

Abstract
Background
Although inducible nitric oxide synthase (iNOS) is known to impart powerful protection against myocardial infarction, the mechanism for this salubrious action remains unclear.
Methods and Results
Adenovirus-mediated iNOS gene transfer in mice resulted, 48-72 h later, in increased expression not only of iNOS protein but also of HO-1 mRNA and protein; HO-2 protein expression did not change. iNOS gene transfer markedly reduced infarct size in wild-type mice, but this effect was completely abrogated in HO-1-/- mice. At 48 h after iNOS gene transfer, NF-κB was markedly activated. In transgenic mice with cardiomyocyte-restricted expression of a dominant negative mutant of IκBα (IκBαS32A,S36A), both the basal HO-1 levels and the upregulation of HO-1 by iNOS gene transfer were suppressed. Chromatin immunoprecipitation analysis of mouse hearts provided direct evidence that NF-κB subunits p50 and p65 were recruited to the HO-1 gene promoter (−468 bp to −459 bp) 48 h after iNOS gene transfer.
Conclusions
This study demonstrates, for the first time, the existence of a close functional coupling between cardiac iNOS and cardiac HO-1: iNOS upregulates HO-1 by augmenting NF-κB binding to the region of the HO-1 gene promoter from −468 bp to −459 bp, and HO-1 then mediates the cardioprotective effects of iNOS. These results also reveal an important role of NF-κB in both basal and iNOS-induced expression of cardiac HO-1. Collectively, the present findings significantly expand our understanding of the regulation of cardiac HO-1 and of the mechanism whereby iNOS exerts its cardioprotective actions.
Keywords: nitric oxide synthase, gene therapy, myocardial infarction, heme oxygenase-1, NF-κB
INTRODUCTION
Extensive evidence indicates that the inducible isoform of nitric oxide (NO) synthase (iNOS) is a major cardioprotective protein 1, 2. A large number of studies have shown that cardiac overexpression of iNOS confers a protected phenotype and that iNOS is an obligatory mediator of the infarct-sparing effects of the late phase of preconditioning induced by ischemia and several other stimuli, indicating that upregulation of this enzyme is a common pathway whereby the heart adapts to stress 3. Consistent with these facts, iNOS gene transfer protects the myocardium from ischemia/reperfusion injury 4, 5. Since iNOS has traditionally been viewed as a deleterious enzyme 2, these results may appear puzzling, and raise the question of the mechanism of iNOS-dependent cardioprotection 6. At present, the molecular basis for the salubrious effects of iNOS remains incompletely understood and represents a major unresolved issue in ischemic biology.
One possible explanation for the unexpected beneficial role of iNOS in myocardial ischemia/reperfusion injury is that it might involve the ubiquitously cytoprotective protein heme oxygenase-1 (HO-1). HO is the rate-limiting enzyme in heme catabolism; it catalyzes the breakdown of heme into equimolar amounts of carbon monoxide, biliverdin, and free iron 7. Three mammalian HO isoforms have been identified, one of which, HO-1, is a stress-responsive protein induced by a remarkably vast panoply of stimuli 7-10. Of the metabolites generated by HO-1 catalysis, biliverdin (and bilirubin) has been shown to possess antioxidant activity, while carbon monoxide has been found to exert many salutary effects in various settings including myocardial ischemia 7, 11, 12. Although induction of HO-1 is known to constitute a common adaptive response that increases cellular resistance to oxidative injury and other types of injury 7, 12, 13, the role of HO-1 in the cardioprotection afforded by iNOS gene therapy remains unclear.
HO-1 is regulated primarily at the transcriptional level 7, 14, 15. HO-1 gene expression is mediated through cis-regulatory DNA sequences located in the promoter region 16, a process that frequently involves transcriptional or structural activation of transcription factors and their translocation to the nucleus 17. Among them, the presence of NF-κB binding sequences in the HO-1 gene promoter region implicates NF-κB in the regulation of the HO-1 gene 18, 19. Since we have previously found that cardiac NF-κB is activated by NO in vivo 20, it appears plausible that augmented NO availability may lead to HO-1 gene expression via NF-κB activity.
On the basis of these considerations, we postulated that the cardioprotection afforded by iNOS may be mediated by induction of HO-1 via increased binding of NF-κB to the HO-1 gene promoter. To test this hypothesis, we combined molecular analyses with physiological studies in a well-characterized murine model of infarction 21. We examined three issues: i) whether iNOS gene transfer induces HO-1 expression in the myocardium, ii) if so, whether HO-1 is necessary for the protection afforded by iNOS gene transfer, and iii) whether iNOS regulates HO-1 expression by modulating the access of NF-κB to the HO-1 gene promoter. We used iNOS gene transfer to study the mechanism of iNOS-dependent protection because this approach enabled us to achieve selective upregulation of iNOS without the numerous cellular perturbations associated with ischemic preconditioning or other interventions known to induce this protein 1. Furthermore, we used genetically engineered mice rather than pharmacologic agents. That is, to conclusively establish whether HO-1 plays an obligatory role in the cardioprotection afforded by iNOS gene transfer, we studied mice with targeted disruption of the HO-1 gene (HO-1-/-) 22. To investigate whether the upregulation of HO-1 induced by iNOS gene transfer is mediated by NF-κB activity, we used IκBα mutant transgenic mice with cardiac-specific abrogation of NF-κB activation 23. Finally, to specifically identify NF-κB binding to the region of the HO-1 gene promoter, we developed, for the first time, a technique that enabled us to perform chromatin immunoprecipitation (ChIP) analysis directly on the mouse heart.
METHODS
This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals (DHHS Publications No. 85-23, revised 1996) and with the guidelines of the Animal Care and Use Committee of the University of Louisville, School of Medicine (Louisville, KY). Detailed methods are available in the Online Supplemental Data.
Genetically engineered mice
The HO-1-/- mice employed in this study were generated by Yet et al. 22; colonies were maintained by breeding HO-1-/- males with HO-1-/- females. Offspring were genotyped at the time of weaning by PCR to amplify the wild-type (WT) and mutant alleles of genomic DNA from tail DNA samples. WT littermates were used as controls. The transgenic mice that express a phosphorylation-resistant mutant of IκBα (IκBα S32A,S36A) under the control of a cardiac-specific promoter have been previously described 23; in these mice, expression of the dominant negative mutant IκBα results in cardiomyocyte-restricted inhibition of NF-κB activation 23. IκBα S32A,S36A transgenic mice (IκBα S32A,S36A Tg) were identified by PCR-based DNA screening 23. Non-transgenic (NTg) littermates were used as controls. For all experiments, mice were maintained in microisolator cages under specific pathogen-free conditions in a room with a temperature of 24°C, 55–65% relative humidity, and a 12-h light–dark cycle.
Adenoviral vectors
Recombinant adenoviral vectors, Av3/LacZ and Av3/iNOS, were constructed essentially as previously described 4, 5.
In vivo gene transfer
Anesthetized mice received an intramyocardial injection in the anterior left ventricular (LV) wall of Av3/LacZ or Av3/iNOS . Two or three days later, mice were euthanized for cardiac tissue collection or underwent the infarction protocol summarized below (Fig. 1) 4, 5.
Figure 1.
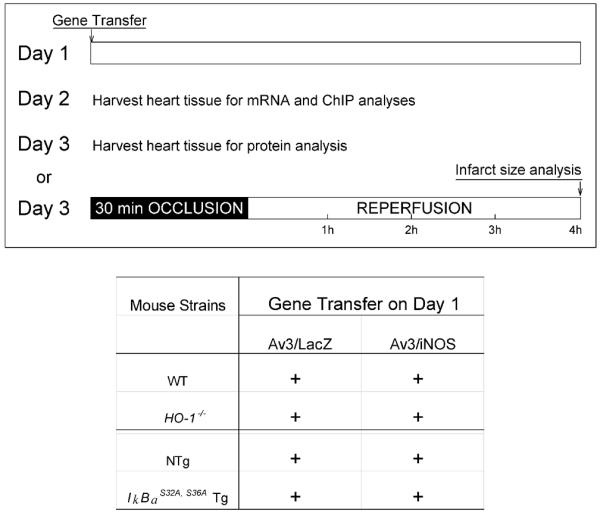
Experimental protocol. On day 1, mice were subjected to intramyocardial injections of Av3/LacZ (LacZ group) or Av3/iNOS (iNOS group). On day 2 or 3, mice were euthanized for cardiac tissue collection or underwent a 30-min coronary occlusion followed by 4 h of reperfusion for the infarct study.
Assessment of infarct size
Myocardial infarction was produced as previously described (Fig. 1) 4, 21, 24.
Western immunoblotting analysis, immunohistochemistry, and bilirubin assay
The methods are described in the Online Supplemental Data.
Reverse-Transcription PCR study
0.1 μg of total RNA was used for the first-strand cDNA synthesis and PCR amplification with the One-Step Platinum Taq reverse-transcription - PCR kit (Invitrogen, San Diego, CA) 25. A 360-bp fragment (for HO-1) or a 494-bp fragment (for GAPDH) was amplified with mouse HO-1 or GAPDH specific primers. Each sample was assayed in duplicate.
ChIP analysis of cardiac tissue
ChIP analysis was performed by using a magnetic bead-based ChIP kit (Active Motif, Carlsbad, CA) according to the manufacturer’s instructions 26. Each sample was assayed in duplicate.
Statistical analysis
Data are reported as means ± SEM. Protein band density was normalized to the corresponding loading control and then to the mean of the corresponding control group 4, 24. All the data are analyzed with a one-way or two-way ANOVA, as appropriate, followed by Student’s t-tests. Because of the small sample sizes, data were also analyzed with nonparametric tests (Kruskal-Wallace test and Mann-Whitney test); since the results were similar to those obtained with one-way or two-way ANOVA and Student’s t-tests, for the sake of simplicity and clarity, the latter (parametric) results are reported herein. A P value < 0.05 was considered statistically significant. All statistical analyses were performed using the SigmaStat software system (3.5V).
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agreed to the manuscript as written.
RESULTS
Exclusions
A total of 105 mice were used for this study: 57 for studies of infarct size, 32 for studies of protein expression, 8 for studies of HO-1 mRNA expression, and 8 for ChIP analyses of NF-κB. Twenty-two mice died during or shortly after the surgical procedure, and 8 were excluded because of technical problems. Thus, a total of 75 mice were included in the final analyses.
Fundamental physiological parameters
Heart rate and body temperature, fundamental physiological parameters that may impact infarct size 21, were similar in all four groups of mice used for studies of coronary occlusion (Table 1). By experimental design 21, 27, rectal temperature remained within a narrow, physiologic range (36.8–37.1°C) in all groups. Five minutes before the 30-min coronary occlusion, the average heart rate in the four groups ranged from 546±21 to 585±19 bpm (P>0.05). Heart rate did not differ significantly among the four groups at any time during the 30-min occlusion or the ensuing reperfusion (Table 1). The size of the region at risk, expressed as a percentage of LV weight, did not differ among the four groups: WT+Av3/LacZ, 41±1%; WT+Av3/iNOS, 41±2%; HO-1-/-+Av3/LacZ, 37±3%; and HO-1-/-+Av3/iNOS, 38±2% (P>0.05).
Table 1.
Rectal temperature and heart rate on the day of the 30-min coronary occlusion
| Occlusion |
Reperfusion |
||||
|---|---|---|---|---|---|
| Groups | Preocclusion | 5 min | 30 min | 5 min | 15 min |
| Temperature (°C) | |||||
| WT + Av3/LacZ (n=8) | 37.0±0.1 | 37.1±0.1 | 37.0±0.1 | 37.0±0.1 | 37.1±0.1 |
| WT + Av3/iNOS (n=7) | 36.8±0.0 | 37.1±0.1 | 37.1±0.1 | 36.8±0.0 | 36.8±0.1 |
| HO-1-/- + Av3/LacZ (n=8) | 36.9±0.1 | 37.1±0.1 | 37.1±0.1 | 37.0±0.1 | 37.0±0.1 |
| HO-1-/- + Av3/iNOS (n=9) | 36.9±0.0 | 37.1±0.0 | 37.0±0.1 | 37.0±0.1 | 36.9±0.1 |
| Heart rate (beats/min) | |||||
| WT + Av3/LacZ (n=8) | 546±21 | 587±21 | 575±21 | 605±20 | 605±17 |
| WT + Av3/iNOS (n=7) | 564±14 | 581±19 | 593±8 | 587±12 | 574±13 |
| HO-1-/- + Av3/LacZ (n=8) | 553±26 | 577±26 | 576±32 | 590±26 | 595±30 |
| HO-1-/- + Av3/iNOS (n=9) | 585±19 | 613±14 | 607±11 | 597±18 | 587±27 |
Data are means ± SEM.
Effects of iNOS gene transfer on HO-1 mRNA, HO-1 protein, and bilirubin
Three days after iNOS gene transfer, immunoblotting revealed a marked increase in myocardial iNOS protein expression (+186% vs. the LacZ group, n=6, P=0.009; Fig. 2). At the same time, the iNOS-transduced myocardial region also exhibited a significant increase in HO-1 protein expression (+263% vs. the LacZ group, n=6, P=0.041; Fig. 2). Consistent with the immunoblotting data, immunohistochemical analysis showed elevated expression of HO-1 (Fig. 3C) and iNOS (Fig. 3F) in the transduced region of the anterior LV wall 3 days after iNOS gene transfer. Figures 3C and 3F illustrate two adjacent sections from the same heart, and show that the spatial distribution of HO-1 immunoreactivity coincided with that of iNOS immunoreactivity. At higher magnification (x300), intense HO-1 (Fig. 3B) and iNOS (Fig. 3E) immunoreactivity can be appreciated in cardiac myocytes, but not in non-myocytes. As illustrated in Fig. 4, at 48 h after iNOS gene transfer there was a marked elevation in cardiac HO-1 mRNA levels (+212%, n=4, vs. LacZ group, n=3; P=0.003). The content of bilirubin, a byproduct of HO-1, was significantly increased in the transduced myocardium 3 days after iNOS gene transfer (0.35±0.04 ng/ug protein [n=4] vs. 0.16±0.01 ng/ug protein [n=6] in the LacZ group; P≤0.001).
Figure 2.

Effects of iNOS gene transfer on myocardial iNOS and HO-1 expression. Data are means ± SEM of experiments performed in duplicate.
Figure 3.

Distribution of HO-1 and iNOS protein expression 3 days after iNOS gene transfer. Panels A -C illustrate HO-1 immunohistochemical staining, whereas panels D-F illustrate iNOS immunohistochemistry. Panels C and F show two adjacent sections from the same heart; note that the spatial distribution of HO-1 immunoreactivity coincides with that of iNOS immunoreactivity. (A, B, D, E, x300; C, F, x7.5; n=4/group).
Figure 4.

Representative RT-PCR analysis of myocardial HO-1 mRNA content 2 days after iNOS gene transfer. Cardiac HO-1 mRNA expression levels increased 2.1-fold, with no changes in cardiac GAPDH mRNA content, in the iNOS group. Data are means ± SEM of experiments performed in duplicate.
Effect of iNOS gene transfer on infarct size in HO-1-/- mice
In WT mice, three days after Av3 injection infarct size was reduced by an average of 50% in the Av3/iNOS-treated vs. the Av3/LacZ-treated group (Fig. 5). In contrast, when HO-1-/- mice received Av3/iNOS, infarct size (47.5±3.4 % of the risk region, n=9) did not differ significantly from that observed in HO-1-/- mice that received Av3/LacZ (51.9±3.3%, n=8; Fig. 5), demonstrating that HO-1 plays an obligatory role in the cardioprotection afforded by iNOS gene transfer. After LacZ gene transfer, infarct size was similar in HO-1-/- and WT mice (Fig. 5), implying that HO-1 does not modulate ischemia/reperfusion injury under basal conditions.
Figure 5.

Effect of ablation of the HO-1 gene on the infarct-sparing effect of iNOS gene therapy. Myocardial infarct size is expressed as a percentage of the region at risk. Open circles represent individual mice, whereas solid circles represent means ± SEM.
Effect of iNOS gene transfer on NF-κB nuclear translocation
Nuclear proteins extracted from the transduced myocardium were assayed for the presence of the active p50 or p65 subunit of NF-κB in the nuclear fraction. Quantitative analysis of Western immunoblotting demonstrated increased nuclear content of NF-κB at 48 h after injection of Av3/iNOS, both with regard to the p50 subunit (+58.1±1.3%, n=3, vs. the LacZ group, n=3; P≤0.001; Fig. 6) and to the p65 subunit (+52.1±1.9%, n=3, vs. the LacZ group, n=3; P≤0.001, Fig. 6).
Figure 6.

iNOS gene transfer produces translocation of NF-κB subunits p50/p65 to the nucleus. Forty-eight hours after iNOS gene transfer, there was significant elevation of p50 and p65 in the nuclear fraction. Data are means ± SEM of experiments performed in duplicate.
Effect of iNOS gene transfer on HO-1 protein expression in IκBαS32A,S36A mutant Tg mice
Consistent with the results reported above, NTg mice that received Av3/iNOS exhibited robust expression of HO-1 in the transduced myocardium (Fig. 7). On average, injection of Av3/iNOS in NTg mice resulted in a 2.2-fold increase in HO-1 protein content vs. Av3/LacZ-treated NTg mice. In contrast, myocardial HO-2 protein expression did not change after iNOS gene transfer (n=3, Fig. 7). Interestingly, HO-1 immunoreactivity was weaker in the transduced myocardium of IκBαS32A,S36A mutant Tg mice treated with Av3/LacZ as compared with NTg mice under the same conditions (− 60% vs. the NTg + Av3/LacZ group, n=3; P=0.019; Fig. 7) as well as in IκBαS32A,S36A mutant Tg mice not subjected to iNOS gene transfer vs. the corresponding NTg mice (− 38%, n=3; online Fig. 1), suggesting that NF-κB modulates cardiac HO-1 protein expression under basal conditions. Although in IκBαS32A,S36A mutant Tg mice subjected to iNOS gene transfer HO-1 was still upregulated relative to IκBαS32A,S36A mutant Tg mice treated with Av3/LacZ, the levels of HO-1 expression were much lower than in NTg mice treated with Av3/iNOS (− 56% vs. the NTg + Av3/iNOS group, n=3; P=0.036; Fig. 7), indicating that NF-κB plays an essential role not only in basal cardiac HO-1 protein expression but also in iNOS-dependent induction of HO-1. The increase in HO-1 in IκBαS32A,S36A Tg mice treated with Av3/iNOS (Fig. 7) likely reflects the multifactorial nature of HO-1 regulation14, 18 and the activation by NO of transcription factors other than NF-κB. In principle, it may also reflect synthesis of HO-1 in non-cardiac myocytes (since the mutant IκBα is expressed selectively in cardiac myocytes 23). However, as indicated above, immunohistochemical analysis confirmed that the induction of HO-1 by iNOS gene transfer occurred in cardiac myocytes (Fig. 3).
Figure 7.

Effect of disruption of NF-κB activation (by cardiac-specific expression of a mutant [IκBαS32A,S36A]) on basal levels of cardiac HO-1 protein expression and upregulation of HO-1 by iNOS gene therapy. NTg, nontransgenic mice; IκBα S32A,S36A Tg, IκBα S32A,S36A transgenic mice. Data are means ± SEM of experiments performed in duplicate.
Effect of iNOS gene transfer on NF-κB binding to the HO-1 gene promoter
To further investigate the mechanism whereby iNOS modulates HO-1, we used a ChIP method that enabled us to directly assess the effect of iNOS gene transfer on the upstream regulatory sequences of the HO-1 gene in cardiac tissue. As shown in Fig. 8, ChIP analysis demonstrated that, 48 h after iNOS gene transfer, NF-κB was recruited to the HO-1 gene promoter (specifically, to the region from −468 bp to −459 bp), as evidenced by the presence of both the p50 subunit (+230% vs. the LacZ group, n=3; P≤0.001) and the p65 subunit (+179% vs. the LacZ group, n=3; P≤0.001), a finding consistent with the observation that iNOS enhances HO-1 mRNA levels (Fig. 4). These results suggest that an NF-κB binding element located in the −468 bp to −459 bp region of the murine HO-1 gene promoter is involved in the transcriptional activation of the HO-1 gene in response to iNOS gene transfer.
Figure 8.

Chromatin immunoprecipitation analysis of NF-κB binding to the HO-1 gene promoter 48 h after iNOS gene transfer. Data are means ± SEM of experiments performed in duplicate.
DISCUSSION
Although studies of ischemic PC 1, 27 and iNOS gene transfer 4, 5, 24 have clearly shown that iNOS imparts powerful protection against myocardial infarction, the mechanism of this salubrious action remains unclear. iNOS is upregulated by numerous and diverse stimuli, and thus appears to be a ubiquitous cardioprotective protein 2. Of similar potential importance is HO-1, another cytoprotective enzyme that has been recognized to play a critical role in response to various forms of stress including myocardial ischemia 28-31. At present, virtually nothing is known regarding the interaction between these two major protective systems in the myocardium and the molecular mechanism whereby iNOS modulates cardiac HO-1. The present study provides considerable new information relevant to these issues.
Our salient findings can be summarized as follows: i) iNOS gene transfer upregulates not only the cardiac levels of iNOS protein, but also those of HO-1 mRNA and protein, indicating that, in the heart, HO-1 is coupled to iNOS; ii) targeted disruption of the HO-1 gene completely abrogates the infarct-sparing effects of iNOS gene transfer, demonstrating that HO-1 is a necessary mediator of iNOS-dependent cardioprotection; iii) iNOS gene transfer promotes translocation of NF-κB to the nucleus and its binding to a specific element in the promoter region of the HO-1 gene, as demonstrated by both Western immunoblotting and ChIP analysis of cardiac tissue, suggesting that the molecular mechanism whereby iNOS controls HO-1 expression involves transcriptional activation of HO-1 via an NF-κB-dependent pathway; iv) cardiac-specific abrogation of NF-κB activation via expression of a dominant negative mutant of IκBα (IκBαS32A,S36A) suppresses the HO-1 upregulation elicited by iNOS gene transfer and also diminishes the basal levels of HO-1 expression, demonstrating that NF-κB is essential for cardiac HO-1 protein expression under basal conditions as well as for the induction of HO-1 by iNOS.
Previous investigations have shown that iNOS and HO-1 exert a multitude of cytoprotective effects 1, 2, 27-33. To our knowledge, however, this is the first study to demonstrate the existence of a tight coupling between cardiac iNOS and cardiac HO-1, two inducible proteins that play a critical role in the response of the heart to ischemia and other forms of stress. This is also the first study to identify NF-κB activation as a key mechanism that mediates iNOS-dependent modulation of HO-1 in the heart.
Role of HO-1 in the cardioprotection afforded by iNOS
Mounting evidence indicates that HO-1plays an important cytoprotective role 28, 33-35. This enzyme has been found to have beneficial effects in a wide variety of pathologic conditions, such as inflammation, atherosclerosis, and ischemia/reperfusion injury 28, 33, 34. In non-cardiac tissues, there is evidence that HO-1 is regulated, among other factors, by NO 36, 37, and on this basis we postulated that iNOS may activate HO-1 in the heart. Although iNOS can be induced by many stimuli, including ischemic preconditioning 2, we elected to utilize iNOS gene transfer to study iNOS-dependent modulation of HO-1 because this approach results in selective upregulation of iNOS (Figs. 2, 3) (and thus in a sustained increase in myocardial NO production) without the confounding effects of the multifarious cellular perturbations and changes in gene expression that are associated with ischemia/reperfusion or with pharmacologic manipulations 1. As a consequence, the effect of NO on cardiac HO-1 can be assessed independently of other cellular changes and in the setting of a relatively steady-state NO generation. Indeed, we 4 have previously demonstrated that, concomitant with the elevation of iNOS protein expression, NOx levels are significantly increased 3 days after iNOS gene transfer in the transduced myocardium but not in the serum, indicating that the source of NO is cardiac. Our present findings that the increased myocyte expression of iNOS was associated with increased myocyte expression of HO-1 protein (Figs. 2 and 3) and mRNA (Fig. 4) and with increased content of bilirubin (a byproduct of HO-1), and that iNOS and HO-1 were colocalized in the same myocardial region that received iNOS gene transfer (Fig. 3), reveal a heretofore unrecognized coupling between the iNOS and HO-1 systems in the heart, even in the absence of ischemic stress or other pathological conditions.
The mere fact that iNOS expression is associated with HO-1 upregulation, however, does not necessarily imply a functional role of HO-1 in iNOS-dependent effects, as HO-1 upregulation may simply be an epiphenomenon. Clearly, elucidation of the role of HO-1 in iNOS-dependent protection requires inhibition of HO-1 activity. This could be achieved pharmacologically, but the utility of HO-1 inhibitors is limited by their relative lack of specificity 38, 39. Consequently, we used a molecular-genetic approach by studying mice with targeted disruption of the HO-1 gene. The similarity in infarct size between WT and HO-1-/- mice after Av3/LacZ administration (Fig.5) implies that HO-1 does not play a significant cardioprotective role under basal conditions, possibly because of its low level of expression in normal myocardium (Fig. 7). However, the fact that HO-1 gene knockout ablated the infarct-sparing effects of iNOS gene transfer (Fig. 5) provides conclusive evidence that the upregulation of HO-1 is necessary for the acquisition of ischemic tolerance afforded by iNOS and that HO-1 is an obligatory mediator of iNOS-dependent protection. Thus, an increase in iNOS in itself (in the absence of ischemia or other stimuli) is sufficient to induce myocardial HO-1 expression in vivo, revealing a new aspect of the regulation of cardiac HO-1. Based on these observations, we propose that a close functional coupling exists between cardiac iNOS and cardiac HO-1 and that the induction of HO-1 is a critical mechanism responsible for the cardioprotective effects of iNOS. Inasmuch as iNOS appears to be a common mediator of the protection induced by various types of preconditioning 1, this concept implies that HO-1 plays a major role in these adaptations as well.
Role of NF-κB in iNOS-dependent upregulation of HO-1
The mechanism whereby iNOS induces expression of HO-1 in the heart is unknown. One of the major transcription factors known to govern HO-1 expression is NF-κB 40, which has also been implicated in the cardioprotection afforded by iNOS gene therapy 24. At present, however, nothing is known regarding whether NF-κB is involved in iNOS-dependent modulation of cardiac HO-1.
NF-κB is most commonly a heterodimer of p50 and p65 and is maintained in an inactive state in the cytoplasm by IκBα 41. In response to various stresses, phosphorylation of the serine residues at positions 32 and 36 results in degradation of IκBα, which allows NF-κB to translocate to the nucleus and activate NF-κB-dependent genes 23, 41. To overcome the limitations inherent in pharmacologic manipulations of NF-κB, we have created a transgenic mouse that expresses cardiac-specifically a dominant negative mutant IκBα protein in which both serine residues 32 and 36 are replaced by alanine (IκBα S32A,S36A) 23. These IκBα S32A,S36A mutant Tg mice exhibit normal cardiac development, morphology, and histology 23, 24. The effectiveness of NF-κB suppression is demonstrated by the fact that tumor necrosis factor-α and lipopolysaccaride, two of the most potent stimuli known to activate NF-κB 42, fail to increase its nuclear levels in IκBα S32A,S36A mutant Tg mice 23, 24.
To gain insights into the role of NF-κB in iNOS-dependent modulation of HO-1, we first examined the nuclear content of NF-κB at 48 h after iNOS gene transfer. Our finding of increased nuclear content of p50 and p65 (Fig. 6) is consistent with our previous observation that iNOS gene transfer results in increased phosphorylation of IκBα at serine residues 32 and 36 and increased NF-κB DNA binding activity in the nuclear fraction 24. The finding that cardiomyocyte-restricted abrogation of NF-κB activation in IκBαS32A,S36A mutant Tg mice given Av3/LacZ significantly reduced the basal levels of cardiac HO-1 protein expression as compared with NTg mice given Av3/LacZ (Fig. 7) implies that NF-κB modulates cardiac HO-1 protein expression under basal conditions. The observation that the upregulation of HO-1 protein expression by iNOS gene transfer in NTg mice was blocked in IκBαS32A,S36A mutant Tg mice (Fig. 7) demonstrates that NF-κB is obligatorily involved in this process. Thus, taken together, these results reveal that NF-κB plays an essential role not only in the basal cardiac expression of HO-1 but also in the upregulation of HO-1 by iNOS. Our previous finding that the reduction in infarct size afforded by iNOS gene therapy is abolished in IκBαS32A,S36A mutant Tg mice 24 indicates that NF-κB is also essential for iNOS-dependent protection and that abrogation of the expression of NF-κB-dependent genes (among which is HO-1) renders the heart more susceptible to lethal ischemic/reperfusion injury. On the other hand, the fact that HO-2 protein levels were similar in NTg and IκBαS32A,S36A mutant Tg mice regardless of iNOS gene transfer (Fig. 7) indicates that the increased iNOS expression does not affect cardiac HO-2 protein and that NF-κB is not involved in the regulation of HO-2.
The classic NF-κB isoform (a p50 and p65 heterodimer) is capable of binding to the κB DNA binding site, a 10- or 11-bp sequence 43. Our ChIP analysis identifies, in the intact mouse, a specific DNA element (GGGTTTGCCC) located upstream of the transcription initiation site of the mouse cardiac HO-1 gene (from −468 bp to −459 bp) that binds both the p50 and p65 subunits (Fig. 8), providing a molecular substrate for the upregulation of the HO-1 gene by iNOS. To our knowledge, this is the first study to employ ChIP analysis in the mouse heart, a powerful approach for identifying transcription factors associated with specific regions of the target gene promoter 26.
Conclusions
We have used ChIP analysis of intact cardiac tissue combined with a genetically molecular approach in a well-established and physiologically relevant murine model of infarction. With this approach, we have demonstrated, for the first time, that iNOS modulates the expression of HO-1 in the heart by augmenting NF-κB nuclear localization and binding to the HO-1 gene promoter, resulting in increased transcription of the HO-1 gene. Our data indicate that NF-κB activation is important both for the basal levels of cardiac HO-1 protein expression and for iNOS-dependent upregulation of HO-1. We have also shown that HO-1 plays an obligatory role in the cardioprotection afforded by iNOS gene therapy. The present findings significantly expand our understanding of the regulation of cardiac HO-1 and of the mechanism whereby iNOS exerts its cardioprotective effects. The data reveal the existence of an iNOS-HO-1 cardioprotective module in which these proteins effectively function together to limit myocardial ischemia/reperfusion injury.
Commentary.
Although nitric oxide (NO) donors such as nitroglycerin have been used as antianginal and preload-reducing agents for over a century, their cardiovascular effects are still not entirely understood. In 1997, we proposed the NO hypothesis of ischemic preconditioning, which postulates that the cardioprotection afforded by the late phase of preconditioning is underlain by the upregulation of the inducible isoform of NO synthase and the attendant increase in the production of NO. This hypothesis has been subsequently validated and extended to other organs, including kidney and intestine, implying that the upregulation of iNOS is a central axis whereby tissues protect themselves from ischemia. However, the molecular basis responsible for the protective effects of iNOS in the heart remains largely unknown. In the current investigation we have discovered the existence of a close functional coupling between cardiac iNOS and cardiac heme oxygenase-1 (HO-1), another major cytoprotective protein. We found that upregulation of cardiac iNOS via gene transfer leads to increased expression of HO-1, and that this occurs via nuclear translocation of the transcription factor NF-κB and its binding to the promoter of the HO-1 gene. The identification of a coupling between iNOS and HO-1 has significant implications for the fields of gene therapy and ischemia/reperfusion injury, both from a conceptual and a therapeutic standpoint. For example, activation of the iNOS-HO-1 module via pharmacologic means or gene therapy may prove beneficial in patients with ischemic heart disease.
Supplementary Material
ACKNOWLEDGMENTS
None.
FUNDING SOURCES This study was supported in part by NIH grants R01 HL55757, HL-70897, HL-76794, P01HL78825, and P20 RR024489.
Footnotes
DISCLOSURES None.
REFERENCES
- 1.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 2.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 3.Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol. 2007;292:H19–27. doi: 10.1152/ajpheart.00712.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Guo Y, Xuan YT, Lowenstein CJ, Stevenson SC, Prabhu SD, Wu WJ, Zhu Y, Bolli R. Gene therapy with inducible nitric oxide synthase protects against myocardial infarction via a cyclooxygenase-2-dependent mechanism. Circ Res. 2003;92:741–748. doi: 10.1161/01.RES.0000065441.72685.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Q, Guo Y, Tan W, Stein AB, Dawn B, Wu WJ, Zhu X, Lu X, Xu X, Siddiqui T, Tiwari S, Bolli R. Gene therapy with iNOS provides long-term protection against myocardial infarction without adverse functional consequences. Am J Physiol Heart Circ Physiol. 2006;290:H584–589. doi: 10.1152/ajpheart.00855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 8.Deng YM, Wu BJ, Witting PK, Stocker R. Probucol protects against smooth muscle cell proliferation by upregulating heme oxygenase-1. Circulation. 2004;110:1855–1860. doi: 10.1161/01.CIR.0000142610.10530.25. [DOI] [PubMed] [Google Scholar]
- 9.Visner GA, Lu F, Zhou H, Liu J, Kazemfar K, Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation. 2003;107:911–916. doi: 10.1161/01.cir.0000048191.75585.60. [DOI] [PubMed] [Google Scholar]
- 10.Immenschuh S, Hinke V, Ohlmann A, Gifhorn-Katz S, Katz N, Jungermann K, Kietzmann T. Transcriptional activation of the haem oxygenase-1 gene by cGMP via a cAMP response element/activator protein-1 element in primary cultures of rat hepatocytes. Biochem J. 1998;334(Pt 1):141–146. doi: 10.1042/bj3340141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, Dawn B, Motterlini R, Bolli R. Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1649–1653. doi: 10.1152/ajpheart.00971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182. doi: 10.1165/rcmb.2006-0333TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakao A, Choi AM, Murase N. Protective effect of carbon monoxide in transplantation. J Cell Mol Med. 2006;10:650–671. doi: 10.1111/j.1582-4934.2006.tb00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jadhav A, Torlakovic E, Ndisang JF. Interaction Among Heme Oxygenase, Nuclear Factor-{kappa}B, and Transcription Activating Factors in Cardiac Hypertrophy in Hypertension. Hypertension. 2008 doi: 10.1161/HYPERTENSIONAHA.108.114801. [DOI] [PubMed] [Google Scholar]
- 15.Geraldes P, Yagi K, Ohshiro Y, He Z, Maeno Y, Yamamoto-Hiraoka J, Rask-Madsen C, Chung SW, Perrella MA, King GL. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/PI3-kinase/AKT-2 pathway. J Biol Chem. 2008 doi: 10.1074/jbc.M807036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill-Kapturczak N, Voakes C, Garcia J, Visner G, Nick HS, Agarwal A. A cis-acting region regulates oxidized lipid-mediated induction of the human heme oxygenase-1 gene in endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:1416–1422. doi: 10.1161/01.ATV.0000081656.76378.A7. [DOI] [PubMed] [Google Scholar]
- 17.Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a “molecular wrecking ball” to a “mesmerizing” trigger of cellular events. Biochem Biophys Res Commun. 2005;338:568–577. doi: 10.1016/j.bbrc.2005.08.121. [DOI] [PubMed] [Google Scholar]
- 18.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci U S A. 1994;91:5987–5991. doi: 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alam J, Cai J, Smith A. Isolation and characterization of the mouse heme oxygenase-1 gene. Distal 5′ sequences are required for induction by heme or heavy metals. J Biol Chem. 1994;269:1001–1009. [PubMed] [Google Scholar]
- 20.Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ, Bolli R. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–1109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 21.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol. 1998;275:H1375–1387. doi: 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–29. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawn B, Xuan YT, Marian M, Flaherty MP, Murphree SS, Smith TL, Bolli R, Jones WK. Cardiac-specific abrogation of NF- kappa B activation in mice by transdominant expression of a mutant I kappa B alpha. J Mol Cell Cardiol. 2001;33:161–173. doi: 10.1006/jmcc.2000.1291. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Guo Y, Tan W, Ou Q, Wu WJ, Sturza D, Dawn B, Hunt G, Cui C, Bolli R. Cardioprotection afforded by inducible nitric oxide synthase gene therapy is mediated by cyclooxygenase-2 via a nuclear factor-kappaB dependent pathway. Circulation. 2007;116:1577–1584. doi: 10.1161/CIRCULATIONAHA.107.689810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duh D, Saksida A, Petrovec M, Dedushaj I, Avsic-Zupanc T. Novel one-step real-time RT-PCR assay for rapid and specific diagnosis of Crimean-Congo hemorrhagic fever encountered in the Balkans. J Virol Methods. 2006;133:175–179. doi: 10.1016/j.jviromet.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Nie L, Vazquez AE, Yamoah EN. Identification of Transcription Factor-DNA Interactions Using Chromatin Immunoprecipitation Assays. Methods Mol Biol. 2009;493:311–322. doi: 10.1007/978-1-59745-523-7_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci U S A. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi AM. Heme oxygenase-1 protects the heart. Circ Res. 2001;89:105–107. [PubMed] [Google Scholar]
- 29.Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278:H643–651. doi: 10.1152/ajpheart.2000.278.2.H643. [DOI] [PubMed] [Google Scholar]
- 30.L’Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, Barsacchi R, Paolicchi A, Masiello P, Drummond GS, McClung JA, Abraham NG. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293:H3532–3541. doi: 10.1152/ajpheart.00826.2007. [DOI] [PubMed] [Google Scholar]
- 31.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolli R, Li QH, Tang XL, Guo Y, Xuan YT, Rokosh G, Dawn B. The late phase of preconditioning and its natural clinical application--gene therapy. Heart Fail Rev. 2007;12:189–199. doi: 10.1007/s10741-007-9031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, Dzau VJ, Lee ME, Perrella MA. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ Res. 2001;89:168–173. doi: 10.1161/hh1401.093314. [DOI] [PubMed] [Google Scholar]
- 34.Stocker R, Perrella MA. Heme oxygenase-1: a novel drug target for atherosclerotic diseases? Circulation. 2006;114:2178–2189. doi: 10.1161/CIRCULATIONAHA.105.598698. [DOI] [PubMed] [Google Scholar]
- 35.Dawn B, Bolli R. HO-1 induction by HIF-1: a new mechanism for delayed cardioprotection? Am J Physiol Heart Circ Physiol. 2005;289:H522–524. doi: 10.1152/ajpheart.00274.2005. [DOI] [PubMed] [Google Scholar]
- 36.Datta PK, Lianos EA. Nitric oxide induces heme oxygenase-1 gene expression in mesangial cells. Kidney Int. 1999;55:1734–1739. doi: 10.1046/j.1523-1755.1999.00429.x. [DOI] [PubMed] [Google Scholar]
- 37.Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, Bach FH, Otterbein LE. Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med. 2003;198:1707–1716. doi: 10.1084/jem.20031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinobe RT, Vlahakis JZ, Vreman HJ, Stevenson DK, Brien JF, Szarek WA, Nakatsu K. Selectivity of imidazole-dioxolane compounds for in vitro inhibition of microsomal haem oxygenase isoforms. Br J Pharmacol. 2006;147:307–315. doi: 10.1038/sj.bjp.0706555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiesel P, Patel AP, Carvajal IM, Wang ZY, Pellacani A, Maemura K, DiFonzo N, Rennke HG, Layne MD, Yet SF, Lee ME, Perrella MA. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ Res. 2001;88:1088–1094. doi: 10.1161/hh1001.091521. [DOI] [PubMed] [Google Scholar]
- 40.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 41.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 42.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL. IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.