

Abstract
Background
Hypertrophic cardiomyopathy is a genetic disease characterized by cardiac hypertrophy, myocyte disarray, interstitial fibrosis, and left ventricular (LV) dysfunction. We have proposed that hypertrophy and fibrosis, the major determinants of mortality and morbidity, are potentially reversible. We tested this hypothesis in β-myosin heavy chain–Q403 transgenic rabbits.
Methods and Results
We randomized 24 β-myosin heavy chain–Q403 rabbits to treatment with either a placebo or simvastatin (5 mg · kg−1 · d−1) for 12 weeks and included 12 nontransgenic controls. We performed 2D and Doppler echocardiography and tissue Doppler imaging before and after treatment. Demographic data were similar among the groups. Baseline mean LV mass and interventricular septal thickness in nontransgenic, placebo, and simvastatin groups were 3.9±0.7, 6.2±2.0, and 7.5±2.1 g (P<0.001) and 2.2±0.2, 3.1±0.5, and 3.3±0.5 mm (P=0.002), respectively. Simvastatin reduced LV mass by 37%, interventricular septal thickness by 21%, and posterior wall thickness by 13%. Doppler indices of LV filling pressure were improved. Collagen volume fraction was reduced by 44% (P<0.001). Disarray was unchanged. Levels of activated extracellular signal-regulated kinase (ERK) 1/2 were increased in the placebo group and were less than normal in the simvastatin group. Levels of activated and total p38, Jun N-terminal kinase, p70S6 kinase, Ras, Rac, and RhoA and the membrane association of Ras, RhoA, and Rac1 were unchanged.
Conclusions
Simvastatin induced the regression of hypertrophy and fibrosis, improved cardiac function, and reduced ERK1/2 activity in the β-myosin heavy chain–Q403 rabbits. These findings highlight the need for clinical trials to determine the effects of simvastatin on cardiac hypertrophy, fibrosis, and dysfunction in humans with hypertrophic cardiomyopathy and heart failure.
Keywords: hypertrophy, fibrosis, genetics, simvastatin, heart failure
Cardiovascular disease is the single most common cause of death in the Western world.1 Heart failure, the predominant long-term outcome of all forms of cardiovascular disease, accounts for ≈450 000 deaths per year in the United States alone.1 Cardiac hypertrophy and interstitial fibrosis, the common responses of the heart to all forms of injury, are the major determinants of morbidity and mortality from cardiovascular disease.2,3
Familial hypertrophic cardiomyopathy (HCM), a genetic model of cardiac hypertrophy and fibrosis,4 is the most common cause of sudden cardiac death in the young and a major cause of heart failure in elderly.5 Hypertrophy and fibrosis, as in acquired cardiovascular disease, are also the primary determinants of mortality and morbidity in HCM.6,7 The molecular genetic basis of HCM has been elucidated, and >100 mutations in 9 genes encoding sarcomeric proteins have been identified.4 Experimental studies4 in conjunction with studies in humans8 have provided significant insight into the pathogenesis of HCM and have led us and others to propose that the primary abnormality in HCM is impaired myocardial mechanical function.9 Accordingly, increased myocyte stress, which is imparted by the mutant contractile proteins, activates “stress-responsive” intracellular signaling molecules in a manner similar to pressure-overload, thus provoking the transcriptional machinery to induce hypertrophy and fibrosis. Thus, hypertrophy and fibrosis in HCM are “secondary phenotypes” and are potentially reversible.
We generated a transgenic rabbit model for HCM by the cardiac-restricted expression of β-myosin heavy chain (MyHC)-glutamine 403 (Q403), which is known to cause HCM in humans.10 The β-MyHC-Q403 rabbits fully recapitulate the phenotype of human HCM and exhibit cardiac hypertrophy, interstitial fibrosis, myocyte disarray, and cardiac dysfunction,11,12 and they serve as a desirable model to determine the effects of therapies targeted at specific pathways to reverse cardiac hypertrophy, fibrosis, and dysfunction. Recently, 3-hydroxy-3-methyglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) have been shown to inhibit angiotensin II–mediated myocyte hypertrophy13,14 and to block intracellular signaling molecules implicated in cardiac hypertrophy.15-17 Thus, we determined the effects of simvastatin, a pleiotropic HMG-CoA reductase inhibitor, on cardiac hypertrophy, fibrosis, and dysfunction in the β-MyHC-Q403 transgenic rabbits.
Methods
The mutant β-MyHC-Q403 transgenic rabbits were generated as described previously, and their phenotypic characteristics were the same as those published previously.11,12 A total of 24 adult β-MyHC-Q403 transgenic rabbits were matched for age and sex and randomized to either treatment with a placebo or simvastatin. Twelve age- and sex-matched nontransgenic littermates were included as controls. Baseline M-mode, 2D, Doppler echocardiography and tissue Doppler imaging were performed in all rabbits. The primary end point was regression of left ventricular (LV) mass, as detected by 2D echocardiography. The secondary end points were changes in interventricular septal thickness, posterior wall thickness, cardiac function, interstitial fibrosis, and myocyte disarray. Simvastatin was mixed with rabbit high-fiber diet #5326 (prepared by Purina Test diet) and was fed to rabbits at a concentration of 5 mg · kg−1 · d−1, which is considered a safe dose in rabbits,18 for 12 weeks. Follow-up echocardiographic studies were performed at the completion of the study. Echocardiographic images were obtained and analyzed as described previously.11,12
Detection and Quantification
Fibrillar Collagen
Interstitial collagen volume fraction was determined as described previously.19 In brief, 5-μm-thick myocardial sections were stained with collagen-specific Sirius red F3BA and were analyzed by an investigator who was blinded to the groups, in 10 randomly selected fields per section, in 10 sections per rabbit, and in 12 rabbits per group in a random fashion by computerized planimetry. Perivascular and epimysial collagens were excluded. To confirm the results of picrosirius red staining, 5 additional thin sections were stained with Masson trichrome and analyzed for collagen volume fraction.
Myocyte Disarray
Myocyte disarray was detected and quantified as described previously, with some modifications.19 Each myocardial section was divided into 50 fields of approximately equal size, and the presence or absence of disarray in each field was scored. Number of fields per section showing disarray was computed as percent showing disarray (per section) and the total percent disarray was determined in 8 sections per rabbit (400 fields per rabbit). Areas of myocardium at the junctions of interventricular septum with the ventricles and sections near the blood vessels, trabeculations, and papillary muscles were excluded.
Expression of Skeletal α-Actin
The expression of skeletal α-actin, a marker of secondary cardiac hypertrophy, was detected by Northern blotting. In brief, 20-μg aliquots of total RNA extracts were loaded onto formaldehydeagarose gels, electrophoresed, and transferred to nylon membranes.11 A 502-bp fragment of the rabbit skeletal α-actin gene was amplified by polymerase chain reaction. Oligonucleotide primers (forward primer: 5’TCATGGTCGGTATGGGTCAGA3’; reverse primer: 5’CCTCATAAATGGGCACGTTG3’) were designed on the basis of the sequence of human skeletal α-actin (GenBank accession number M20543). The probe was labeled with [32P]dCTP and hybridized to membranes. Signals were detected by autoradiography and quantified by spot densitometry.
Signaling Kinases and Molecules
Expression levels of total and phosphorylated extracellular signal-regulated kinase (ERK) 1/2, p38, and p70S6 kinase were detected by immunoblotting using pan-specific and phospho-specific antibodies. Expression levels of total and phosphorylated Jun N-terminal kinases (JNKs) were detected by immunoblotting after immunoprecipitation with specific antibodies. All primary antibodies were used at a concentration of 1:200, and secondary antibodies were used at concentrations of 1:1000 to 1:2000.
Activation of Ras was detected by selective-affinity precipitation of Ras-GTP with immobilized Raf-1 (Ras binding domain), and Ras detection was determined by blotting with a pan-isoform–specific Ras antibody. Activation of Rac was determined by selective affinity precipitation of Rac-GTP with immobilized p21 activated kinase 1 (Rac binding domain). A positive control assay, composed of Ras activation after the timed exposure of cultured mink lung epithelial cells to 10% fetal calf serum, was included.
Subcellular Fractionation and Localization of Ras and RhoA
To determine the degree of membrane association of Ras, RhoA, and Rac1, we performed subcellular fractionation of heart tissue. For each sample, 300 mg of tissue was homogenized in 14 mL of hypotonic buffer (10 mmol/L Tris-HCl [pH 7.5], 1.0 mmol/L MgCl2, 0.5 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 10 μg/mL aprotinin). The lysate was then transferred to a polycarbonate ultracentrifuge tube and spun at 100 000g for 90 minutes at 4°C. The supernatant fluid (representing the cytosolic fraction) was transferred to a new microcentrifuge tube; the pellet (representing the membrane fraction) was resuspended in 1 mL of hypotonic buffer, to which was added 1% sodium dodecyl sulfate. A total of 15 μg of the cellular and membrane fractions were fractionated on SDS-PAGE gels and analyzed by Western blotting using monoclonal anti-Ras and anti-RhoA (Upstate Biotechnology Inc).
Statistical Analysis
Continuous variables were expressed as mean±SD. Differences among the groups were compared by ANOVA for phenotypes with equal variance and by Kruskal-Wallis test for those with unequal variance. Differences at baseline and follow-up in each group were compared by paired t tests.
Results
Phenotype in the Mutant β-MyHC-Q403 Transgenic Rabbits
There were no significant differences in the mean age, male/female ratio, body weight, heart rate, or blood pressure between the β-MyHC-Q403 transgenic and nontransgenic rabbits (Table 1). However, as shown previously, LV mass, interventricular septal thickness, posterior wall thickness, and left atrial size were increased significantly in the β-MyHC-Q403 rabbits compared with nontransgenic ones (Table 1). Similarly, Doppler indices of LV filling pressures, early (E) and late (A) mitral inflow velocities, E/A ratio, and E/early diastolic velocity (Ea) ratio were increased significantly and isovolumic relaxation time was decreased. Furthermore, myocardial systolic and diastolic velocities were reduced significantly. Collagen volume fraction and the extent of myocyte disarray were increased in the β-MyHC-Q403 rabbits (Table 1).
TABLE 1.
Baseline Characteristics and Cardiac Phenotypes in the β-MyHC-Q403 Transgenic Rabbits
| Nontransgenic | β-MyHC-Q403 | P | |
|---|---|---|---|
| n | 12 | 24 | |
| Baseline characteristics | |||
| Sex, male/female | 9/3 | 17/7 | 0.79 |
| Age, mo | 27.6±2.7 | 30.0±7 | 0.13 |
| Body weight, kg | 3.7±0.5 | 3.9±0.4 | 0.13 |
| Heart rate, bpm | 145±14 | 143±15 | 0.83 |
| Systolic blood pressure, mm Hg | 74±8 | 73±7 | 0.84 |
| Phenotype of cardiac hypertrophy and function | |||
| LV mass, g | 3.9±0.7 | 6.8±2.1 | 0.001 |
| IVST, mm | 2.2±0.2 | 3.2±0.5 | 0.001 |
| PWT, mm | 2.3±0.3 | 3.0±0.6 | 0.001 |
| LA size, mm | 7.1±0.6 | 8.8±2.0 | 0.010 |
| LVEDD, mm | 14.4±1.4 | 14.7±1.3 | 0.64 |
| FS | 0.37±0.7 | 0.29±0.3 | 0.003 |
| Mitral inflow E velocity, cm/s | 47±6 | 54±8 | 0.004 |
| E/A ratio | 1.8±0.4 | 2.4±0.9 | 0.019 |
| IVRT, ms | 45.3±6 | 39.2±6 | 0.014 |
| E/Ea | 5.4±1.3 | 8.9±2.7 | <0.001 |
| Septal Sa | 7.1±1.6 | 4.8±1.1 | <0.001 |
| Septal Ea | 7.3±1.5 | 4.7±1.7 | <0.001 |
| Histological phenotypes | |||
| Collagen volume fraction, % | 3.6±1.2 | 9.6±2.2 | <0.001 |
| Myocyte disarray, % | 5.7±1.8 | 12.0±4.1 | 0.003 |
IVST indicates interventricular septal thickness; PWT, posterior wall thickness; LVEDD, left ventricular end-diastolic dimension; FS, fractional shortening; Sa, systolic velocity; Ea, early diastolic velocity; IVRT, isovolumic relaxation time.
Expression levels of activated ERK1/2 were increased by ≈2-fold in the β-MyHC-Q403 rabbits compared with non-transgenic rabbits (Figure 1). Levels of total ERK1/2 were unchanged. We detected no significant increase in the levels of total and activated p38, JNKs, Ras, Rac1, or p70S6 kinase, a downstream target of phosphatidylinositol 3-kinase, in the β-MyHC-Q403 rabbits (Figures 1 and 2A). In addition, the membrane association of Ras, Rac1, and RhoA was not significantly changed (Figure 2B).
Figure 1.
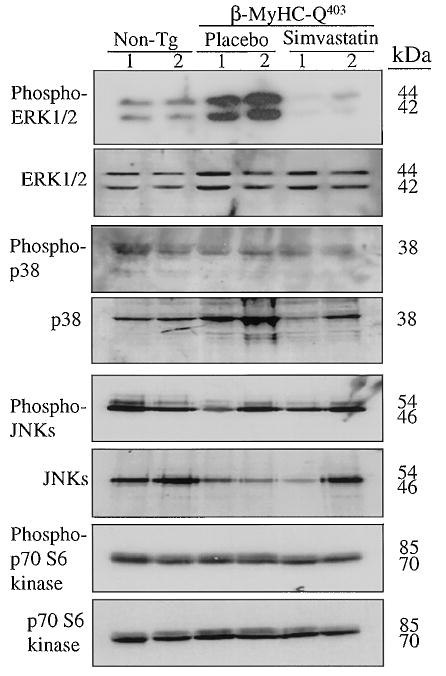
Levels of activated and total stress-responsive intra-cellular signaling kinases in the heart of β-MyHC-Q403 transgenic rabbits. Each lane represents a cardiac protein extract from one rabbit, and 2 rabbits per group are shown. Immunoblots for ERK1/2, p38, JNK, and p70S6 kinase are shown. The upper blot in each set of panels represents levels of phosphorylated kinases, and the lower panel represents levels of total kinases. Non-tg indicates nontransgenic.
Figure 2.
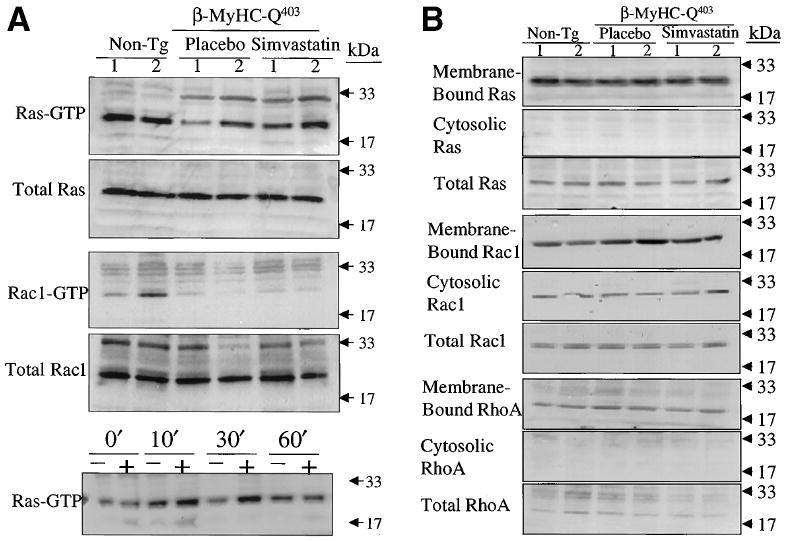
GTP-bound and membrane association of Ras, Rac1, and RhoA. A, Activation Ras and Rac1. Each lane represents cardiac protein extract from one rabbit. Upper blot in each set of panels shows levels of GTP-bound Ras and Rac1, and the lower blot shows levels of total Ras and Rac-1. The bottom panel represents a positive control showing activation of Ras after stimulation of mink lung epithelial cells with 10% fetal calf serum at the specified time points. Molecular size markers are indicated on the right of each panel. B, Membrane-bound, soluble, and total Ras, Rac1, and RhoA. Non-tg indicates nontransgenic.
Effects of Simvastatin on LV Hypertrophy, Fibrosis, and Function
There were no significant differences in mean age, male/female ratio, body weight, heart rate, blood pressure, or cardiac phenotype, including LV mass, interventricular septal thickness, posterior wall thickness, LV end-diastolic diameter, LV end-systolic diameter, mitral inflow and pulmonary venous Doppler velocities, and tissue Doppler indices, at baseline between the placebo and simvastatin groups (Table 2). Therefore, the placebo and simvastatin groups were well matched.
TABLE 2.
Indices of Cardiac Hypertrophy and Function at Baseline and After Treatment With Simvastatin
|
β-MyHC-Q403 |
||||
|---|---|---|---|---|
| Nontransgenic | Placebo | Simvastatin | P* | |
| n | 12 | 12 | 12 | NA |
| Sex, male/female | 9/3 | 8/4 | 9/3 | 0.87 |
| Age, mo | 27.6±2.7 | 30.3±6.3 | 1.5±7.7 | 0.29 |
| Body weight, g | 3.7±0.5 | 4.0±0.4 | 3.8±0.4 | 0.14 |
| Heart rate, bpm | 145±14 | 147±18 | 139±14 | 0.49 |
| Systolic blood pressure, mm Hg | 74±8 | 74±5 | 72±8 | 0.93 |
| Indices of cardiac hypertrophy and size | ||||
| LV mass, g | ||||
| Baseline | 3.9±0.7 | 6.2±2.0 | 7.5±2.1 | <0.001 |
| Follow-up | 3.6±0.8 | 5.9±2.6 | 4.4±1.6 | 0.017 |
| Change | 0.25±0.8 | 0.26±2.5 | 2.8±2.0 | 0.004 |
| P (paired t test) | 0.37 | 0.74 | 0.001 | |
| IVST, mm | ||||
| Baseline | 2.2±0.2 | 3.1±0.5 | 3.3±0.5 | <0.001 |
| Follow-up | 2.2±0.2 | 2.8±0.7 | 2.6±0.4 | 0.019 |
| Change | 0.0±0.2 | 0.3±0.8 | 0.7±0.6 | 0.033 |
| P (paired t test) | 0.90 | 0.18 | 0.006 | |
| PWT, mm | ||||
| Baseline | 2.3±0.3 | 3.0±0.5 | 3.0±0.7 | 0.003 |
| Follow-up | 2.2±0.3 | 3.2±0.8 | 2.4±0.6 | 0.001 |
| Change | 0.0±0.2 | −0.2±0.7 | 0.4±0.6 | 0.026 |
| P (paired t test) | 0.29 | 0.30 | 0.036 | |
| LVEDD, mm | ||||
| Baseline | 14.5±1.4 | 14.7±1.3 | 16.0±2.1 | 0.053 |
| Follow-up | 14.0±1.7 | 14.3±1.3 | 13.8±2.2 | 0.08 |
| Change | 0.4±1.8 | 0.36±1.4 | 2.11±3.3 | 0.14 |
| P (paired t test) | 0.43 | 0.40 | 0.06 | |
| Indices of left ventricular filling pressure | ||||
| Mitral inflow E/A ratio | ||||
| Baseline | 1.8±0.4 | 2.2±0.5 | 2.6±1.2 | 0.075 |
| Follow-up | 1.8±0.3 | 2.0±0.3 | 1.7±0.4 | 0.31 |
| Change | 0.03±0.3 | 0.2±0.5 | 0.9±1.3 | 0.043 |
| P (paired t test) | 0.63 | 0.15 | 0.048 | |
| IVRT, ms | ||||
| Baseline | 45.2±6.5 | 37.8±6.2 | 40.4±6.0 | 0.024 |
| Follow-up | 45.1±4.5 | 38.2±5.8 | 43.5±6.0 | 0.014 |
| Change | 0.17±4.2 | −0.36±4.0 | −3.3±2.9 | 0.082 |
| P (paired t test) | 0.89 | 0.77 | 0.004 | |
| Ar-A duration, ms | ||||
| Baseline | −2.1±16.0 | 4.2±11.1 | 13.5±16.6 | 0.043 |
| Follow-up | −5.3±11.2 | 1.0±7.5 | −8±15.2 | 0.196 |
| Change | 3.3±6.7 | 3.2±6.2 | 22.5±23.7 | 0.004 |
| P (paired t test) | 0.12 | 0.12 | 0.010 | |
| Septal E/Ea | ||||
| Baseline | 6.7±1.7 | 12.4±4.3 | 13.2±6.3 | 0.002 |
| Follow-up | 6.4±1.0 | 10.7±3.8 | 7.4±2.4 | 0.002 |
| Change | 0.2±0.9 | 1.7±5.0 | 6.3±5.2 | 0.004 |
| P (paired t test) | 0.38 | 0.28 | 0.002 | |
| Lateral E/Ea | ||||
| Baseline | 5.4±1.3 | 8.6±2.8 | 9.1±2.7 | 0.001 |
| Follow-up | 4.8±0.9 | 8.6±3.7 | 5.2±1.5 | 0.001 |
| Change | 0.6±1.0 | 0.0±1.5 | 4.2±3.2 | <0.001 |
| P (paired t test) | 0.054 | 1.0 | 0.002 | |
| Indices of global cardiac function and myocardial relaxation and contraction | ||||
| FS, % | ||||
| Baseline | 37.0±7.2 | 29.0±3.5 | 30.0±4.3 | 0.001 |
| Follow-up | 36.6±7.2 | 27.9±4.4 | 33.0±4.4 | 0.003 |
| Change | 0.5±5.6 | 1.1±4.7 | −1.5±4.4 | 0.46 |
| P (paired t test) | 0.76 | 0.46 | 0.28 | |
| Septal Sa, cm/s | ||||
| Baseline | 7.1±1.6 | 4.5±0.7 | 5.1±1.4 | <0.001 |
| Follow-up | 7.2±1.2 | 5.1±1.1 | 6.7±1.0 | <0.001 |
| Change | −0.04±1.0 | −0.56±0.9 | −1.6±1.8 | 0.020 |
| P (paired t test) | 0.89 | 0.053 | 0.013 | |
| Septal Ea, cm/s | ||||
| Baseline | 7.3±1.7 | 4.5±1.2 | 4.9±1.8 | <0.001 |
| Follow-up | 6.8±1.0 | 5.1±1.6 | 6.2±1.6 | 0.025 |
| Change | 0.5±1.0 | −0.6±1.7 | −1.5±1.8 | 0.015 |
| P (paired t test) | 0.11 | 0.27 | 0.021 | |
| Lateral Sa, cm/s | ||||
| Baseline | 9.0±1.1 | 6.2±1.2 | 6.2±2.0 | <0.001 |
| Follow-up | 8.4±0.9 | 6.5±0.9 | 8.3±2.0 | 0.002 |
| Change | 0.53±0.83 | −0.28±1.1 | −2.1±2.7 | 0.003 |
| P (paired t test) | 0.05 | 0.39 | 0.026 | |
| Lateral Ea, cm/s | ||||
| Baseline | 8.9±1.5 | 6.5±1.7 | 6.5±1.6 | 0.001 |
| Follow-up | 9.2±1.4 | 6.3±1.6 | 8.7±1.8 | <0.001 |
| Change | −0.3±1.9 | 0.19±0.57 | −2.3±2.9 | 0.016 |
| P (paired t test) | 0.59 | 0.29 | 0.025 | |
Values are mean±SD. Ar-A duration indicates time interval between pulmonary venous atrial reverse wave and antegrade mitral inflow. Other abbreviations as in Table 1.
P values are derived by comparing variables among the 3 groups by ANOVA or Kruskal-Wallis test.
Treatment with simvastatin reduced mean LV mass by 37%, interventricular septal thickness by 21%, posterior wall thickness by 15%, and LV end-diastolic diameter by 13% (Table 1). Concordant with regression of cardiac hypertrophy, Doppler indices of LV filling pressure, namely E/A ratio (reduced 35%), isovolumic relaxation time (improved 7.5%), septal E/Ea ratio (reduced 48%), and lateral E/Ea (reduced 46%) were improved significantly, indicating a lower LV filling pressure. Similarly, the time interval between the atrial reverse wave and antegrade mitral flow was reduced (by 167%), indicating a lower left atrial pressure.20 Myocardial systolic and diastolic velocities at both corners of mitral annulus also showed significant improvement. Collectively, Doppler data indicate a significant reduction in the LV filling pressure and improvement in myocardial contractile and relaxation properties.
Interstitial collagen volume fraction was reduced by 44% in the simvastatin group (nontransgenic, 3.6±1.2%; placebo, 9.6±2.2%; simvastatin, 5.4±1.5% of the myocardium; P=0.001). Representative micrographs of Masson trichrome staining are shown in Figure 3. Myocyte disarray comprised 5.7±1.8%, 12.0±4.1%, and 10.2±2.2% of the myocardium in nontransgenic, placebo, and simvastatin groups, respectively (placebo versus simvastatin, P=0.295).
Figure 3.
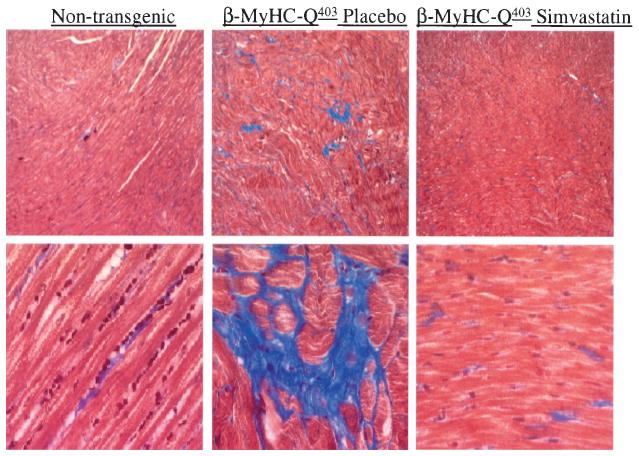
Masson trichrome staining. The upper panels show low magnification (×40) and the lower panels high magnification (×400) of thin myocardial sections.
Expression of the skeletal α-actin was increased in the placebo group compared with nontransgenic rabbits, and treatment with simvastatin reduced its expression >3-fold (Figure 4).
Figure 4.
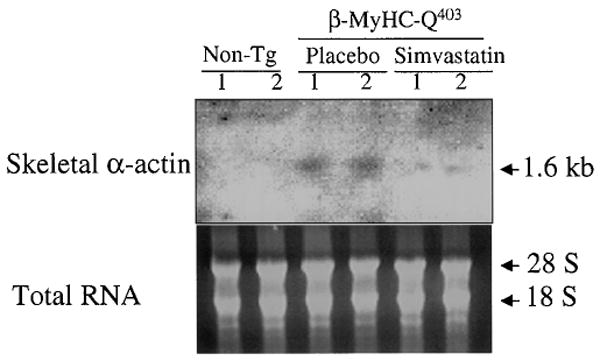
Northern blot showing expression of skeletal α-actin. Non-tg indicates nontransgenic.
Effects of Simvastatin on Intracellular Signaling Kinases and Molecules
Treatment with simvastatin significantly reduced activated ERK1/2 to levels less than those in the nontransgenic rabbits (Figure 2A). Simvastatin had no significant effect on total and activated p38, JNKs, Ras, Rac1, and p70S6 kinase or the membrane association of Ras, Rac1, and RhoA in the mutant β-MyHC-Q403 rabbits (Figure 2).
Discussion
We generated a transgenic rabbit model of human HCM by cardiac-restricted expression of β-MyHC-Q403, which is known to cause HCM in humans.10 The adult β-MyHC-Q403 rabbits fully recapitulate the phenotype of human HCM and exhibit cardiac hypertrophy, myocyte disarray, interstitial fibrosis, increased LV filling pressure, and reduced myocardial contraction and relaxation velocities.11,12 We performed a randomized study and treated the mutant β-MyHC-Q403 rabbits with a placebo or simvastatin for 12 weeks. Treatment with simvastatin reduced LV mass, wall thickness, and filling pressure significantly. In addition, collagen volume fraction was reduced by ≈50%, but the extent of myocyte disarray was unchanged. Levels of activated ERK1/2 were increased in the mutant β-MyHC-Q403 rabbits and were reduced to less than normal after treatment with simvastatin. The expression of skeletal α-actin, a marker of secondary hypertrophy, which was increased in the β-MyHC-Q403, was reduced after treatment with simvastatin. Thus, simvastatin induced the regression of cardiac hypertrophy and fibrosis, improved LV filling pressures, and reduced the levels of activated stress-responsive ERK1/2 in a transgenic rabbit model of human HCM.
The study was randomized, and there were no significant differences in the baseline demographic and echocardiographic phenotypes between the placebo and simvastatin groups. We acquired the data and performed extensive phenotypic characterization without knowledge of the group assignment. The results were concordant for strong beneficial effects of simvastatin on molecular (reduction in phospho-ERK1/2 and expression of skeletal α-actin), histological (reduction of fibrosis), structural (LV mass and wall thickness), and functional (reduction of LV filling pressures and improvement of myocardial contraction and relaxation velocities) phenotypes. Similarly, significant improvement in indices of hypertrophy and LV filling pressure at follow-up (compared with baseline) were detected only in the simvastatin group. The observed beneficial effects of simvastatin in the β-MyHC-Q403 rabbits are in accord with the effects of statins on the prevention of angiotensin II–induced myocyte hypertrophy13 and pressure-overload–induced hypertrophy in rats.14 The dose of simvastatin used in this study is considered safe18 and similar to a previously used dose of 3.6 mg · kg−1 · d−1, which was shown to induce regression of cardiac hypertrophy in load-induced hypertrophy.14 The above dose is higher than the conventional dose of simvastatin used in humans (up to 80 mg/d) and, thus, whether the observed results could be extended to human patients with HCM and heart failure needs to be explored.
The mechanism(s) by which simvastatin induces the regression of hypertrophy and fibrosis and improves cardiac function is likely to involve downregulating the levels of activated ERK1/2, the predominant stress-responsive intra-cellular signaling kinase involved in modulating cardiac hypertrophy.21 Simvastatin, an HMG-CoA reductase inhibitor, has pleiotropic effects that interact with the effects of renin-angiotensin aldosterone system pathways. Simvastatin has also been shown to induce the regression of load-induced cardiac hypertrophy by reducing the activity of the angiotensin-converting enzyme and the cardiac content of angiotensin II.14 Simvastatin inhibits the synthesis of isoprenoid intermediates of cholesterol biosynthesis farnesylpyrophosphate and geranylgeranylpyrophosphate, which are used in the post-translation modification of the Ras and the Rho family of proteins, respectively, by isopropenyl transferases.22 This modification step is considered essential for maturation, membrane localization, and the subsequent activation of small GTP-binding proteins.23
We found no significant differences in the activation and membrane association of Ras, Rac1 and RhoA. However, the lack of a significant increase in activation of the selected small GTP-binding proteins in the heart of β-MyHC-Q403 rabbits does not necessarily exclude their involvement in the pathogenesis of HCM. It may reflect the nature of the stimulus, which is chronic and relatively low, in contrast to in vitro cell culture experiments, in which the stimulus is acute and strong. It is also possible that Ras-independent mechanisms are involved in the activation of ERK1/2 and the effects of simvastatin in the heart of β-MyHC-Q403 rabbits. For example, G-protein–coupled receptor agonists can activate ERK1/2 through the protein kinase A/Rap1-B/Raf pathway and through the activation of phospholipase C. Activated phospholipase C catalyzes the hydrolysis of phosphatidyl-inositol bisphosphate into diacyl-glycerol and inositol trisphosphate. The latter induces calcium release from the endoplasmic reticulum and, together with diacyl-glycerol, activates protein kinase C.
Protein kinase C and Ca2+ lie upstream of a family of mitogen-activated protein kinases and plays a critical role in activating ERK1/2.24-26 Other pathways, including mitogen-activated protein (MAP) kinase kinases (MKKKs) such as Nck-interacting kinase and adaptor proteins such as Grb2, could also activate (MAP/ERK kinase kinase [MEKK] 1)/(MAP/ERK kinase 1/2) and, subsequently, ERK1/2.27 Furthermore, scaffolding or anchoring proteins such as 14-3-3, by regulating protein-protein interactions and subcellular localization, could activate the components of the MEKK1 pathway.27 Moreover, complex interactions between mitogen-activated protein kinase, Ca2+-calmodulin/calcineurin, and β1-integrin pathways exists that could lead to the activation of ERK1/2. Therefore, a variety of upstream regulators, including Ras-independent pathways, could activate ERK1/2. Extensive investigation of signaling pathways and the mechanism(s) by which simvastatin suppresses the activation of ERK1/2 in the β-MyHC-Q403 rabbits require additional investigations.
Hypertrophy and fibrosis are the major clinical and pathological phenotypes of HCM, which is the most common cause of sudden cardiac death in the young.5 None of the existing pharmacological therapies for HCM induces a regression of hypertrophy and fibrosis or reduces mortality.28 Similarly, myomectomy and nonsurgical septal ablation are considered palliative therapies that are limited to patients who have significant septal hypertrophy and resting outflow tract obstruction. Therefore, our findings in a transgenic rabbit model that fully recapitulates the phenotype of HCM in humans is the first pharmacological intervention to reverse the underlying pathology, ie, cardiac hypertrophy, fibrosis, and dysfunction. Because the penetrance of the causal mutations in HCM is age-dependent, our findings also raise the possibility of an early intervention to prevent the development of cardiac phenotype in HCM. Furthermore, because hypertrophy and fibrosis are the common responses of the heart to all forms of injury and are major determinants of mortality and morbidity,2,3 our findings could have broader implications for the treatment and prevention of all forms of cardiovascular disease. We note that simvastatin has a well-established safety profile, and it has been used extensively in humans. Thus, our results highlight the need for a clinical trial to determine the potential salutary effects of simvastatin in humans with HCM or heart failure.
In summary, we have shown that simvastatin, a pleiotropic HMG-CoA reductase inhibitor, induces the regression of cardiac hypertrophy and fibrosis, reduces levels of activated stress-responsive signaling kinases, and improves LV filling pressures in a transgenic rabbit model that fully recapitulates the phenotype of human HCM. These findings, if confirmed in humans, could provide a new option for the treatment and prevention of cardiac hypertrophy, fibrosis, and dysfunction in HCM and in all forms of cardiovascular disease.
Acknowledgments
Supported by grants from the National Heart, Lung, and Blood Institute, Specialized Centers of Research (P50-HL42267-01), grant HL-42550, and an Established Investigator Award (9640133N) from the American Heart Association National Center, Dallas, Texas.
References
- 1.Heart and Stroke Statistical Update. Dallas, Tex: American Heart Association; 2001. [April 1, 2001]. Available at: http://www.americanheart.org/statistics/pdf/HSSTATS2001_1.0.pdf. [Google Scholar]
- 2.Haider AW, Larson MG, Benjamin EJ, et al. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- 3.Assayag P, Carre F, Chevalier B, et al. Compensated cardiac hypertrophy: arrhythmogenicity and the new myocardial phenotype, I: fibrosis. Cardiovasc Res. 1997;34:439–444. doi: 10.1016/s0008-6363(97)00073-4. [DOI] [PubMed] [Google Scholar]
- 4.Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes: clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. [PubMed] [Google Scholar]
- 6.Shirani J, Pick R, Roberts WC, et al. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 7.Spirito P, Maron BJ. Relation between extent of left ventricular hypertrophy and occurrence of sudden cardiac death in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1990;15:1521–1526. doi: 10.1016/0735-1097(90)92820-r. [DOI] [PubMed] [Google Scholar]
- 8.Nagueh SF, Bachinski L, Meyer D, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with familial hypertrophic cardiomyopathy and provides a novel means for an early diagnosis prior to an independent of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58–60. doi: 10.1016/s0140-6736(99)06187-5. [DOI] [PubMed] [Google Scholar]
- 10.Watkins H, Rosenzweig A, Hwang DS, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 11.Marian AJ, Wu Y, Lim DS, et al. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest. 1999;104:1683–1692. doi: 10.1172/JCI7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagueh SF, Kopelen HA, Lim DS, et al. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102:1346–1350. doi: 10.1161/01.cir.102.12.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oi S, Haneda T, Osaki J, et al. Lovastatin prevents angiotensin II-induced cardiac hypertrophy in cultured neonatal rat heart cells. Eur J Pharmacol. 1999;376:139–148. doi: 10.1016/s0014-2999(99)00282-4. [DOI] [PubMed] [Google Scholar]
- 14.Luo JD, Zhang WW, Zhang GP, et al. Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clin Exp Pharmacol Physiol. 1999;26:903–908. doi: 10.1046/j.1440-1681.1999.03165.x. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez-Perera O, Perez-Sala D, Soria E, et al. Involvement of Rho GTPases in the transcriptional inhibition of preproendothelin-1 gene expression by simvastatin in vascular endothelial cells. Circ Res. 2000;87:616–622. doi: 10.1161/01.res.87.7.616. [DOI] [PubMed] [Google Scholar]
- 16.Park HJ, Galper JB. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors up-regulate transforming growth factor-beta signaling in cultured heart cells via inhibition of geranylgeranylation of RhoA GTPase. Proc Natl Acad Sci U S A. 1999;96:11525–11530. doi: 10.1073/pnas.96.20.11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guijarro C, Blanco-Colio LM, Ortego M, et al. 3-Hydroxy-3-methylglutaryl coenzyme a reductase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ Res. 1998;83:490–500. doi: 10.1161/01.res.83.5.490. [DOI] [PubMed] [Google Scholar]
- 18.Gerson RJ, MacDonald JS, Alberts AW, et al. Animal safety and toxicology of simvastatin and related hydroxy-methylglutaryl-coenzyme A reductase inhibitors. Am J Med. 1989;87:28S–38S. doi: 10.1016/s0002-9343(89)80596-0. [DOI] [PubMed] [Google Scholar]
- 19.Lim DS, Lutucuta S, Bachireddy P, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagueh SF, Lakkis NM, Middleton KJ, et al. Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation. 1999;99:254–261. doi: 10.1161/01.cir.99.2.254. [DOI] [PubMed] [Google Scholar]
- 21.Sugden PH, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 22.Grunler J, Ericsson J, Dallner G. Branch-point reactions in the biosynthesis of cholesterol, dolichol, ubiquinone and prenylated proteins. Biochim Biophys Acta. 1994;1212:259–277. doi: 10.1016/0005-2760(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 23.Hori Y, Kikuchi A, Isomura M, et al. Post-translational modifications of the C-terminal region of the rho protein are important for its interaction with membranes and the stimulatory and inhibitory GDP/GTP exchange proteins. Oncogene. 1991;6:515–522. [PubMed] [Google Scholar]
- 24.Zou Y, Komuro I, Yamazaki T, et al. Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J Biol Chem. 1996;271:33592–33597. doi: 10.1074/jbc.271.52.33592. [DOI] [PubMed] [Google Scholar]
- 25.Craddock BL, Hobbs J, Edmead CE, et al. Phosphoinositide 3-kinase-dependent regulation of IL-3-induced proliferation: involvement of mitogen-activated protein kinases, SHP2 and Gab2. J Biol Chem. doi: 10.1074/jbc.M009098200. In press. [DOI] [PubMed] [Google Scholar]
- 26.Lorimer IA, Lavictoire SJ. Activation of extracellular-regulated kinases by normal and mutant EGF receptors. Biochim Biophys Acta. 2001;1538:1–9. doi: 10.1016/s0167-4889(00)00129-4. [DOI] [PubMed] [Google Scholar]
- 27.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 28.Marian AJ. Hypertrophic cardiomyopathy. In: Rackel RE, editor. Conn’s Current Therapy. Philadelphia: Saunders; 2000. pp. 286–287. [Google Scholar]