD. Regulation of Id3 cell cycle function by Cdk-2-dependent phosphorylation. Molecular & Cellular Biology. 1997;17:6815–6821. doi: 10.1128/mcb.17.12.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley EC, Girardi M, Owen MJ, Hayday AC. Alpha beta and gamma delta T cells can share a late common precursor. Current Biology. 1995;5:659–669. doi: 10.1016/s0960-9822(95)00131-x. [DOI] [PubMed] [Google Scholar]
- Engel I, Johns C, Bain G, Rivera RR, Murre C. Early thymocyte development is regulated by modulation of e2a protein activity. J Exp Med. 2001;194:733–746. doi: 10.1084/jem.194.6.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel I, Murre C. E2A proteins enforce a proliferation checkpoint in developing thymocytes. Embo J. 2004;23:202–211. doi: 10.1038/sj.emboj.7600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felices M, Yin CC, Kosaka Y, Kang J, Berg LJ. Tec kinase Itk in gammadeltaT cells is pivotal for controlling IgE production in vivo. Proc Natl Acad Sci U S A. 2009;106:8308–8313. doi: 10.1073/pnas.0808459106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe AI, Krueger A, Gounari F, Zuniga-Pflucker JC, von Boehmer H. Differential synergy of Notch and T cell receptor signaling determines alphabeta versus gammadelta lineage fate. J Exp Med. 2006;203:1579–1590. doi: 10.1084/jem.20060474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Peydro M, de Yebenes VG, Toribio ML. Sustained Notch1 signaling instructs the earliest human intrathymic precursors to adopt a gammadelta T-cell fate in fetal thymus organ culture. Blood. 2003;102:2444–2451. doi: 10.1182/blood-2002-10-3261. [DOI] [PubMed] [Google Scholar]
- Haks MC, Belkowski SM, Ciofani M, Rhodes M, Lefebvre JM, Trop S, Hugo P, Zuniga-Pflucker JC, Wiest DL, Carleton M, et al. Low activation threshold as a mechanism for ligand-independent signaling in pre-T cells. J Immunol. 2003;170:2853–2861. doi: 10.4049/jimmunol.170.6.2853. [DOI] [PubMed] [Google Scholar]
- Haks MC, Lefebvre JM, Lauritsen JP, Carleton M, Rhodes M, Miyazaki T, Kappes DJ, Wiest DL. Attenuation of gammadeltaTCR signaling efficiently diverts thymocytes to the alphabeta lineage. Immunity. 2005;22:595–606. doi: 10.1016/j.immuni.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Havran WL, Chien YH, Allison JP. Recognition of self antigens by skin-derived T cells with invariant gamma delta antigen receptors. Science. 1991;252:1430–1432. doi: 10.1126/science.1828619. [DOI] [PubMed] [Google Scholar]
- Hayday AC, Pennington DJ. Key factors in the organized chaos of early T cell development. Nature Immunology. 2007;8:137–144. doi: 10.1038/ni1436. [DOI] [PubMed] [Google Scholar]
- Hayes SM, Li L, Love PE. TCR signal strength influences alphabeta/gammadelta lineage fate. Immunity. 2005;22:583–593. doi: 10.1016/j.immuni.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Hernandez-Hoyos G, Alberola-Ila J. A Notch so simple influence on T cell development. Semin Cell Dev Biol. 2003;14:121–125. doi: 10.1016/s1084-9521(02)00180-5. [DOI] [PubMed] [Google Scholar]
- Huang Z, Nie L, Xu M, Sun XH. Notch-induced E2A degradation requires CHIP and Hsc70 as novel facilitators of ubiquitination. Mol Cell Biol. 2004;24:8951–8962. doi: 10.1128/MCB.24.20.8951-8962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J Exp Med. 2006;203:1329–1342. doi: 10.1084/jem.20060268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, Steinman L, Saito T, Locksley RM, Davis MM, et al. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity. 2008;29:90–100. doi: 10.1016/j.immuni.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltsova EK, Ciofani M, Benezra R, Miyazaki T, Clipstone N, Zuniga-Pflucker JC, Wiest DL. Early growth response 1 and NF-ATc1 act in concert to promote thymocyte development beyond the beta-selection checkpoint. J Immunol. 2007;179:4694–4703. doi: 10.4049/jimmunol.179.7.4694. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, Chin DM, Bacus SS, Stark GR, Gudkov AV. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. Embo J. 1997;16:1391–1400. doi: 10.1093/emboj/16.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreslavsky T, Garbe AI, Krueger A, von Boehmer H. T cell receptor-instructed alphabeta versus gammadelta lineage commitment revealed by single-cell analysis. J Exp Med. 2008;205:1173–1186. doi: 10.1084/jem.20072425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees RK, Ferrero I, MacDonald HR. Tissue-specific segregation of TCRgamma delta+ NKT cells according to phenotype TCR repertoire and activation status: parallels with TCR alphabeta+NKT cells. Eur J Immunol. 2001;31:2901–2909. doi: 10.1002/1521-4141(2001010)31:10<2901::aid-immu2901>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Smith SW, McLaughlin KA, Schwartz LM, Osborne BA. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature. 1994;367:281–284. doi: 10.1038/367281a0. [DOI] [PubMed] [Google Scholar]
- Melichar HJ, Narayan K, Der SD, Hiraoka Y, Gardiol N, Jeannet G, Held W, Chambers CA, Kang J. Regulation of gammadelta versus alphabeta T lymphocyte differentiation by the transcription factor SOX13. Science. 2007;315:230–233. doi: 10.1126/science.1135344. [DOI] [PubMed] [Google Scholar]
- Miyazaki T. Two distinct steps during thymocyte maturation from CD4-CD8- to CD4+CD8+ distinguished in the early growth response (Egr)-1 transgenic mice with a recombinase-activating gene-deficient background. Journal of Experimental Medicine. 1997;186:877–885. doi: 10.1084/jem.186.6.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol. 2005;6:1079–1086. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- Nie L, Xu M, Vladimirova A, Sun XH. Notch-induced E2A ubiquitination and degradation are controlled by MAP kinase activities. Embo J. 2003;22:5780–5792. doi: 10.1093/emboj/cdg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordentlich P, Lin A, Shen CP, Blaumueller C, Matsuno K, Artavanis-Tsakonas S, Kadesch T. Notch inhibition of E47 supports the existence of a novel signaling pathway. Mol Cell Biol. 1998;18:2230–2239. doi: 10.1128/mcb.18.4.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Sato S, Frederick JP, Sun XH, Zhuang Y. Impaired immune responses and B-cell proliferation in mice lacking the Id3 gene. Mol Cell Biol. 1999;19:5969–5980. doi: 10.1128/mcb.19.9.5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington DJ, Silva-Santos B, Shires J, Theodoridis E, Pollitt C, Wise EL, Tigelaar RE, Owen MJ, Hayday AC. The inter-relatedness and interdependence of mouse T cell receptor gammadelta+ and alphabeta+ cells. Nat Immunol. 2003;4:991–998. doi: 10.1038/ni979. [DOI] [PubMed] [Google Scholar]
- Pereira P, Zijlstra M, McMaster J, Loring JM, Jaenisch R, Tonegawa S. Blockade of transgenic gamma delta T cell development in beta 2-microglobulin deficient mice. EMBO Journal. 1992;11:25–31. doi: 10.1002/j.1460-2075.1992.tb05023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Xia M, Hu J, Hicks E, Iyer A, Xiong N, August A. Enhanced development of CD4+ {gamma}{delta} T cells in the absence of Itk results in elevated IgE production. Blood. 2009 doi: 10.1182/blood-2008-12-196345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke F, Wilson A, Mancini SJ, MacDonald HR. Notch regulation of lymphocyte development and function. Nat Immunol. 2004;5:247–253. doi: 10.1038/ni1045. [DOI] [PubMed] [Google Scholar]
- Reynaud-Deonauth S, Zhang H, Afouda A, Taillefert S, Beatus P, Kloc M, Etkin LD, Fischer-Lougheed J, Spohr G. Notch signaling is involved in the regulation of Id3 gene transcription during Xenopus embryogenesis. Differentiation. 2002;69:198–208. doi: 10.1046/j.1432-0436.2002.690413.x. [DOI] [PubMed] [Google Scholar]
- Rivera RR, Johns CP, Quan J, Johnson RS, Murre C. Thymocyte selection is regulated by the helix-loop-helix inhibitor protein, Id3. Immunity. 2000;12:17–26. doi: 10.1016/s1074-7613(00)80155-7. [DOI] [PubMed] [Google Scholar]
- Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A. 2006;103:9976–9981. doi: 10.1073/pnas.0603728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Santos B, Pennington DJ, Hayday AC. Lymphotoxin-mediated regulation of gammadelta cell differentiation by alphabeta T cell progenitors. Science. 2005;307:925–928. doi: 10.1126/science.1103978. [DOI] [PubMed] [Google Scholar]
- Ueda-Hayakawa I, Mahlios J, Zhuang Y. Id3 restricts the developmental potential of gammadelta lineage during thymopoiesis. J Immunol. 2009;182:5306–5316. doi: 10.4049/jimmunol.0804249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes BJ, Cado D, Robey E. Notch activity influences the alphabeta versus gammadelta T cell lineage decision. Cell. 1997;88:833–843. doi: 10.1016/s0092-8674(00)81929-7. [DOI] [PubMed] [Google Scholar]
- Wojciechowski J, Lai A, Kondo M, Zhuang Y. E2A and HEB are required to block thymocyte proliferation prior to pre-TCR expression. J Immunol. 2007;178:5717–5726. doi: 10.4049/jimmunol.178.9.5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Schwartz R, Engel I, Murre C, Kersh GJ. Interplay between RORgammat, Egr3, and E proteins controls proliferation in response to pre-TCR signals. Immunity. 2006;24:813–826. doi: 10.1016/j.immuni.2006.03.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.