

Abstract
Hypertrophic cardiomyopathy (HCM), the most common cause of sudden cardiac death in the young, is characterized by a diverse array of cardiac phenotypes evolving over several decades. We have developed transgenic rabbits that fully recapitulate the phenotype of human HCM and provide for the opportunity to delineate the sequence of evolution of cardiac phenotypes, and thus, the pathogenesis of HCM.
We determined evolution of biochemical, molecular, histological, structural and functional phenotypes at 4 age-periods in 47 β-myosin heavy chain-glutamine (MyHC-Q)-403 transgenic rabbits. Ca+2 sensitivity of myofibrillar ATPase activity was reduced very early and in the absence of other discernible phenotypes. Myocyte disarray also occurred early, prior to, and independent of hypertrophy and fibrosis. The latter phenotypes evolved predominantly during puberty in conjunction with activation of stress-related signaling kinases. Myocardial contraction and relaxation velocities were decreased early despite normal global cardiac function and in the absence of histological phenotype. Global cardiac function declined with aging, while left atrial size was increased along with Doppler indices of left ventricular filling pressure.
Thus, Ca+2 sensitivity of myofibrillar ATPase activity is a primary phenotype expressed early and independent of the ensuing phenotypes. Pathogenesis of myocyte disarray, which exhibits age-independent penetrance, differs from those of hypertrophy and fibrosis, which show age-dependent expression. Myocardial dysfunction is an early marker that predicts subsequent development of hypertrophy. These findings in an animal model that recapitulates the phenotype of human HCM, implicate involvement of multiple independent mechanisms in the pathogenesis of cardiac phenotypes in HCM.
Keywords: Transgenic animal models, Cardiomyopathy, Hypertrophy, Genetics, ATPase, Echocardiography, Tissue Doppler
1. Introduction
HCM is a relatively common disease [1] caused by mutations in contractile sarcomeric proteins [2,3]. HCM is diagnosed clinically by the presence of unexplained cardiac hypertrophy and pathologically by myocyte hypertrophy, disarray and interstitial fibrosis [4]. HCM is a common cause of sudden cardiac death (SCD) in the young [5] and a major cause of diastolic heart failure in the elderly [2]. Cardiac hypertrophy [6], myocyte disarray [7] and interstitial fibrosis [8] are considered major determinants of clinical outcome and risk of SCD in HCM.
Progress in elucidation of molecular genetics of HCM in humans has been dramatic and over 200 mutations in 11 genes have been identified [3,9]. However, the pathogenesis of HCM and temporal evolution of its diverse phenotypes are largely unknown. Efforts to identify the initial defect(s) and to determine the sequence of evolution of cardiac phenotypes in humans have been hampered by the slow evolution of phenotypes over several decades and the practical difficulties, such as performing serial histological studies. Genetic animal models have been generated in an attempt to unravel the pathogenesis of HCM and to develop specific therapies. While in vitro and in vivo functional studies have provided significant insight and have implicated a number of abnormalities (reviewed in [10]), the results, at least regarding mutations in the myosin heavy chain (MyHC) have been compounded by the composition of the sarcomeric MyHC isoforms, since differences in the sarcomeric protein composition is known to affect myofibrillar ATPase activity and acto-myosin molecular mechanics [11,12]. We have developed a transgenic rabbit model [13] by cardiac-restricted expression of mutant β-MyHC-glutamine 403 (-Q403), known to cause HCM in humans [14]. The β-MyHC-Q403 transgenic rabbit model offer several advantages over mouse models that partly pertain to the composition of sarcomeric proteins. In contrast to mice in which α-MyHC predominates, the β-MyHC is the predominant sarcomeric protein in the rabbit and human hearts [11,12]. The β-MyHC-Q403 transgenic rabbits exhibit cardiac hypertrophy, myocyte disarray, interstitial fibrosis and normal global systolic function [15] and fully recapitulate the phenotype of HCM in humans. Therefore, we determined the sequence of evolution of cardiac phenotypes in a large number of β-MyHC-Q403 transgenic rabbits over a 4-year period in an attempt to gain insight into the pathogenesis of cardiac phenotype and the primary defect imparted by the β-MyHC-Q403 mutation in in vivo.
2. Material and methods
The Institutional Review Board of Baylor College of Medicine approved the study protocol.
2.1. β-MyHC-Q403 transgenic rabbits
Generation of the β-MyHC-Q403 transgenic rabbits and characterization of initial cardiac phenotype have been published [15,16]. We compared the cardiac phenotype in 47 transgenic and 47 non-transgenic rabbits, categorized into 4 age groups (cross-sectional study). The first group was comprised of rabbits (transgenic and their non-transgenic littermates) younger than 6 months of age (pre-puberty period [17]). The second group included 6 –18 months old rabbits, which is the period of accelerated growth and early adulthood; the third included 19 – 30 months rabbits (approximate end of reproductive life); and the fourth group was comprised of rabbits older than 30 months of age (considered old age [17]). The above categorization is in accord with the phenotype in human patients who exhibit accelerated cardiac hypertrophic response during puberty [18] and the observation that HCM also could evolve into dilated cardiomyopathy in humans, often in older age [2]. All rabbits underwent echocardiography including Tissue Doppler Imaging (TDI) followed by histological and molecular studies.
In the next set of studies, to determine evolution of cardiac phenotypes in the same rabbits over a 1-year period (longitudinal follow up study), we performed serial studies in 24 adult mutant and 24 adult non-transgenic rabbits, matched for age and gender, at 18 and 30 months of age. We performed serial echocardiography and Tissue Doppler Imaging (TDI) at the baseline and follow up and upon completion of the follow up echocardiograms, rabbits were euthanized for histological and molecular studies.
2.2. Echocardiographic studies and tissue Doppler imaging
Echocardiographic and tissue Doppler images were obtained and analyzed as previously published [16,19].
2.3. Myofibrillar protein isolation and measurement of ATPase activity
Myofibrillar protein was isolated from hearts of 3-5 transgenic and 3-5 non-transgenic rabbits in each age group by treatment with Triton-X 100 per a protocol described by Solaro et al. [20]. In brief, myocardial tissue was minced and homogenized using Polytron in relaxing buffer (4mM phosphocreatine, 1mM ATP, 50 mM 2,3-butanedione monoxime, 1mM DTT, 1mM benzamidine HCl, 0.1 mM PMSF, 1% Triton–X and proteinase inhibitors). The homogenate was precipitated by centrifugation and the pellet was resuspended in standard buffer (75 mM KCl, 10 mM imidazole pH 7.2, 2mM MgCl2, 2 mM EGTA and 1 mM NaN3) with 2% Triton-X. Following 2 cycles of homogenization and centrifugation, Triton-X was removed and the final pellet was resuspended in K-60 buffer containing 60 mM KCl, 20 mM MOPS and 2 mM MgCl2. Protein concentration was determined by Bradford assay.
Myofibrillar Ca+2 activated ATPase activity was measured essentially using a protocol provided by Dr. RJ Solaro (University of Illinois) with minimal modifications. In brief, ATPase activities were determined by measuring release of inorganic phosphate (Pi) from ATP by myofibrillar proteins in triplicate sets for each Ca+2 concentration. The reaction begun by the adding 2 mM ATP to each tube containing 100 μg of myofibrillar proteins and incubating for 5 minutes at 30 °C. The reaction was stopped by adding 250 μl of 10% ice-cold trichloroacetic acid to each tube. The final ATPase assay products were centrifuged at 6000 rpm on a bench top centrifuge at 4 °C for 10 minutes and 100 μL of each supernatant was used for the Pi Carter assay. Final Pi concentration was determined at 625 nm using a spectrophotometer.
2.4. Detection and quantification of signaling kinases and molecules
Expression levels of total and phosphorylated ERK1/2, p38, and JNKs kinases and activation of Ras, Rac and RhoA and their membrane association were detected as described [19].
2.5. Detection and quantification of fibrillar collagen
Interstitial collagen volume fraction (CVF) was determined by computerized planimetry in 10 randomly selected fields per section and in 10 sections per rabbit by an investigator blinded to the groups, as described [19].
2.6. Detection and quantification of myocyte disarray
Myocyte disarray was detected and quantified in 50 approximately equal-size fields per section and 8 sections per rabbits as described [19].
2.7. Quantification of myocyte cross-sectional area
Myocyte cross sectional area (CSA) was determined using a published method with modifications [21]. To define the boundaries of myocytes, fresh frozen thin myocardial sections were stained with a monoclonal anti-laminin antibody (Sigma, Saint Louis Missouri) at a concentration of 1:50. The secondary antibody was Texas red streptavidin-conjugate goat anti-mouse antibody at a concentration of 1:200. Nuclei were stained with Hoechst dye. Quantitative indirect immunofluorescence was used to calculate total area occupied by the myocytes. The number of myocytes was counted in each field and the mean CSA of myocytes per field was determined. The mean myocyte CSA was determined in 5 fields per section and 8 sections (average 40 fields) per rabbits. Myocyte CSA was calculated in 5000 to 6000 myocyte per each age group. Experiments were performed without knowledge of the group assignment.
2.8. Statistical analysis
Continuous variables were expressed as mean ± SD. Differences among the groups were compared by analysis of variance (equal variance) and by Kruskal–Wallis test (unequal variance). Differences at the baseline and follow up in each group were compared by paired t-test. Correlation between histological phenotype and echocardiographic phenotypes were analyzed by Pearson correlation.
3. Results
3.1. Cardiac phenotypes in the β-MyHC-Q403 transgenic rabbits
Cardiac phenotypes were compared between 47 transgenic (all from 2 lines with similar expression levels of the transgene protein and indistinguishable phenotype between lines [13]) and 47 age- and gender-matched non-transgenic rabbits. As shown previously, albeit in a smaller number of rabbits [13,19], the β-MyHC-Q403 transgenic rabbits exhibited cardiac and myocyte hypertrophy, myocyte disarray, interstitial fibrosis, myocardial dysfunction but preserved global systolic function and higher left ventricular filling pressure, as detected by Doppler and TDI indices. Figure 1 shows examples of echocardiographic, gross pathological, and histological phenotypes in a 4-year old β-MyHC-Q403 transgenic rabbit. Individual data points according to age for selective phenotypes, namely, septal thickness, left ventricular mass, septal contraction velocity (Sa), septal relaxation velocity (Ea), E/Ea (a Doppler index of left ventricular filling pressure [22], myocyte CSA, myocyte disarray and CVF in transgenic and non-transgenic rabbits are shown in Figure 2.
Fig. 1.
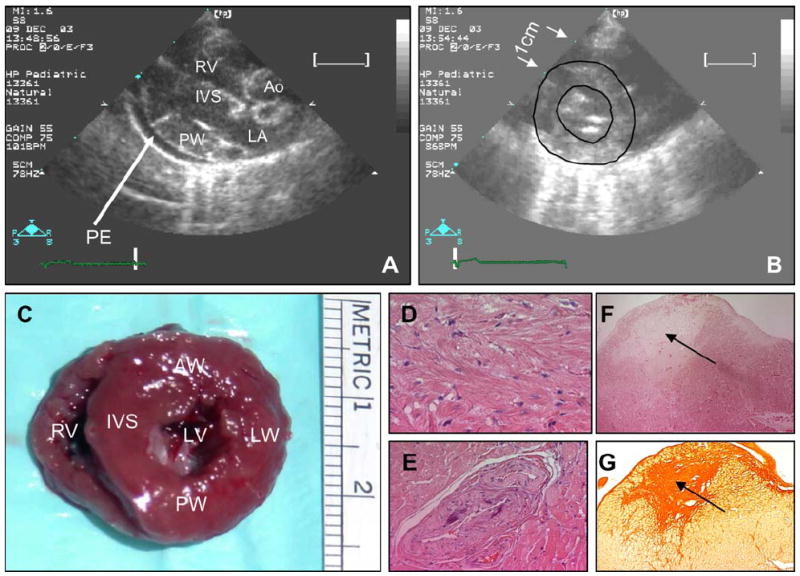
Cardiac phenotypes in a 4-year old transgenic rabbit. Panel A shows an end-diastolic parasternal long-axis view of the right ventricle (RV), interventricular septum (IVS), posterior wall (PW), proximal ascending aorta (Ao), left atria (LA) and pericardial effusion (PE). Severe hypertrophy of IVS and PW (5-6 mm) and a moderate to large size pericardial effusion are notable. Panel B shows short-axis view of the left ventricle at papillary muscle level (endocardial and epicardial boundaries are outlined); Panel C shows gross pathological cross-section of the heart at the level of papillary muscle, exhibiting significant concentric hypertrophy (arrows mark 1 cm distance). Panel D show H&E staining of thin myocardial sections, depicting myocyte disarray; Panel E shows hyperplasia/hypertrophy of media of a small intramural coronary artery, also observed in human HCM. Panels F shows an area of myocardial necrosis at the tip of a papillary muscle (indicated by an arrow) and Panel G shows Sirius red stained thin myocardial section showing severe replacement fibrosis in the area corresponding to myocardial infarction on Panel F.
Fig. 2.
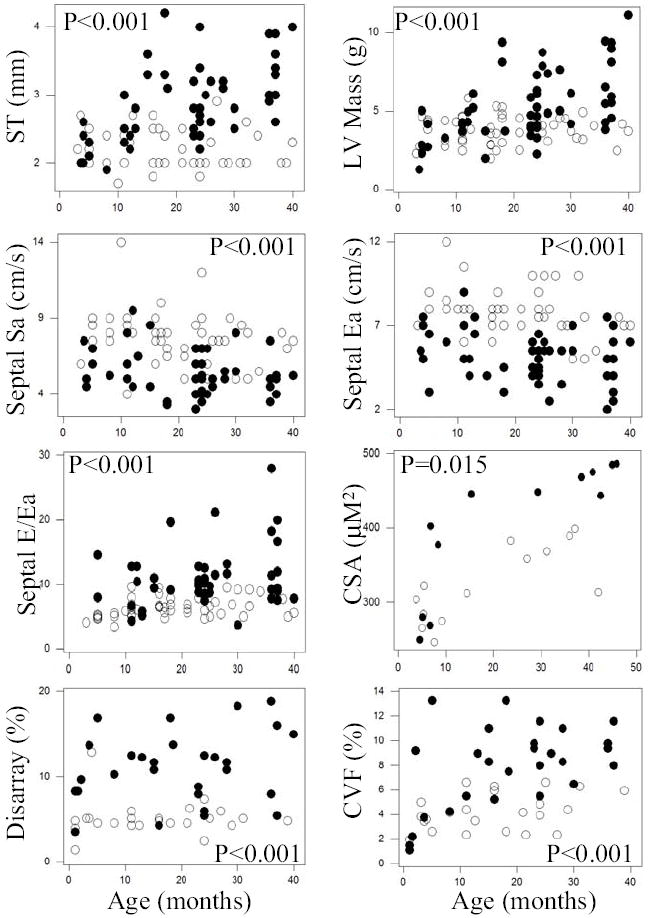
Differences in selected cardiac phenotypes in the entire groups of transgenic (full circles) and non-transgenic (open circles) rabbits. Individual data points for septal thickness (ST), left ventricular mass (LV Mass), septal contraction (Septal Sa) and relaxation (septal Ea) velocities, ratio of mitral inflow E velocity to septal relaxation velocity (Septal E/Ea, an index of left ventricular filling pressure), myocyte cross sectional area (CSA), myocyte disarray and collagen volume fraction (CVF) are shown and compared. The p values reflect comparisons of the mean and SD of the two groups.
To detect potential gender-dependent differences in the phenotypic expression of β-MyHC-Q403, echocardiographic and histological phenotypes were compared between males and female rabbits. Overall, there were no significant differences in the phenotypic expression between male and female transgenic rabbits with the exception of CVF, which was greater in females (10.40 ± 2.05% vs. 6.64 ± 2.05, P=0.001).
3.2. Ca+2 sensitivity of myofibrillar ATPase activity
Myofibrillar protein extracts were pure and of excellent quality (Figure 3). Myofibrillar ATPase activity in different concentrations of Ca+2 in transgenic and non-transgenic rabbits in each age group are shown in Figure 3. ATPase activity of myofibrillar proteins isolated from the hearts of mutant transgenic rabbits were less sensitive to Ca+2 concentrations in all four age groups as compared to that in the corresponding non-transgenic rabbits.
Fig. 3.
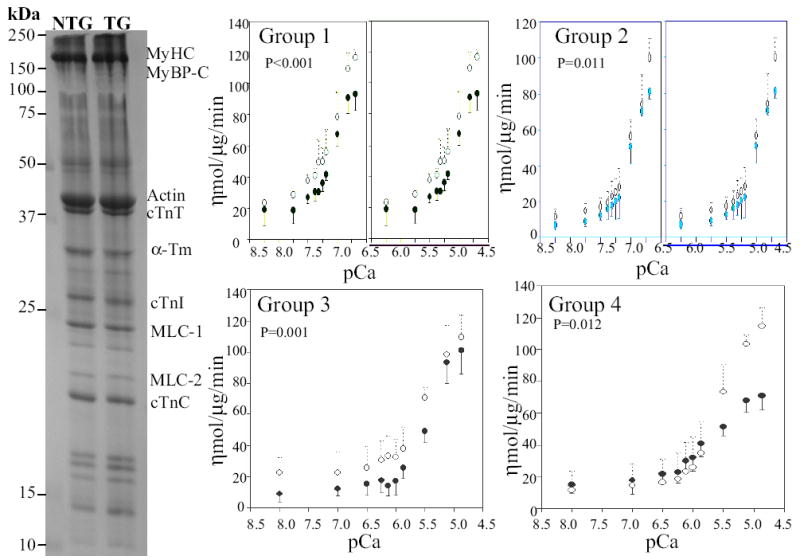
Isolated myofibrillar proteins and Ca+2 sensitivity of myofibrillar ATPase activity. The blot shows myofibrillar protein extracts following polyacrylamide gel electrophoresis and staining with Coomassie blue. Molecule size is indicated and major myofibrillar proteins are identified. pCa+2-ATPase activity curves in transgenic (solid circles) and non-transgenic (open circles) in each age group are depicted. As shown, myofibrillar ATPase activity at each Ca+2 concentration was lower in transgenic rabbits in all four age groups as compared to non-transgenics.
3.3. Activation of selected signaling molecules
The study was restricted primarily to analysis of expression of MAPK pathway, a ubiquitous pathway commonly involved in a variety of cardiovascular pathology [23]. Expression levels of activated ERK1/2 were not significantly different in transgenic and non-transgenic rabbits in the first 6 months of life. However, it was increased by approximately 2-fold in the heart of adult (> 6 months old) mutant transgenic rabbits as compared to non-transgenics (Figure 4). Other signaling kinases that were studied were p38 and JNKs and GTP-bound Ras, -Rac and -RhoA that were not significantly different between transgenic and non-transgenic rabbits.
Fig. 4.
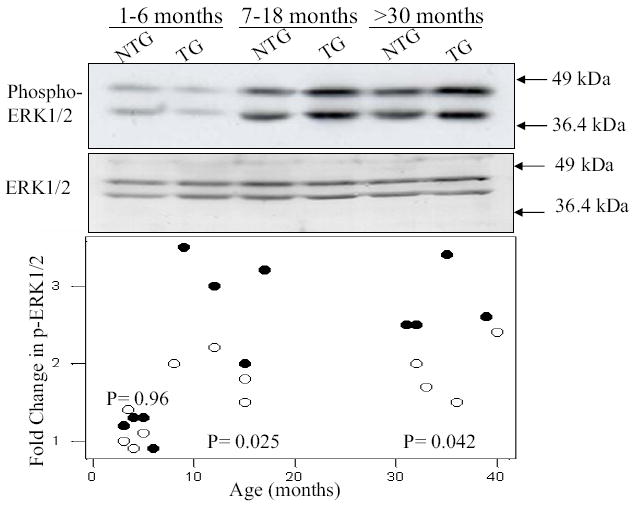
Immunoblots and relative expression levels of ERK1/2 in age-matched transgenic and non-transgenic rabbits. Blots show phosphorylated (upper blot) and total ERK1/2 (lower blot) in three age groups in non-transgenic (NTG) and transgenic (TG) rabbits. The lower panel depicts bar graphs showing relative expression levels of ERK1.2 in non-transgenic (n=4 per age group) and transgenic rabbits (n=4 per age group) at different age groups. The p values reflect differences in the relative expression levels of phosphorylated ERK1/2 in transgenic rabbits as compared to corresponding non-transgenic groups.
3.4. Echocardiographic phenotypes in transgenic and non-transgenic rabbits at different age groups
Baseline characteristics and echocardiographic indices of cardiac hypertrophy and function at different age groups in transgenic and non-transgenic rabbits are shown in Table 1. There was no significant cardiac hypertrophy prior to adulthood, while mean septal thickness, posterior wall thickness and left ventricular mass were increased in adult transgenic rabbits (7-18 months old and older). Myocardial systolic contraction (Sa) and relaxation velocities (Ea) at the septal corner of mitral annulus, as detected by TDI, were significantly reduced early in life and remained low throughout. Indices of global systolic function, such as fractional shortening were similar between 1-6 and 7-18 months old transgenic and non-transgenic rabbits but were reduced significantly in older (19-30 and > 30 months old) transgenic rabbits. The E/Ea ratios at the lateral corner of mitral annulus were consistent with higher filling pressures in transgenic rabbits in all age groups. Transgenic and non-transgenic rabbits exhibited digressing patterns of isovolumic relaxation time (IVRT) with aging. IVRT was longer in 1-6 months old transgenic rabbits as compared to non-transgenics. However, IVRT gradually shortened because of gradual increase in left ventricular filling pressure in transgenic rabbits. In contrast, in non-transgenic rabbits IVRT gradually prolonged from 35 msec in 1-6 months rabbits to 47 msec in > 30 months rabbits (P=0.003).
Table 1.
TEMPORAL EVOLUTION OF ECHOCARDIOGRAPHIC PHENOTYPE IN β-MyHC-Q403 TRANSGENIC RABBITS
| Age Groups (Months) | |||||
|---|---|---|---|---|---|
| 1-6 | 7-18 | 19 – 30 | >30 | p* | |
| N | 10 | 12 | 15 | 10 | |
| Age (months) | |||||
| Non-Tg | 3.3 ± 1.8 | 13.6 ± 3.5 | 24.5 ± 2.7 | 33.1 ± 2.4 | <0.001 |
| β-MyHC-Q403 | 3.3 ± 1.4 | 13.1 ± 3.0 | 24.2 ± 2.0 | 35.1 ± 3.1 | <0.001 |
| p** | 0.93 | 0.62 | 0.72 | 0.15 | |
| Gender (M/F) | |||||
| Non-Tg | 5/5 | 6/6 | 9/6 | 6/4 | 0.93 |
| β-MyHC-Q403 | 6/4 | 7/5 | 9/6 | 7/3 | 0.94 |
| p** | 0.65 | 0.68 | 1.0 | 0.64 | |
| Body Weight (Kg) | |||||
| Non-Tg | 2.1 ± 1.6 | 3.5 ± 0.5 | 3.7 ± 0.4 | 3.4 ± 0.3 | <0.001 |
| β-MyHC-Q403 | 2.7 ± 0.6 | 3.8 ± 0.5 | 3.8 ± 0.4 | 3.8 ± 0.4 | <0.001 |
| p** | 0.35 | 0.13 | 0.89 | 0.16 | |
| Heart rate (bpm) | |||||
| Non-Tg | 161.5 ± 26.1 | 146.2 ± 18.0 | 143.8 ± 17.3 | 143.0 ± 7.1 | 0.23 |
| β-MyHC-Q403 | 170.6 ± 27.9 | 151.7 ± 22.1 | 144.4 ± 16.0 | 142.6 ± 15.6 | 0.007 |
| p** | 0.53 | 0.46 | 0.92 | 0.94 | |
| IVST (mm) | |||||
| Non-Tg | 2.4 ± 0.3 | 2.2± 0.3 | 2.1 ± 0.3 | 2.5 ± 0.1 | 0.11 |
| β-MyHC-Q403 | 2.3 ± 0.2 | 2.8 ± 0.7 | 2.9 ± 0.4 | 3.2 ± 0.5 | 0.001 |
| p** | 0.77 | 0.009 | <0.001 | <0.001 | |
| PWT (mm) | |||||
| Non-Tg | 2.1 ± 0.3 | 2.2 ± 0.3 | 2.2 ± 0.3 | 2.3 ± 0.4 | 0.48 |
| β-MyHC-Q403 | 2.2 ± 0.4 | 2.8 ± 0.7 | 3.0 ± 0.5 | 3.0 ± 0.7 | 0.004 |
| p** | 0.52 | 0.009 | <0.001 | 0.015 | |
| LVEDD (mm) | |||||
| Non-Tg | 13.8 ± 2.6 | 13.9 ± 1.9 | 14.9 ± 1.7 | 13.9 ± 1.4 | 0.51 |
| β-MyHC-Q403 | 12.3 ± 2.5 | 13.8 ± 3.0 | 13.8 ± 2.6 | 15.2 ± 1.9 | 0.06 |
| p** | 0.29 | 0.95 | 0.14 | 0.07 | |
| LVESD (mm) | |||||
| Non-Tg | 8.0 ± 1.7 | 8.8 ± 1.8 | 9.0 ± 1.3 | 8.9 ± 1.6 | 0.69 |
| β-MyHC-Q403 | 7.7 ± 2.2 | 9.3 ± 2.6 | 9.5 ± 2.0 | 10.6 ± 1.8 | 0.024 |
| p** | 0.80 | 0.54 | 0.38 | 0.038 | |
| LV Mass (g) | |||||
| Non-Tg | 3.4 ± 0.9 | 3.6 ± 1.0 | 4.0 ± 0.6 | 3.8 ± 0.8 | 0.81 |
| β-MyHC-Q403 | 3.2 ± 1.2 | 5.0 ± 1.9 | 5.3 ± 1.8 | 6.9 ± 2.3 | 0.001 |
| p** | 0.42 | 0.069 | 0.024 | 0.002 | |
| FS (%) | |||||
| Non-Tg | 37.8 ± 9.1 | 36.9 ± 6.5 | 38.6 ± 5.9 | 36.3 ± 7.3 | 0.84 |
| β-MyHC-Q403 | 37.1 ± 11.8 | 33.4 ± 7.2 | 31.7 ± 3.9 | 31.0 ± 5.3 | 0.16 |
| p** | 0.89 | 0.17 | <0.001 | 0.07 | |
| Septal Sa (cm/s) | |||||
| Non-Tg | 7.7 ± 1.2 | 8.1 ± 1.7 | 7.8 ± 1.9 | 7.1 ± 1.3 | 0.33 |
| β-MyHC-Q403 | 5.6 ± 1.1 | 5.9 ± 2.0 | 5.1 ± 1.2 | 5.2 ± 1.2 | 0.37 |
| p** | 0.012 | 0.005 | <0.001 | 0.009 | |
| Septal Ea (cm/s) | |||||
| Non-Tg | 8.2 ± 0.8 | 7.9 ± 1.4 | 8.3 ± 1.1 | 7.1 ± 1.6 | 0.11 |
| β-MyHC-Q403 | 5.8 ± 1.5 | 5.4 ± 1.8 | 4.8 ± 1.0 | 5.0 ± 1.7 | 0.36 |
| p** | 0.004 | 0.001 | <0.001 | 0.017 | |
| Lateral Sa (cm/s) | |||||
| Non-Tg | 9.7 ± 2.7 | 10.6 ± 2.0 | 9.2 ± 1.3 | 8.8 ± 1.2 | 0.049 |
| β-MyHC-Q403 | 7.5 ± 1.0 | 7.3 ± 2.0 | 7.0 ± 2.2 | 6.3 ± 1.5 | 0.43 |
| p** | 0.11 | <0.001 | <0.001 | 0.001 | |
| Lateral Ea (cm/s) | |||||
| Non-Tg | 8.8 ± 2.2 | 10.1 ± 1.7 | 10.3 ± 1.4 | 8.4 ± 1.2 | 0.026 |
| β-MyHC-Q403 | 7.6 ± 2.1 | 7.3 ± 1.4 | 6.7 ± 1.8 | 6.3 ± 1.3 | 0.21 |
| p** | 0.30 | <0.001 | <0.001 | 0.003 | |
| Mitral inflow E/A ratio | |||||
| Non-Tg | 1.6 ± 0.4 | 1.8 ± 0.6 | 1.9 ± 0.5 | 1.6 ± 0.3 | 0.35 |
| β-MyHC-Q403 | 1.5 ± 0.2 | 2.0 ± 1.3 | 1.8 ± 0.5 | 2.2 ± 0.7 | 0.26 |
| p** | 0.54 | 0.63 | 0.67 | 0.005 | |
| IVRT (ms) | |||||
| Non-Tg | 34.8 ± 3.9 | 42.2 ± 5.7 | 41.9 ± 3.8 | 47.0 ± 7.5 | 0.003 |
| β-MyHC-Q403 | 41.9 ± 3.7 | 42.1 ± 8.5 | 42.8 ± 8.1 | 39.9 ± 5.7 | 0.72 |
| p** | 0.008 | 0.96 | 0.65 | 0.056 | |
| Septal E/Ea | |||||
| Non-Tg | 4.8 ± 0.4 | 6.5 ± 1.4 | 6.5 ± 1.3 | 6.9 ± 1.8 | 0.048 |
| β-MyHC-Q403 | 11.3 ± 5 | 9.8 ± 4.4 | 10.9 ± 2.9 | 12.4 ± 6.6 | 0.58 |
| p** | 0.30 | 0.034 | <0.001 | 0.017 | |
| Lateral E/Ea | |||||
| Non-Tg | 4.5 ± 0.7 | 5.0 ± 0.9 | 5.3 ± 1.4 | 5.7 ± 1.5 | 0.25 |
| β-MyHC-Q403 | 7.4 ± 3.1 | 6.8 ± 2.1 | 8.2 ± 2.8 | 8.7 ± 2.9 | 0.3 |
| p** | 0.03 | 0.016 | <0.001 | 0.005 | |
p* values (on the right column) are derived comparing variables among the four groups by ANOVA or Kruskal-Wallis test and p** reflects comparisons of transgenic and non-transgenics ar each time point t test or Kruskal-Wallis test.
Abbreviations: Non-Tg: Non-transgenic; MyHC: Myosin heavy chain; Q: Glutamine; M/F: Male/Female; bpm: beat per minute; LV mass: left ventricular mass; IVST: Interventricular septal thickness; PWT: Posterior wall thickness; LVEDD: Left ventricular end diastolic dimension; FS: Fractional shortening; Sa: Systolic velocity; Ea: Early diastolic velocity; IVRT: Isovolumic relaxation time.
3.5. Histological phenotypes in transgenic and non-transgenic rabbits at different age groups
Myocyte CSA, CVF, and extent of myocyte disarray at different age groups are shown in Table 2 and representative examples are presented in Figure 5. Myocyte CSA was not significantly different between 1-6 months old transgenic and non-transgenic rabbits. In contrast, myocyte CSA was increased in 7-18 months old and older rabbits as compared to corresponding non-transgenic rabbits. Myocyte disarray was increased by approximately 3-fold in 1-6 months old transgenic rabbits, indicating that myocyte disarray occurred early and prior to left ventricular hypertrophy and fibrosis. Extent of myocyte disarray remained unchanged over time and comprised approximately 10-15% of the myocardium. In contrast, interstitial fibrosis showed an age-dependent penetrance. It was 2- to 3-fold higher in transgenic rabbits older than 6 months of age as compared to corresponding non-transgenic rabbits. In both transgenic and non-transgenic rabbits CVF was increased with aging, however, the change was 3-fold greater in the mutant transgenic group.
Table 2.
TEMPORAL EVOLUTION OF HISTOLOGICAL PHENOTYPES IN β-MyHC-Q403 TRANSGENIC RABBITS
| Age Groups (Months) | |||||
|---|---|---|---|---|---|
| 1-6 | 7-18 | 19-30 | >30 | p | |
| N | 7 | 9 | 9 | 5 | |
| Myocyte Disarray (%) | |||||
| Non-Tg | 4.2 ± 1.4 | 4.8 ± 0.6 | 5.1 ± 1.6 | 5.0 ± 0.2 | 0.52 |
| β-MyHC-Q403 | 10.5 ± 4.4 | 12.4 ± 2.3 | 11.9 ± 3.0 | 12.6 ± 5.2 | 0.53 |
| P | 0.009 | 0.001 | 0.001 | 0.01 | |
| CVF (%) | |||||
| Non-Tg | 2.8 ± 1.5 | 4.6 ± 1.6 | 4.2 ± 1.3 | 6.1 ± 0.3 | 0.03 |
| β-MyHC-Q403 | 5.0 ± 4.6 | 9.6 ± 3.4 | 10.2 ± 1.8 | 11.1 ± 1.9 | 0.02 |
| P | 0.41 | 0.025 | <0.001 | 0.025 | |
| Myocyte CSA (μm2)* | |||||
| Non-Tg | 278 ± 20 | 290 ± 32 | 369 ± 11 | 365 ± 46 | 0.009 |
| β-MyHC-Q403 | 248 ± 12 | 405 ± 34 | 482 ± 12 | 475 ± 22 | <0.001 |
| p | 0.10 | 0.009 | 0.008 | 0.034 | |
p values on the right column are derived comparing variables among the four groups by ANOVA or Kruskal-Wallis test and those at each column by t test or Kruskal-Wallis test.
Abbreviations: as in Table 1; CVF: Collagen volume fraction; CSA: Cross sectional area
N = 3-4 rabbits per group
Fig. 5.
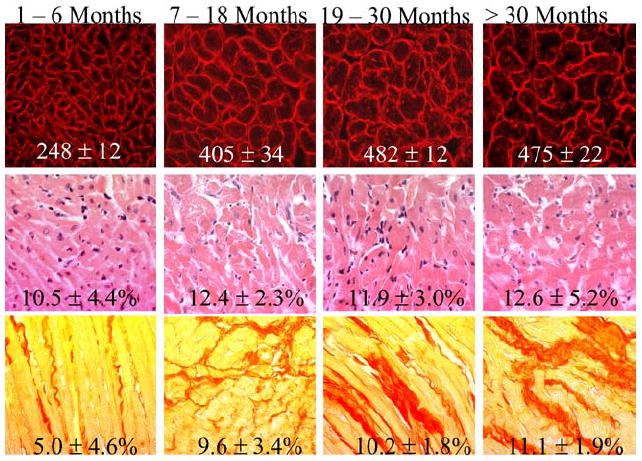
Representative examples of histological phenotypes at four age groups in the cross-sectional study. The upper panels show immunofluorescent thin myocardial sections stained with anti-laminin antibody defining myocyte cross sectional area. Mean values and SD of myocyte cross sectional area of over 5,000 myocytes per each age group is depicted at the bottom of each figure. The middle panels show H&E stained myocardial sections showing myocyte disarray. Percent of myocardium showing myocyte disarray (mean values and SD) was calculated in 4,000 to 6,000 high magnification (x400) fields per each age group and is depicted. The lower panels show examples of picrosirius red stained myocardial sections used to calculate percent of myocardium stained positive for collagen. Mean values and SD, calculated in 10,000 to 15,000 high magnification (x400) fields per each age group, are depicted.
3.6. Lack of an association between histological phenotypes and reduced myocardial contraction and relaxation velocities
To determine whether histological and morphological phenotypes were responsible for the observed myocardial dysfunction, we analyzed correlation between the extent of myocyte disarray, interstitial fibrosis, septal thickness and left ventricular mass with septal Sa, lateral Sa, septal Ea and lateral Ea. There was no significant correlation between reduced myocardial contraction and relaxation velocities and the extent of myocyte disarray, CVF, and left ventricular mass in transgenic rabbits. Development of hypertrophy was associated with further reduction of septal Sa (F=5.8, P=0.02) and septal thickness accounted for 8.2% of variance of septal Sa. Notably, septal Sa was also reduced in 1-6 months old transgenic rabbits despite a normal septal thickness and there was no correlation between septal or posterior wall thickness or left ventricular mass and reduced myocardial contraction and relaxation velocities in this group (correlation between septal thickness and septal Sa: n=10, R2= 1.4%, F= 1.2, P=0.37). Similarly, reduced myocardial contraction and relaxation velocities did not correlate with histological indices in 1-6 months old transgenic rabbits.
3.7. Progression of cardiac hypertrophy and deterioration of global systolic dysfunction in rabbits undergone serial echocardiography
To determine the evolution of phenotypes in the same rabbits, we performed serial echocardiography and TDI at two time points of 18 and 30 months of age. The results, shown in Table 3, were remarkable for further increase in the interventricular septal thickness, posterior wall thickness, left ventricular mass and left atrial diameter during the follow up period, while fractional shortening, septal Sa and lateral Sa were reduced progressively with aging. In addition, Doppler indices of left ventricular filling pressure reflected a significantly higher pressure in the older rabbits (Table 3). Myocyte disarray and CVF were increased by 3-fold in 30-month old mutant transgenic compared to the non-transgenic rabbits. Likewise, myocyte CSA was increased by 30%. In the non-transgenic rabbits, there were significant interval changes in septal Sa and lateral Sa, which were reduced with aging as compared to the baseline values. Otherwise, there were no other significant changes.
Table 3.
SERIAL CHANGES IN ECHOCARDIOGRAPHIC AND HISTOLOGICAL PHENOTYPES IN β-MyHC-Q403 TRANSGENIC RABBITS
| Non-Transgenic Baseline | Follow up | β-MyHC-Q403 Baseline | Follow up | |
|---|---|---|---|---|
| N | 24 | 24 | 24 | 24 |
| Age (months) | 11.4 ± 4.2 | 24.7 ± 4.2 | 17.9 ± 7.1 | 30.9 ± 7.1 |
| Gender (M/F) | 13/11 | 13/11 | 17/7 | 17/7 |
| Body Weight (Kg) | 3.4 ± 0.3 | 3.8 ± 0.5 | 3.6 ± 0.3 | 3.9 ± 0.4 |
| Heart rate (bpm) | 148 ± 18 | 144 ± 14 | 145 ± 14 | 143 ± 16 |
| IVST (mm) | 2.2 ± 0.3 | 2.2 ± 0.3 | 2.8 ± 0.4 | 3.2 ± 0.5 a,b |
| PWT (mm) | 2.2 ± 0.3 | 2.3 ± 0.3 | 2.89 ± 0.4 | 3.0 ± 0.6b |
| LVEDD (mm) | 14.0 ± 1.8 | 14.5 ± 1.7 | 13.1 ± 2.8 | 15.4 ± 1.8 a |
| LVESD (mm) | 8.6 ± 1.7 | 9.1 ± 1.5 | 8.4 ± 2.3 | 10.9 ± 1.6 a |
| LV Mass (g) | 3.7 ± 0.9 | 3.9 ± 0.6 | 4.4 ± 1.3 | 6.8 ± 2.1 a,b |
| LA Diameter (mm) | 7 ± 0.6 | 7.2 ± 0.6 | 7.4 ± 1.6 | 8.8 ± 2.0 a,b |
| FS (%) | 37.8 ± 7.4 | 37.4 ± 6.3 | 36.0 ± 8.6 | 29.5 ± 3.9 a,b |
| Septal Sa (cm/s) | 8.2 ± 1.7 | 7.1 ± 1.3 c | 5.9 ± 1.6 | 4.8 ± 1.1 a,b |
| Septal Ea (cm/s) | 8.0 ± 1.4 | 7.7 ± 1.4 | 5.3 ± 1.5 | 4.7 ± 1.5b |
| Lateral Sa (cm/s) | 10.9 ± 2.0 | 9.0 ± 1.2 c | 7.6 ± 2.1 | 6.2 ± 1.6 a,b |
| Lateral Ea (cm/s) | 10.1 ± 1.5 | 9.8 ± 1.6 | 6.8 ± 1.5 | 6.5 ± 1.6b |
| Mitral inflow E Vel. (cm/s) | 48.0 ± 6.4 | 50.9 ± 8.9 | 45.7 ± 9.2 | 53.9 ± 7.7 a |
| Mitral inflow E/A ratio | 1.8 ± 0.6 | 1.8 ± 0.5 | 1.7 ± 0.3 | 2.4 ± 0.9 a |
| IVRT (ms) | 40.7 ± 6.6 | 43.3 ± 5.6 | 44.8 ± 7.9 | 39.2 ± 6.1 a |
| Septal E/Ea | 6.2 ± 1.5 | 6.8 ± 1.4 | 9.3 ± 2.8 | 12.8 ± 5.3 a,b |
| Lateral E/Ea | 4.9 ± 1.0 | 5.3 ± 12 | 7.2 ± 0.57 | 8.9 ± 0.57 a,b |
| Histological Phenotypes | ||||
| N | 6-10 | 5-10 | ||
| Myocyte Disarray (%) | NA | 4.4 ± 1.3 | NA | 10.6 ± 3.6d |
| CVF (%) | NA | 3.8 ± 1.7 | NA | 8.2 ± 3.6 |
| Myocyte CSA (μm2) | NA | 366 ± 30 | NA | 475 ± 22d |
The baseline values are as published in Patel et al. Circulation 2001.
p <0.05 vs. value obtained in first time point
p <0.05 vs. the corresponding value in non-transgenic littermates
p <0.01 vs. value obtained in first time point in non-transgenic rabbits
p <0.001
3.8. Early myocardial dysfunction predicts subsequent development of cardiac hypertrophy
Echocardiograms performed at 18 months old β-MyHC-Q403 rabbits showed normal wall thickness and left ventricular mass but reduced myocardial contraction and relaxation velocities in 9/24 rabbits. To determine whether reduced myocardial contraction and relaxation velocities predicted development of subsequent hypertrophy, we repeated echocardiography one year later. The results, summarized in Figure 6, were remarkable for the development of significant cardiac hypertrophy, as evidenced by an increase in interventricular septal thickness and left ventricular mass.
Fig. 6.
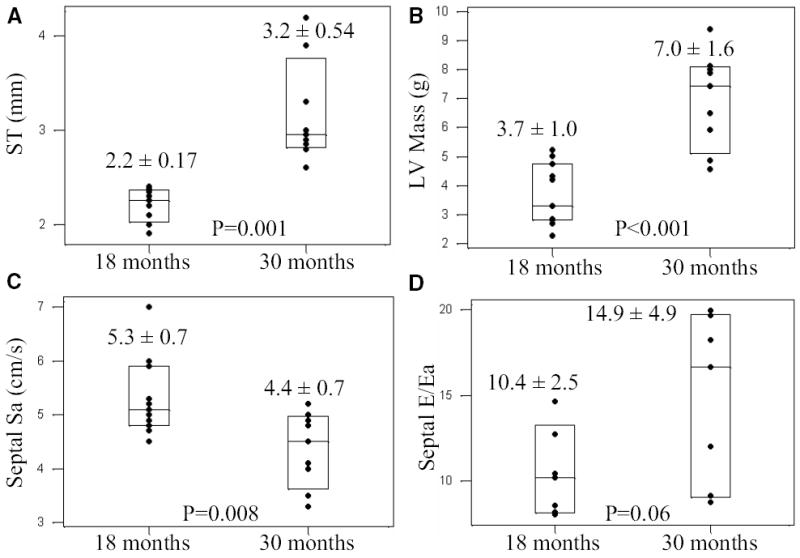
Subsequent development of cardiac hypertrophy in transgenic rabbits with reduced tissue Doppler velocity. Each box-plot shows individual data points, the mean value and the 95% confidence interval. Panels A and B represent septal thickness (ST) and left ventricular mass (LV Mass), respectively, in the same transgenic rabbits at two time points of 18 and 30 months. Panels C and D show septal contraction velocity (Sa) and a Doppler index of left ventricular filling pressure in the same rabbits at 18 and 30 months of age. All rabbits had reduced tissue Doppler velocities and no cardiac hypertrophy at 1 year of age (first box-plot in each panel). ST and LV mass were increased during the follow up period, septal Sa was further reduced and left ventricular filling pressure was mildly increased during the follow up period. The p values reflect paired comparisons of the mean and SD values between the two time points in the same rabbits.
4. Discussion
Several findings of the present study, performed in a large number of transgenic and non-transgenic rabbits over a 4-year period, provide insight into the pathogenesis of HCM and have potential diagnostic implications. The results showed reduced Ca+2 sensitivity of myofibrillar ATPase activity in vivo in an animal model with the cardiac background of β-MyHC, as in humans [11,12] that occurred early and prior to development of other phenotypes. This novel observation implicates impaired myocardial bioenergetics as a primary defect that that could contribute to the pathogenesis subsequent phenotypes [24]. In addition, myocyte disarray exhibited an age-independent penetrance with temporal dissociation from the evolution of hypertrophy and fibrosis. This finding suggests involvement of independent molecular mechanisms in the pathogenesis of disarray, hypertrophy and fibrosis. The finding of subsequent development of cardiac hypertrophy in mutant transgenic rabbits that initially exhibited only myocardial dysfunction, support the potential utility of TDI in early diagnosis of mutation carriers [16,25,26] and raises the possible utility of TDI in early interventions to prevent the evolving phenotypes. Moreover, age-dependent penetrance of cardiac and myocyte hypertrophy and interstitial fibrosis, absent early in life and developed at or after puberty, support the hypothesis that hypertrophy and fibrosis are secondary phenotype probably due to activation of trophic and mitotic factors and related cell signaling molecules in the heart. Finally, gradual deterioration of global ventricular systolic and diastolic functions with aging in conjunction with progressive increase in left ventricular filling pressure per Doppler indices are in accord with the natural evolution of HCM in humans [2]. Collectively, these findings suggest involvement of multiple independent mechanisms in the pathogenesis of diverse cardiac phenotypes in the β-MyHC-Q403 transgenic rabbits that exhibit a phenotype virtually identical to human HCM.
Since myocyte disarray and myocardial dysfunction were both expressed early, we analyzed whether disarray was responsible for reduced myocardial velocities. There was no correlation between two early phenotypes and myocardial contraction and relaxation were also impaired in the very young transgenic rabbits in the absence of myocyte disarray. Thus, it is unlikely that disarray, fibrosis and hypertrophy were responsible for the reduced myocardial contraction and relaxation velocities. As observed in human patients [26], myocardial contraction and relaxation velocities were further reduced upon development and progression of myocyte and cardiac hypertrophy. Accordingly, a modest association between reduced myocardial velocities and interventricular septal thickness was present in the entire group (R2= 8.2%). Our data does not elucidate the basis for myocyte disarray, however, it suggests that pathogenesis of disarray dissociates from that of hypertrophy and fibrosis. The cause for the disarray appears to be inherent in the genetic defect since it classically occurs with defects directly affecting sarcomeric proteins, but not with other forms of hypertrophy. It is plausible that altered regulation of molecules that regulate myocyte alignment, axis formation and cell-to-cell attachment by the primary defect is responsible for myocyte disarray.
The study was restricted to analysis of activation of MAPK pathway, as a representative signaling pathway involved in cardiac hypertrophy. Except for increased activation of ERK1/2 during the development of hypertrophy, no discernible changes in the expression levels of other signaling kinases were detected. The latter may reflect the relative insensitivity of the immunoblotting technique to detect modest differences in the levels of activated signaling molecules in the heart. Unlike cell culture experiments, in which the stimulus is acute, direct and potent, the stimulus for hypertrophy in the intact transgenic rabbit is indirect, chronic and likely to be less potent. Thus, the data should not be interpreted to suggest exclusion of other signaling molecules in the pathogenesis of HCM. Nonetheless, increased levels of activated ERK1/2, commonly activated in models of cardiac hypertrophy [23,27], depict ERK1/2 as potential therapeutic targets for modulating the hypertrophic response in HCM. In humans as in the transgenic rabbit model, there is the period prior to puberty when measures could be implemented to prevent, attenuate or delay the onset of high-risk phenotypes, such as hypertrophy and fibrosis. In this regard, TDI could prove to be a reliable method for early identification of the mutation carriers and initiation of preventive counseling and therapies. Further studies will be required to determine the utility of TDI for implementing preventive measures in human subjects.
Global ventricular function was initially normal despite decreased myocardial contraction and relaxation velocities. However, in older rabbits (19-30 and >30 months) left ventricular fractional shortening was reduced and left ventricular filling pressure, as determined by several Doppler parameters and tissue Doppler indices, was increased. The E/A ratio increased, and acceleration time, deceleration time and IVRT were shortened. These indices are considered reliable indicators of increased left ventricular filling pressure and chamber stiffness in patients and animals with systolic dysfunction [16,22,28,29]. The dimensionless E/Ea index was also increased providing further evidence of increased left ventricular filling pressure [22]. Furthermore, consistent with the above findings, left atrial end-systolic anteroposterior dimension was increased as previously described in humans [28] and murine models [30] of HCM.
In summary, we have characterized temporal evolution of cardiac phenotypes over a 4-year period in a large number of β-MyHC-Q403 transgenic rabbits. The findings showed reduced Ca+2 sensitivity of myofibrillar ATPase activity occurred early and independent of other phenotypes; dissociation of temporal evolution of disarray, hypertrophy and fibrosis; and support the potential utility of TDI in early detection of mutation carriers. Thus, the data suggests involvement of multiple independent mechanisms in the pathogenesis of HCM phenotypes in the β-MyHC-Q403 transgenic rabbits that fully recapitulate the phenotype of human HCM.
Acknowledgments
Supported by grants from the National Heart, Lung, and Blood Institute, Specialized Centers of Research P50-HL54313, RO1 HL68884, an Established Investigator Award (9640133N) from the AHA, National Center, Dallas, Texas and a TexGen grant from Greater Houston Community Foundation.
References
- 1.Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. 2000;102:858–64. doi: 10.1161/01.cir.102.8.858. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–20. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–70. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy--pathology and pathogenesis. Histopathology. 1995;26:493–500. doi: 10.1111/j.1365-2559.1995.tb00267.x. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. [PubMed] [Google Scholar]
- 6.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–85. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 7.Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275–9. doi: 10.1016/s0002-9149(01)01640-x. [DOI] [PubMed] [Google Scholar]
- 8.Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 9.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–80. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 10.Marian AJ, Roberts R. Molecular pathogenesis of cardiomyopathies. In: Sperelakis N, Kurachi Y, Terzic A, Cohen M, editors. Heart Physiology and Pathophysiology. San Diego: Academic Press; 2000. pp. 1045–63. [Google Scholar]
- 11.Schwartz K, Lecarpentier Y, Martin JL, Lompre AM, Mercadier JJ, Swynghedauw B. Myosin isoenzymic distribution correlates with speed of myocardial contraction. J Mol Cell Cardiol. 1981;13:1071–5. doi: 10.1016/0022-2828(81)90297-2. [DOI] [PubMed] [Google Scholar]
- 12.Alpert NR, Brosseau C, Federico A, Krenz M, Robbins J, Warshaw DM. Molecular mechanics of mouse cardiac myosin isoforms. Am J Physiol Heart Circ Physiol. 2002;283:H1446–54. doi: 10.1152/ajpheart.00274.2002. [DOI] [PubMed] [Google Scholar]
- 13.Marian AJ, Wu Y, Lim DS, McCluggage M, Youker K, Yu QT, Brugada R, DeMayo F, Quinones M, Roberts R. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest. 1999;104:1683–92. doi: 10.1172/JCI7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108–14. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 15.Marian AJ, Wu Y, Lim DS, McCluggage M, Youker K, Yu QT, Brugada R, DeMayo F, Quinones M, Roberts R. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest. 1999;104:1683–92. doi: 10.1172/JCI7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagueh SF, Kopelen HA, Lim DS, Zoghbi WA, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102:1346–50. doi: 10.1161/01.cir.102.12.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suckow MA, Douglas FA. The Laboratory Rabbit. Boca Raton, New York: CRC Press; 1997. [Google Scholar]
- 18.Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med. 1986;315:610–4. doi: 10.1056/NEJM198609043151003. [DOI] [PubMed] [Google Scholar]
- 19.Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, Marian AJ. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–24. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–62. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 21.Oh H, Taffet GE, Youker KA, Entman ML, Overbeek PA, Michael LH, Schneider MD. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci U S A. 2001;98:10308–13. doi: 10.1073/pnas.191169098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prunier F, Gaertner R, Louedec L, Michel JB, Mercadier JJ, Escoubet B. Doppler echocardiographic estimation of left ventricular end-diastolic pressure after MI in rats. Am J Physiol Heart Circ Physiol. 2002;283:H346–52. doi: 10.1152/ajpheart.01050.2001. [DOI] [PubMed] [Google Scholar]
- 23.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–81. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 24.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–82. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 25.Nagueh SF, Bachinski L, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with familial hypertrophic cardiomyopathy and provides a novel means for an early diagnosis prior to an independent of hypertrophy. Circulation. 2001;104:128–30. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagueh SF, McFalls J, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation. 2003;108:395–8. doi: 10.1161/01.CIR.0000084500.72232.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamazaki T, Komuro I, Yazaki Y. Signalling pathways for cardiac hypertrophy. Cell Signal. 1998;10:693–8. doi: 10.1016/s0898-6568(98)00036-9. [DOI] [PubMed] [Google Scholar]
- 28.Nagueh SF, Lakkis NM, Middleton KJ, Spencer WH, III, Zoghbi WA, Quinones MA. Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation. 1999;99:254–61. doi: 10.1161/01.cir.99.2.254. [DOI] [PubMed] [Google Scholar]
- 29.Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quinones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527–33. doi: 10.1016/s0735-1097(97)00344-6. [DOI] [PubMed] [Google Scholar]
- 30.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–4. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]