

SUMMARY
Gene transfer has potential as a once-only treatment that reduces viral load, preserves the immune system, and avoids lifetime highly active antiretroviral therapy. This study, the first randomized, double-blind, placebo-controlled, phase II cell-delivered gene transfer clinical trial, was conducted in 74 HIV-1 infected adults who received a tat/vpr specific anti-HIV ribozyme (OZ1) or placebo delivered in autologous CD34+ hematopoietic progenitor cells. There were no OZ1-related adverse events. There was no statistical difference in viral load between the OZ1 and placebo group at the primary end-point (average at weeks 47 and 48) but time weighted areas under the curve from weeks 40-48 and 40-100 were significantly lower in the OZ1 group. Throughout the 100 weeks, CD4+ lymphocyte counts were higher in the OZ1 group. This study provides the first indication that cell-delivered gene transfer is safe and biologically active in HIV patients and can be developed as a conventional therapeutic product.
INTRODUCTION
Highly active anti-retroviral therapy (HAART) has greatly improved the prognosis of individuals infected with HIV-1, but is often associated with toxicities, adverse interactions with other drugs and the emergence of viral resistance1. Results from HIV-1 vaccine studies have been disappointing. In fact, increased viral replication after therapeutic or prophylactic immunization has been reported2. Gene-based therapies, including ribozyme, antisense, aptamer, RNAi, zinc finger nuclease, dominant negative protein, fusion inhibitor, intracellular antibody and viral decoy approaches3-26, have been proposed as a long-lived alternative to small-molecule HAART10,13,20,22,24,26,27. Some of these were shown to be safe in Phase I clinical trials4,12,17,20,28-30. Ribozymes are small catalytic RNA molecules that can be engineered to target specific RNA sequences10,24,26,27,31-33 and offer advantages by virtue of their specificity of target site recognition and cleavage, without any reported “off target” effects10,24,31,34.
OZ1 comprises a Moloney Murine Leukemia Virus-based, replication-incompetent gamma retroviral vector (LNL6) containing a gene that encodes a ribozyme that targets the overlapping vpr and tat reading frames of HIV-132,35. OZ1 inhibits the replication of laboratory and clinical isolates of HIV-1 in vitro. Resistance mutations in the region of HIV-1 targeted by OZ1 were not observed in long term cell culture10,27,32,35.
Two phase I clinical trials have been conducted using either CD4+ T lymphocytes29 or CD34+ hematopoietic stem cells4 to assess the feasibility and safety of ex vivo transduction and re-infusion of OZ1-transduced cells. Both trials demonstrated that the approach was technically feasible and safe. There was no serious adverse event related to the gene transfer process or the gene transfer product during the study period or the subsequent long-term safety follow-up. Safety parameters were assessed in accordance with United States Food and Drug Administration (FDA)/ Center for Biologics Evaluation and Research (CBER) recommendations36,37.
Our concept, tested in this study, is that OZ1-transduced CD34+ hematopoietic progenitor cells would engraft, divide and differentiate in vivo to produce a pool of mature myeloid and lymphoid cells that are protected from productive HIV-1 replication27 This multi-center, phase II, randomized, double-blind, placebo-controlled clinical trial evaluated the safety and efficacy of OZ1. Seventy four (74) HIV-1 infected participants were randomized (1:1) and received OZ1-transduced (n=38) or Control (n=36) CD34+ cells (Fig 1b). Each participant received a single intravenous infusion of autologous CD34+ cells without undergoing myeloablation or any form of bone marrow conditioning. Gene transfer safety parameters were assessed throughout the study in accordance with FDA/CBER guidelines36,37. The protocol included two antiretroviral treatment interruptions to provide positive selective pressure for OZ1-protected cells. The impact of OZ1 on plasma HIV-1 viral load was assessed at the end of the second, eight-week, analytic treatment interruption (the primary endpoint) (Fig 1c). Secondary endpoints of quantitative marking (presence of OZ1 gene) and expression (active RNA form of OZ1) of the gene transfer product, time-weighted area under the curve for viral load (TWAUC), CD4+ T lymphocyte count in absolute and percentage of T lymphocytes (CD4%), presence of HIV-1 proviral DNA and thymic function (T cell receptor excision circles, TREC) were also assessed to week 100. The OZ1 treatment group participants are now enrolled in a long-term safety follow-up protocol.
Figure 1. Ribozyme target site within HIV-1 genome, schema of gene modified cell manufacture and protocol design.
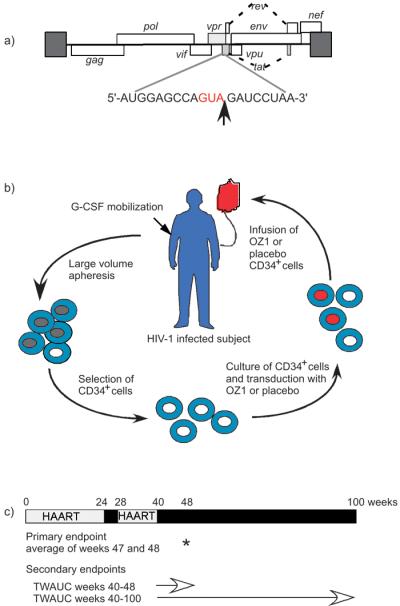
a) The HIV-1 genome is shown together with the target nucleotide sequence. The cleavage site is indicated by an arrow.
b) HIV-1 positive individuals received 30μg/kg/day G-CSF over 5 days. Peripheral blood stem cells were collected by large volume apheresis, using either manual or automatic settings, on days 4 and 5. CD34+ cells were selected and cultured in the presence of Stem Cell Factor and Megakaryocyte Growth & Differentiation Factor (SCF/MGDF) for 30-36 hours to recruit cells into cell cycle in preparation for retroviral transduction with OZ1 or placebo in the presence of RetroNectin, and SCF/MGDF. On Day 8, the gene modified cell product was washed, prepared for infusion and tested for purity, potency and sterility prior to infusion. All participants received their autologous cell product as per the randomization.
c) Participants continued HAART for 24 weeks post infusion before entering a four-week treatment interruption (week 24-28) which was intended to apply selective pressure on any OZ1 containing cells. The analytical treatment interruption commenced after week 40 post infusion. Assessments were conducted weekly during the analytical treatment interruption until week 48 and then monthly until either the resumption of HAART or week 100. Participants were advised to recommence HAART if the protocol defined viral load limits were reached: 500,000 copies/ml (up to week 48) and 100,000 copies/ml (week 48-100). HAART was also recommenced if protocol defined limits for CD4+ lymphocyte counts (<150 cells/μl) were reached or if the participant developed an opportunistic infection. Participants who recommenced HAART were scheduled to attend 3 monthly visits.
RESULTS
Participants and Treatment
Of 107 participants assessed for eligibility between July 2003 and January 2006, 74 were randomized and received an autologous CD34+ cell infusion (USA n=42; Australia n=32). Intention-to-treat (all infused participants; ITT) and per-protocol (all participants who entered the analytic treatment interruption; PP) populations were defined prior to data-base lock and unblinding (Fig 2). Baseline participant demographics, HIV-1 history, were comparable (Table 1). The dose of G-CSF and the volume of the apheresis collection were similar for the two groups (data not shown). Both treatment groups received an equivalent dose of viable CD34+ cells/kg. The infused cell product in the OZ1 treatment group contained a mean of 54.0% OZ1 gene-marked cells, based on 1 vector copy per cell (Table 1). Data were obtained to week 100 unless participants were lost to follow-up beforehand. Participants in the OZ1 group are now enrolled in a separate long-term follow-up protocol which will continue for at least 15 years. To date, the longest follow-up period from the time of infusion of the first participant is 5 years.
Figure 2. Patient Disposition.

At screening, a total of 28 patients were excluded; 21 did not meet eligibility criteria and 7 were excluded for other reasons including intervening adverse events. Of the 78 participants enrolled in the study, 2 were withdrawn prior to randomization and 2 after randomization due to cell processing failures. 3 participants were lost to follow up prior to the primary efficacy endpoint. A total of 9 participants did not enter the analytic treatment interruption.
OZ1 gene marking (DNA) and OZ1 expression (RNA)
OZ1 gene marking (DNA) and expression (RNA) were assessed to measure the frequency of gene containing cells and the production of active ribozyme, respectively. At week 4 post infusion, OZ1 DNA was detected in PBMC from 94% of the participants in the OZ1 treatment group. This percentage decreased to 12% of participants tested at week 48 and to 7% at week 100. Data from peripheral blood cell subsets (CD4+, CD8+ T cells; B cells; monocytes and granulocytes) showed a similar decline in the percentages over time. OZ1 DNA did not reach the quantifiable range of the assay in any blood cell sample at any time point. Limits of detection and quantification for OZ1 DNA were 0.01% and 0.38% of cells analysed, respectively.
OZ1 RNA was detected in PBMC (≥ 1 copy/250ng RNA) in all participants in the OZ1 treatment group at week 1. At this time point, expression was quantifiable in five participants (370 to 600 copies/250 ng of total RNA); expression in the other participants was detectable but not quantifiable. OZ1 RNA was detected in 94% of these participants at week 4 and 29% at week 48 but not quantifiable in any sample. Of the 21 participants who remained off HAART beyond week 48 post infusion, OZ1 RNA was detected in 15 participants. At week 100, OZ1 RNA was detected in 5 out of 12 (42%) participants for whom RNA samples were available and who continued in the treatment interruption to week 100, versus 2 of 19 (11%) participants who had resumed HAART. In some participants, insufficient RNA was available for analysis at this time point. Examples of results for the real-time quantitative PCR RNA assay, and the percentages of participants in which OZ1 DNA and RNA were detected over time have been provided in the Supplementary Material; Supplementary Fig. 1 & 2. Data from peripheral blood cell subsets showed a similar decline in the OZ1 expression over time.
Primary Efficacy End Point
For both intention-to-treat and per-protocol populations, the mean plasma HIV-1 viral load at weeks 47/48 (8 weeks after entering the analytic treatment interruption) was lower in the OZ1 group but the difference was not statistically significant (Table 2). The number of participants with a plasma viral load of less than 4 log10 copies/ml at weeks 47/48 (15/32) was greater in the OZ1 group than in the Control group (5/33) (p=0.009). In the per-protocol OZ1 population, the correlation between the dose of CD34+ cells/kg infused and the viral load at weeks 47/48 was weakly negative (Rho −0.319; p=0.076). There was no such negative correlation in the Control group (Rho +0.188; p=0.312).
Secondary Efficacy End Points
The viral load data from the analytic treatment interruption beginning at week 40 were used to calculate the TWAUC (Fig 3). The TWAUC in the per-protocol population was statistically lower in the OZ1 group (weeks 40-48; median difference −0.34 log10 copies/ml/day, p=0.024 and weeks 40-100 median difference −0.37 log10 copies/ml/day, p=0.034) (Table 2). The number of participants with a TWAUC in the lowest quartile during weeks 40-100 was statistically greater in the OZ1 group (OZ1 n=12; 37.5%, Control n=5; 15.2%, p=0.04). The median times to reach 3, 4 and 5 log10 copies/ml viral load during the analytic treatment interruption, in the per-protocol population, were greater in the OZ1 group. However, a statistically significant difference was demonstrated only for the time to reach to 4 log10 copies/ml viral load (Table 2).
Figure 3. HIV-1 Viral Load: Intention-to-Treat Population.

The mean log10 viral load shown here was determined using real time assay results. As participants recommenced HAART at different times, the n values at the different time points have been included. PCR sequence data of the HIV-1 protease and reverse transcriptase genes were used to generate a virtual phenotype. Similar data was seen for the per-protocol population Only one participant with a mutation associated with resistance to protease inhibitors failed to suppress HIV-1 replication after the recommencement of HAART. screening visit is indicated as scr, ●---●---● OZ1 group, ○- -○- -○ Control group
During the analytic treatment interruption, 17 (45%) participants in the OZ1 group reinitiated HAART compared to 22 (61%) Control participants. For the participants in the Control group, the median time to re-initiation of HAART was 29.4 weeks (n=22, 61%), and for the OZ1 group it was greater than 60 weeks. This difference between treatment groups was not statistically significant.
Both treatment groups displayed peripheral blood lymphodepletion related to G-CSF mobilization and large volume apheresis38-40 (Fig 4 and Supplementary Fig 3. The relationship between viral load and CD4+ count was similar for both treatment groups. For the per-protocol population, the CD4+ lymphocyte counts (mean cells/μl and % of T lymphocytes) were 476 and 27% in the OZ1 group compared to 437 and 24% in the Control group at weeks 47/48 and, 490 and 28% (OZ1) and 441 and 25% (Control) at week 100. Analysis of the percentage of CD4+ and CD8+ T lymphocytes, showed higher CD4 and correspondingly lower CD8 in the OZ1 group (Fig 4a-c). CD4+ lymphocyte recovery was also examined in participants who recommenced HAART. In both treatment groups, once HAART was recommenced, CD4 + T lymphocytes (percentage) increased towards the baseline to week 100 (Fig 4d). The median decrease in CD4+ T lymphocyte count from baseline at week 100 was approximately 100 cells/μl.
Figure 4. T lymphocyte counts over time.



T lymphocyte counts are presented as a mean percentage of CD3+ T-lymphocytes for each treatment group. For panels A & B, only patients who did not resume HAART from week 40 are included in the analysis. An additional Supplementary Figure 4 showing CD4+ T lymphocytes as percentage of CD3+ T-lymphocytes: Intention-To-Treat Population is in the Supplementary Material.
A) Change in CD4 percentage from baseline at screening: Intention-To-Treat Population, B) Change in CD8 percentage from baseline at screening: Intention-To-Treat Population, and C) CD4+ T-lymphocytes as percentage of CD3+ T-lymphocytes only for participants who resumed HAART prior to or at week 52. (At week 100, n=10 in OZ1 treatment group, n=9 in the Control group). The dotted line in the figure represents the baseline percentage. ●---●---● OZ1 group, ○- -○- -○ Control group
Exploratory analysis of the primary end point at weeks 47/48 was performed in the subset of participants who continued to display OZ1 expression in PBMC at any time point beyond week 48. The median plasma viral load in these OZ1 participants (log 3.81, 95% CI median 3.18-4.23; n=15) was significantly lower than in the Control participants (log 4.57, 95% CI median 4.31-4.83; n=33) (p=0.003). The median TWAUC from week 40-48 in these OZ1 participants (3.44 log copies/ml/day, n=15) was significantly lower than in the Control participants (3.93 log copies/ml/day, n=33) (p=0.03) as was the median TWAUC from week 40-100 (3.97 log copies/ml/day, n=15) in comparison to the Control group (4.53 log copies/ml/day, n=33) (p=0.005). Although the HIV-1 viral load in the OZ1 expressors were lower than the non-OZ1 expressors, this difference was not statistically significant.
Safety Evaluations
There was no death or clinically severe cardiovascular, renal or hepatic adverse event reported for randomized participants (Supplementary Table 1). All 3 participants who experienced a serious adverse event were in the Control group. None of these serious adverse events was assessed as being related to G-CSF, leukapheresis, cell infusion or other study procedures. Most participants (99%) experienced at least one G-CSF associated adverse event such as musculoskeletal pain (92%) or headache (32%). Three participants, 2 of whom were in the OZ1 group, developed transient hepatomegaly associated with G-CSF administration; 69% and 77% of all participants had transient elevations of aspartate aminotransferase and alanine aminotransferase respectively, again associated with G-CSF. One participant in the OZ1 group developed a grade 4 elevation of aspartate aminotransferase at week 40, however, it returned to within the normal range by week 48 of the study.
Six participants (OZ1 n=2; Control n=4) experienced asymptomatic grade 3 thrombocytopenia following apheresis. One of the OZ1 participants required a platelet transfusion. Three participants experienced thrombocytopenia associated with the first interruption of HAART. In all 3 cases, platelet counts returned to normal after recommencement of HAART. Three participants (OZ1 n=1; Control n=2) developed grade 3 neutropenia; two recovered to the normal range by the next visit. One participant had two episodes of grade 3 neutropenia (0.62 and 0.65 × 103/μl), at week 20 and week 64 post-infusion after which time neutrophil counts returned to baseline levels. Severe adverse events associated with community acquired infections were reported in both groups (OZ1 n=2, 5.3%, Control n=3, 8.3%) and consisted of cases of influenza, oral herpes simplex and tonsillitis. Mild to moderate cases of oral candidiasis (total n=4) and oral hairy leukoplakia (total n=2) developed during periods of antiretroviral interruption equally in participants from both groups. CD4+ lymphocyte counts at the time of these events ranged from 342-467 cells/μl and 19-36% of T lymphocytes.
No predominant integration site (LAM-PCR) or replication competent retrovirus was detected at any time point. No hematopoietic cell clonal expansion or other event suggestive of insertional mutagenesis was observed. The OZ1 target sequence in the HIV-1 plasma RNA was assessed over time. Individual variation in nucleotide sequence was detected in 9 of 63 participants where sequence data was available from more than one visit (OZ1 n=5/35, 14.3%; Control n=4/28, 14.3%). None of these changes was a modification at the ribozyme recognition site to prevent cleavage, nor suggested the evolution of resistant virus.
DISCUSSION
The present study is the largest cell-delivered gene therapy trial conducted to dateand the only randomised controlled phase II study of a potential cell-delivered gene therapeutic. Although the primary efficacy endpoint was not reached, HIV-1 viral loads were consistently lower in the OZ1 group for all analyses. Statistically significant differences were observed for the number of participants with less than 4 log10 copies/ml viral load at week 47/48, the per-protocol TWAUC week 40-48 and week 40-100, the number of participants in the lowest quartile of TWAUC week 40-100, and time to 4 log10 copies/ml viral load in the analytic treatment interruption. The impact of OZ1 on HIV-1 viral load is further supported by the correlation with OZ1 expression beyond week 48 and the correlation with CD34+ cell dose. The CD4+ and CD8+ T lymphocyte count data were consistent with the HIV-1 viral load differences between the treatment groups. Target site sequencing showed no evidence for the development of resistance to OZ1 during this 100-week study, which could be due to the low percentage of gene-marked cells and/or the production of a less fit virus due to mutation(s) in the highly conserved region targeted by OZ1 32.
Mathematical modeling undertaken prior to this clinical study (manuscript submitted; GPS, GC Fanning, JLM, LAE, SMP, JMM) predicted that during the analytic treatment interruption, OZ1 recipients would experience an initial increase in HIV-1 viral load followed by the establishment of a lower set point. The model predicted that the establishment of OZ1 CD34+ cells in the bone marrow at 10-20% of total CD34+ cell population could reduce viral load by 0.5 log10 in one year. In this phase II study, bone marrow aspiration was not performed because it is invasive and would present additional risk, particularly for the control participants; hence the percentage engraftment is not known. Based on the frequency of cells containing OZ1 (0.01% to 0.38%) in the peripheral blood, it can be inferred that engraftment was substantially lower than 10-20%. Previous studies have also shown that in the absence of strong selective pressure, peripheral blood reconstitution with gene-containing cells is limited 41-44. Given the engraftment and gene expression results in this study, the biological effect of OZ1 was greater than predicted by the modelling. Candidate mechanisms for this additional effect include an impact of OZ1 on cell to cell transmission in the lymphatic system; compartmentalisation such that the number of OZ1-containing cells in the peripheral blood is not representative of the survival of OZ1-containing cells in sequestered discrete foci (eg the bone marrow and lymphatic system, in particular the GALT);and protection of particular cell sub-populations such as antigen-presenting cells (including dendritic cells and macrophages) or HIV-specific CD4+ T lymphocytes
Improvements in mobilisation, CD34+ cell collection and transduction compared to the phase I study8 resulted in a mean transduced CD34+ cell dose of 5 × 106/kg and an increase in the frequency of OZ1-containing cells in the peripheral blood (approximate 2 log10). The peripheral blood lymphodepletion observed here has been described after large volume apheresis38,39. As with those studies, the protracted recovery appears to be limited to the T lymphocyte population. This may reflect slow thymic production and/or shortened lifespan of mature T-lymphocytes. The protracted lymphocyte recovery in this study was exacerbated during the antiretroviral treatment interruptions. In the future, modifications would aim to increase the dose, improve homing to the bone marrow, engraftment and differentiation through the myeloid and lymphoid compartments of transduced CD34+ cells and to develop a more potent construct.
In this phase II study, as for the phase I studies 4,29, no safety concerns associated with OZ1 gene transfer were identified. All serious adverse events were unrelated to study procedures or study drug infusion. Importantly, no death, AIDS-defining event, severe infection, clinically significant cardiovascular, renal or hepatic event was reported in randomized participants.
The increased morbidity and mortality associated with the cessation of HAART observed during the SMART study became known during the course of this study45. Specific discussions regarding the safety of the treatment interruptions in the OZ1 protocol were conducted with the Data Safety Monitoring Board and the Institutional Review Boards. The strict entry criteria met by all participants, the stringent monitoring and the provision for investigators to reintroduce HAART at any stage, distinguish this OZ1 trial from the SMART study. Future HIV-1 gene therapy trials could be designed to study participants prior to the initiation of HAART, obviating the need for treatment interruptions.
This study supports our concept that OZ1 cell-delivered gene therapy is safe, and has efficacy, albeit modest. It shows the potential of the gene therapy approach for the treatment of HIV-1 and represents a major advance in the field
METHODS
Participants and Blinding
Seventy four (74) early stage, HIV-1 infected participants aged from 18 to 45 were randomly allocated to receive either OZ1-transduced or Control-treated CD34+ cells. Each participant received a single intravenous infusion of autologous cells without myeloablation or other conditioning. The inclusion and exclusion criteria are described in the Supplementary Information. The Sponsor, Investigators, site staff, laboratory staff and participants were all blinded to the treatment group assignment.
OZ1 and Control Manufacture
OZ1 is an LNL6-based retroviral vector containing a ribozyme encoded by the DNA sequence 5′-TTA GGA TCC TGA TGA GTC CGT GAG GAC GAA ACT GGC TC-3′. Manufacturing was performed by BioReliance Corporation, Rockville, MD under Good Manufacturing Practice conditions. The OZ1 gene transfer product was harvested as the supernatant of AM-12/RRz2 retrovirus-producer cells; a media only Control was also produced.
Protocol Design
This 100-week, 45-visit study recruited participants whose viral load was suppressed by HAART. The study design included 2 periods of antiretroviral treatment interruption (weeks 24-28 and weeks 40-48) (Fig 1). The second antiretroviral treatment interruption, termed the analytic treatment interruption, was allowed to continue until week 100 unless protocol-defined limits were reached. At any stage in the protocol, the Investigator could advise a participant to reinitiate therapy.
Participants were treated for 5 consecutive days with G-CSF (30μg/kg/day) to mobilize CD34+ cells which were harvested by large volume apheresis (approximately 20 liters) on both days 4 and 5 (Fig 1b). The isolation, culture and transduction of the CD34+ cells are described in the Supplementary Information. The day of infusion was set as Day 0.
HIV-1 viral load testing was performed using the Roche Amplicor HIV-1 Monitor Assay (range 400-750,000 copies/ml) in real time. Efficacy parameters were assessed both in real time, and in batch testing by a single laboratory of stored frozen plasma samples.
Safety evaluations were performed on all participants who were randomized. Blood samples were taken throughout the study for analysis of the sequence of the tat/vpr RNA in the region targeted by OZ1. HIV-1 genotyping was performed at the end of each treatment interruption by PCR sequencing of the protease and reverse transcriptase genes (Virco Virtual Phenotype, Belgium). Testing for the emergence of a predominant OZ1-containing clone was conducted on PBMC by LAM-PCR integration analysis at Cincinnati Children's Hospital Medical Center46 at 6-monthly intervals. Replication competent retrovirus (RCR) was evaluated by PCR using primers specific for the amphotropic retroviral envelope.
The study was conducted in accordance with International Conference on Harmonization/Good Clinical Practice (ICH/GCP). Approval for the study was obtained from the appropriate Institutional Review Boards, Institutional Biosafety Committees, the US Food and Drug Administration (Centre for Biologics Evaluation Review), National Institutes of Health (Recombinant DNA Advisory Committee) and the Therapeutic Goods Administration and the Office of Gene Technology Regulator in Australia. The study was listed on a public clinical trial registry at www.clinicaltrials.gov; NCT00074997.
Statistical Methods
No statistical analysis was performed on the baseline data. For the analyses of efficacy, two populations were defined. The intention-to-treat population (ITT) was pre-defined in the Statistical Analysis Plan as all participants who were randomized and received the cell infusion. The per-protocol population (PP) was defined as all participants who completed the first 4 week treatment interruption, recommenced HAART and entered the analytic treatment interruption. Formal group comparisons of continuous primary and secondary parameters were performed using the Wilcoxon Rank Sum test with 95% confidence intervals provided for the medians and the difference in the medians (calculated using PROC STATXACT in SAS and the Hodges-Lehman estimate). Binary endpoints were analyzed using Fisher's Exact test, with 95% confidence intervals. Participants for whom data were unavailable, either due to non-compliance with the protocol or withdrawal prior to the analytic treatment interruption, were included in formal comparisons by being ranked as having the joint-worst outcome for continuous endpoints or being categorized as failures for binary endpoints. Participants who recommenced HAART prior to week 48 had an imputed value, the last value recorded, assigned and carried forward to week 48. Analyses were also performed without imputation.
The primary end point, the mean plasma viral load at weeks 47/48 (log10 copies/ml), was derived by taking the mean per participant of the log10 copies/ml viral load value at each of weeks 47 and 48. The TWAUC was calculated using linear trapezoidal integration on the logged data. For time to resumption of HAART, participants who did not start the analytic treatment interruption were included in analyses with a time of zero, while participants who did not resume HAART prior to week 100 had the time to resumption of HAART included in analyses as a censored observation and statistical significance assessed using the log rank test.
In all analyses, statistical significance was taken at the two-sided 5% level, with no adjustment for multiple secondary parameters and analyses.
Supplementary Material
Acknowledgements
The following clinicians, research nurses and coordinators contributed significantly to the trial; Shikha Agrawal, Rafael Amado, Colin Anderson, Pat Cain, Robert Cordova, Geraldine Dolan, Charles Farthing, Eileen Glutzer, Maricela Gonzales, Michael Harbour, Natalie Hyland, Andre Lebel, Sunny Lundy, Karen Macrae, Samantha Miller, Jane Norris, Laura O'Donnell, Leigh Pearce, Jacinta Perram, Vanessa Rees, Robyn Richardson, Jega Sarangapany, Debbie Slamowitz, Robyn Vale, Glaucia Vasquez and Andrew Zolopa. We also acknowledge the contribution of the leukapheresis collection and cell processing staff, Willie Quan for assistance with Quality Assurance and Andra Miller of the Biologics Group with regulatory aspects. Simon Margrie, Quintiles Pty Limited, played a key role in development of the statistical analysis plan and in the statistical analyses. Cherise Ang and Andrew King assisted with the preparation of the manuscript. We acknowledge Wayne Gerlach, Denis Wade, Jim Peacock, Rob de Feyter and Leigh Farrell for their vital role during the early stages of development of OZ1. We sincerely thank the participants who enrolled in this intensive study, and the members of the Data Safety Monitoring Board. The UCLA CFAR and UCLA GCRC were supported by National Institutes of Health Grants AI28697 and M01-RR00865.
REFERENCES
- 1.Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med. 2008;358:1590–602. doi: 10.1056/NEJMra0706737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watkins DI, Burton DR, Kallas EG, Moore JP, Koff WC. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat Med. 2008;14:617–21. doi: 10.1038/nm.f.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltimore D. Gene therapy. Intracellular immunization. Nature. 1988;335:395–396. doi: 10.1038/335395a0. [DOI] [PubMed] [Google Scholar]
- 4.Amado RG, et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum Gene Ther. 2004;15:251–62. doi: 10.1089/104303404322886101. [DOI] [PubMed] [Google Scholar]
- 5.An DS, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A. 2007;104:13110–5. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson J, et al. Safety and efficacy of a lentiviral vector containing three anti-HIV genes--CCR5 ribozyme, tat-rev siRNA, and TAR decoy--in SCID-hu mouse-derived T cells. Mol Ther. 2007;15:1182–8. doi: 10.1038/sj.mt.6300157. [DOI] [PubMed] [Google Scholar]
- 7.Bahner I, et al. Comparison of trans-dominant inhibitory mutant human immunodeficiency virus type 1 genes expressed by retroviral vectors in human T lymphocytes. J Virol. 1993;67:3199–207. doi: 10.1128/jvi.67.6.3199-3207.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cagnon L, Rossi J. Retroviral delivery and anti-HIV testing of hammerhead ribozymes. Methods Mol Biol. 1997;74:451–7. doi: 10.1385/0-89603-389-9:451. [DOI] [PubMed] [Google Scholar]
- 9.Dropulic B, Elkins DA, Rossi JJ, Sarver N. Ribozymes: use as anti-HIV therapeutic molecules. Antisense Res Dev. 1993;3:87–94. doi: 10.1089/ard.1993.3.87. [DOI] [PubMed] [Google Scholar]
- 10.Fanning G, Amado R, Symonds G. Gene therapy for HIV/AIDS: the potential for a new therapeutic regimen. J Gene Med. 2003;5:645–53. doi: 10.1002/jgm.436. [DOI] [PubMed] [Google Scholar]
- 11.Kohn DB. Gene therapy using hematopoietic stem cells. Curr Opin Mol Ther. 1999;1:437–42. [PubMed] [Google Scholar]
- 12.Kohn DB, et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood. 1999;94:368–71. [PubMed] [Google Scholar]
- 13.Li M, Li H, Rossi JJ. RNAi in combination with a ribozyme and TAR decoy for treatment of HIV infection in hematopoietic cell gene therapy. Ann N Y Acad Sci. 2006;1082:172–9. doi: 10.1196/annals.1348.006. [DOI] [PubMed] [Google Scholar]
- 14.Lo AS, Zhu Q, Marasco WA. Intracellular antibodies (intrabodies) and their therapeutic potential. Handb Exp Pharmacol. 2008:343–73. doi: 10.1007/978-3-540-73259-4_15. [DOI] [PubMed] [Google Scholar]
- 15.Marasco WA, LaVecchio J, Winkler A. Human anti-HIV-1 tat sFv intrabodies for gene therapy of advanced HIV-1-infection and AIDS. J Immunol Methods. 1999;231:223–38. doi: 10.1016/s0022-1759(99)00159-3. [DOI] [PubMed] [Google Scholar]
- 16.Mautino MR, Morgan RA. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mRNA. Hum Gene Ther. 2000;11:2025–37. doi: 10.1089/10430340050143444. [DOI] [PubMed] [Google Scholar]
- 17.Morgan RA, et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum Gene Ther. 2005;16:1065–74. doi: 10.1089/hum.2005.16.1065. [DOI] [PubMed] [Google Scholar]
- 18.Morris KV, Rossi JJ. Lentivirus-mediated RNA interference therapy for human immunodeficiency virus type 1 infection. Hum Gene Ther. 2006;17:479–86. doi: 10.1089/hum.2006.17.479. [DOI] [PubMed] [Google Scholar]
- 19.Rossi JJ. The application of ribozymes to HIV infection. Curr Opin Mol Ther. 1999;1:316–22. [PubMed] [Google Scholar]
- 20.Strayer DS, et al. Current status of gene therapy strategies to treat HIV/AIDS. Mol Ther. 2005;11:823–42. doi: 10.1016/j.ymthe.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Taylor JA, et al. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Mol Ther. 2008;16:46–51. doi: 10.1038/sj.mt.6300335. [DOI] [PubMed] [Google Scholar]
- 22.von Laer D, Hasselmann S, Hasselmann K. Gene therapy for HIV infection: what does it need to make it work? J Gene Med. 2006;8:658–67. doi: 10.1002/jgm.908. [DOI] [PubMed] [Google Scholar]
- 23.Zahn RC, et al. Efficient entry inhibition of human and nonhuman primate immunodeficiency virus by cell surface-expressed gp41-derived peptides. Gene Ther. 2008 doi: 10.1038/gt.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarver N, et al. Ribozymes as potential anti-HIV-1 therapeutic agents. Science. 1990;247:1222–5. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- 25.Schambach A, et al. Towards hematopoietic stem cell-mediated protection against infection with human immunodeficiency virus. Gene Ther. 2006;13:1037–47. doi: 10.1038/sj.gt.3302755. [DOI] [PubMed] [Google Scholar]
- 26.Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–54. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macpherson JL, Ely JA, Sun LQ, Symonds GP. Ribozymes in gene therapy of HIV-1. Front Biosci. 1999;4:D497–505. doi: 10.2741/macpherson. [DOI] [PubMed] [Google Scholar]
- 28.Levine BL, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–7. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macpherson JL, et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J Gene Med. 2005;7:552–64. doi: 10.1002/jgm.705. [DOI] [PubMed] [Google Scholar]
- 30.Podsakoff GM, et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells. Mol Ther. 2005;12:77–86. doi: 10.1016/j.ymthe.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 31.Sun LQ, Ely JA, Gerlach W, Symonds G. Anti-HIV ribozymes. Mol Biotechnol. 1997;7:241–51. doi: 10.1007/BF02740815. [DOI] [PubMed] [Google Scholar]
- 32.Sun LQ, Wang L, Gerlach WL, Symonds G. Target sequence-specific inhibition of HIV-1 replication by ribozymes directed to tat RNA. Nucleic Acids Res. 1995;23:2909–13. doi: 10.1093/nar/23.15.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun LQ, et al. Resistance to human immunodeficiency virus type 1 infection conferred by transduction of human peripheral blood lymphocytes with ribozyme, antisense, or polymeric trans-activation response element constructs. Proc Natl Acad Sci U S A. 1995;92:7272–6. doi: 10.1073/pnas.92.16.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossi JJ. Ribozyme therapy for HIV infection. Adv Drug Deliv Rev. 2000;44:71–8. doi: 10.1016/s0169-409x(00)00085-5. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, et al. Preclinical characterization of an anti-tat ribozyme for therapeutic application. Hum Gene Ther. 1998;9:1283–91. doi: 10.1089/hum.1998.9.9-1283. [DOI] [PubMed] [Google Scholar]
- 36.Food & Drug Administration, C.B.E.R. Guidance for Industry . Gene therapy clinical trials - observing subjects for delayed adverse events. US Department of Health and Humand Services; Rockville, MD: 2006. [Google Scholar]
- 37.Food & Drug Administration, C.B.E.R. Guidance for Industry . Supplemental guideances on testing for replication competent retrovirus in retroviral vector based gene therapy products and during follow-up of patients in clinical trials using retroviral vectors. US Department of Health and Humand Services; Rockville, MD: 2006. [DOI] [PubMed] [Google Scholar]
- 38.Cavallaro AM, et al. Three to six year follow-up of normal donors who received recombinant human granulocyte colony-stimulating factor. Bone Marrow Transplant. 2000;25:85–9. doi: 10.1038/sj.bmt.1702072. [DOI] [PubMed] [Google Scholar]
- 39.Nicolini FE, et al. Long-term persistent lymphopenia in hematopoietic stem cell donors after donation for donor lymphocyte infusion. Experimental Hematology. 2004;32:1033–1039. doi: 10.1016/j.exphem.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 40.Novotny J, et al. Sustained decrease of peripheral lymphocytes after allogeneic blood stem cell aphereses. Br J Haematol. 1998;100:695–7. doi: 10.1046/j.1365-2141.1998.00629.x. [DOI] [PubMed] [Google Scholar]
- 41.Malech HL, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci U S A. 1997;94:12133–12138. doi: 10.1073/pnas.94.22.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aiuti A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–3. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 43.Hacein-Bey-Abina S, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 44.Ott MG, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 45.The Strategies for Management of Antiretroviral Therapy (SMART) Study Group CD4+ Count-Guided Interruption of Antiretroviral Treatment. N Engl J Med. 2006;355:2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt M, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051–7. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.