

Abstract
Glycosylation produces a diverse and abundant repertoire of glycans, which are collectively known as the glycome. Glycans are one of the four fundamental macromolecular components of all cells, and are highly regulated in the immune system. Their diversity reflects their multiple biological functions that encompass ligands for proteinaceous of receptors known as lectins. Since the discovery that selectins and their glycan ligands are important for the regulation of leukocyte trafficking, it has been shown that additional features of the vertebrate immune system are also controlled by endogenous cellular glycosylation. This Review focuses on the emerging immunological roles of the mammalian glycome.
Glycosylation is the enzymatic process that produces glycosidic linkages of saccharides to other saccharides, proteins and lipids, and is probably as ancient as life itself. Unicellular and multicellular organisms depend on glycosylation to produce monomeric and multimeric glycan linkages that are essential for cell viability and normal function1–4. The resulting glycome encompasses a diverse and abundant repertoire of glycans, which are one of the four fundamental macromolecular components of all cells (together with nucleic acids, proteins and lipids) (FIG. 1). Glycans have important biological functions in protein maturation and turnover, cell adhesion and trafficking, and receptor binding and activation5–8.
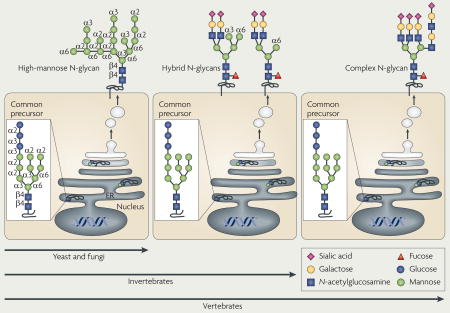
Figure 1. The types of mammalian glycan.

Glycan structures of the six classes of secretory glycan (N-glycans, hyaluronan, O-glycans, glycolipids, glycosylphosphatidylinositol (GPI) anchor and glycosaminoglycans) and the single intracellular glycan O-linked β-N-acetylglucosamine (O-GlcNAc) are shown. Representative examples of each type are indicated using the symbol nomenclature for monosaccharides (see key). Multiple and multi-antennary examples of the predominant mature, complex-type N-glycans are shown, including the high-mannose N-glycan that is found attached to some glycoproteins. Examples of core 1–4 O-glycans are depicted, as well as O-mannose, O-fucose and O-glucose structures. Core 5–7 O-glycans are not shown. The GPI anchor and examples of the glycosaminoglycans and glycolipids are also depicted. In all examples, glycan linkages are identified by the anomeric configuration (α orβ) of the donor saccharide following its linkage to the ring position (1–6) of the glycan acceptor. Et–P denotes a phosphoethanolamine linkage. NS, 2S, 4S and 6S denote the sulphation positions of the glycosaminoglycan chains. Asn, asparagine; Ser, serine; Thr, threonine.
Glycosylation is prominent in the lumen of the endoplasmic reticulum (ER) and in the Golgi apparatus. The cellular repertoire of glycans that are produced by glycosylation in these organelles of the secretory pathway reflects the combinatorial expression of subsets of glycosyltransferase and glycosidase enzymes, of which there are more than 200 in the mammalian genome. The formation and breakdown of glycans are regulated at several levels in the cell. One of the mechanisms involves transcriptional regulation of the genes that encode these enzymes, but others include access to substrates and molecular interactions that alter enzyme localization in the lumen of the ER and Golgi2,9,10. Changes in the glycome can occur in response to environmental and genetic stimuli, and are frequently associated with the acquisition of altered cellular phenotypes1–10.
Glycosylation also occurs among proteins in the cytoplasm and nucleus through the actions of the Ogt glycosyltransferase, which produces a reversible O-linked β-N-acetylglucosamine (O-GlcNAc) post-translational modification11. O-GlcNAc linkages are important in numerous physiological processes and disease. In contrast to this intracellular glycosidic bond that is formed by Ogt, secretory glycosylation in the ER and Golgi produces a large structural repertoire, including oligomeric glycan linkages that are presented at the cell surface and in extracellular compartments. Intracellular glycosylation has been reviewed elsewhere and is not discussed further in this Review12.
Glycosylation can substantially modify the structure and function of proteins by steric influences involving intermolecular and intramolecular interactions and can mediate the production of glycan ligands for lectins (FIG. 2). Lectins are proteins with glycan-binding activity that were first described in plants but subsequently have been found in all cells, from microorganisms to humans13–15. Lectin–glycan binding is a means of molecular recognition, which organisms can use to identify and decode the biological information that is present in their own cellular glycome as well as the glycomes of other organisms.
Figure 2. Basic mechanisms of glycan function in the immune system.

Glycans can function as ligands for lectins (a) and as steric elements that alter molecular interactions at the cell surface and between extracellular compartments (b). Interactions can involve the same cell (cis) or different cells (trans), and can be either homotypic or heterotypic, and intramolecular or intermolecular. It is often difficult to distinguish steric effects of glycans from alterations in lectin–glycan binding. This figure is idealized and not drawn to scale.
The extracellular positioning of the secretory glycome and its phylogenetic variation between organisms (BOX 1) allows glycans to be involved in pathogen–host interactions16. Glycan linkages of the mammalian host are an abundant and attractive target for pathogens to establish an infection through lectin binding. Mammals have developed a phyloglycomic recognition system, which includes a subset of Toll-like receptors (TLRs) and C-type lectins, to detect the glycans of lower organisms as a means of non-self discrimination and immunological activation17. These pattern-recognition receptors and their responses to non-vertebrate glycans are not considered further in this Review.
Box 1. The extracellular glycome is phylogenetically distinct.
The glycome, which is the repertoire of glycans that bear glycosidic linkages, is produced by the enzymatic process of glycosylation. The glycome of a cell at any given time reflects the cell’s ‘portfolio’ of functioning glycosyltransferases and glycan-modifying enzymes. The combinatorial and sequential function of glycosyltransferases and glycosidases in the secretory pathway contribute to the diversity of cellular glycans and provide a means for the regulation of glycan formation and turnover. In the lumen of the endoplasmic reticulum (ER) and Golgi apparatus, resident glycosyltransferases participate in the stepwise incorporation of saccharides into glycan chains and branches. As proteins and lipids transit through the secretory pathway, they are differentially modified by glycosyltransferases and glycosidases, which add and remodel glycan linkages, thereby promoting glycan diversification. Other enzymes, including sulphotransferases, can further modify these nascent glycan linkages. The resulting ‘mature’ glycans exist alone or attached to proteins and lipids, and are mainly transported to the cell surface and to extracellular compartments.
Glycan complexity has increased with evolution (see figure), such that there has been a decrease in the use of mannose linkages and the addition of new glycosidic bonds that bear different saccharides, including sialic acid. This is particularly evident when comparing the N-glycan biosynthetic pathway in lower eukaryotes (yeast and fungi), invertebrates and vertebrates. The increased complexity of the glycome through phylogeny has led to the formation of glycan-dependent epitopes that distinguish stem cells, different cell lineages and cellular activation states, and that can be diagnostic of inherited genetic diseases and prognostic in cancer174–176.
The development and function of the mammalian immune system are associated with restructuring of the cellular glycome. The immune-cell glycome is altered during cell differentiation, activation and apoptosis, and these alterations are being linked to both homeostatic and disease mechanisms of immune-cell functions, including immune-cell trafficking and differentiation, antigen- and cytokine-receptor activation, auto-immunity, and the induction of leukocyte apoptosis18–21. This Review discusses the roles of mammalian secretory glycosylation in the immune system as established by in vivo studies that combine knowledge of the glycan biosynthetic pathways with genetic and cellular research approaches.
Cell trafficking
Of the immunological functions that have been ascribed to mammalian glycosylation, perhaps the best understood involves the glycan ligands of the lectins E-selectin, L-selectin and P-selectin22,23. The selectins and their ligands form a cell–cell adhesion system that operates mainly between leukocytes and endothelial cells. This system is essential for the tethering and rolling of circulating leukocytes on the vascular cell wall, which promotes subsequent firm binding by the integrins and the transmigration of leukocytes through the endothelium and into the surrounding tissue24–26. This mechanism of cell trafficking is responsible for the colonization of lymphoid organs by lymphocytes during development and allows leukocytes to migrate into lymphoid organs or inflamed tissues during an immune response. Each selectin binds a particular glycan ligand, such as sialyl-Lewis-X (sLex) for E- and P-selectin, and 6-sulpho-sLex for L-selectin. The binding interactions between selectins and their ligands can occur between many different cell types, including between lymphoid and non-lymphoid, as well as non-immune, cells26,27.
Regulation of selectins and their ligands
Various cellular mechanisms regulate selectin function. Selectin expression, for example, is normally restricted to endothelial cells, platelets and leukocytes: E- and P-selectin expression is induced on the vascular endothelium during inflammation, P-selectin expression is also induced on platelets and L-selectin is expressed on the surface of leukocytes28. In addition, expression of the glycoproteins that ‘carry’ (that is, are scaffolds for the construction of ) the selectin ligands sLex and 6-sulpho-sLex can be similarly regulated, thereby altering the abundance and cellular localization of sLex and 6-sulpho-sLex. The glycoproteins that carry selectin ligands continue to be discovered and include P-selectin glycoprotein ligand 1 (PSGL1; also known as SELPG), E-selectin ligand 1 (ESL1), CD34, CD44, glycosylation-dependent cell-adhesion molecule 1 (GlyCAM1) and αMβ2-integrin 23,29,30. Another mechanism that controls selectin function involves the combinatorial enzymatic activities of different Golgi-resident glycosyltransferases and sulphotransferases that contribute to the synthesis of sLex and 6-sulpho-sLex. These enzymes are tightly regulated in different cell types and can therefore contribute to the diverse functions of selectins in the immune system.
The synthesis of selectin ligands requires a set of enzymes, including fucosyltransferases, sialyltransferases, galactosyltransferases, glucosaminyltransferases and sulphotransferases, that act in a specific order on an underlying glycan chain as it transits through the Golgi apparatus. The expression of these enzymes can be regulated in response to intracellular signalling and transcription-factor induction31–33. The sLex and 6-sulpho-sLex glycans exist in vivo mainly on the O-glycan chains of glycoproteins, and their production depends on the expression of the fucosyltransferases Fut4 and Fut7. The absence of Fut7 or Fut4, and in particular of both enzymes together, markedly attenuates lymphocyte homing to peripheral lymph nodes and myeloid-cell trafficking in inflammation34–36.
The α2,3-sialyltransferases ST3Gal4 and ST3Gal6 seem to participate in the formation of selectin ligands in vivo. Studies of mice that lack ST3Gal4 have revealed a partial deficiency in selectin ligands, with a marked effect on leukocyte homing and trafficking in inflammation, which was not observed in mice that lack ST3Gal1, ST3Gal2 or ST3Gal3 (REFS 37–39). The in vivo effect of ST3Gal6 deficiency has yet to be reported.
The contribution of O-glycans in carrying selectin ligands in vivo has been investigated in mice that lack core 2 N-acetylglucosaminyltransferase 1 (GCNT1) activity. GCNT1 is one of three glycosyltransferases that construct the core 2 O-glycan branch, which is a type of O-glycan structure that is commonly found on O-glycoproteins. GCNT1 deficiency strongly attenuated E- and P-selectin-mediated inflammation involving myeloid cells, but had relatively little effect on the lymphoid system, including the formation of secondary lymphoid organs and adaptive immune responses40. Subsequent studies of GCNT1-deficient mice detected a subtle but significant defect in the formation of L-selectin ligands in cells that form the high endothelial venules of some lymph nodes, which was associated with decreased kinetics of peripheral lymphocyte homing41,42. The finding that GCNT1 deficiency did not severely affect L-selectin ligands and lymphoid trafficking was addressed by the discovery of an endogenous β1,3-glucosaminyltransferase. In endothelial cells, this enzyme compensates for the lack of core 2 O-glycan branches and extends the core 1 O-glycan branch to provide a scaffold on which L-selectin ligands can also be produced43. Moreover, with loss of both this core-1-extension enzyme and GCNT1, L-selectin ligands were observed on N-glycan chains, which seem to contribute to a robust lymphocyte homing response in the absence of these two glycosyltransferases44. Compensation was also observed between the glycan sulphotransferases that are required for 6-sulpho-sLex synthesis, namely GlcNAc6ST1 and GlcNAc6ST2. In contrast to deficiency of a single enzyme, deficiency of both sulphotransferases led to almost complete loss of L-selectin-ligand formation on the high endothe-lial venules of lymph nodes, together with a marked decrease in lymphocyte homing and immune function45,46. The different defects of innate and adaptive immune responses that are associated with these profiles of selectin-ligand deficiency reflect the combinatorial and regulated expression of glycosyltransferases and glycan sulphotransferases that construct the E-, L- and P-selectin ligands. This modulation of ligand-expression profiles has progressed our understanding of the complexity of selectin function in the mammalian immune response.
Selectin-targeted therapy
Selectin-mediated binding promotes cell–cell adhesion that induces intracellular signalling and could therefore be exploited in therapeutic approaches. Once engaged by their cognate glycan ligands, the selectins recruit tyrosine kinases, such as FYN and spleen tyrosine kinase (SYK), to their cytoplasmic domains, thereby inducing intracellular tyrosine phosphorylation, activating mitogen-activated protein kinases and promoting integrin activation47–50. Pharmacological intervention that is focused on inhibiting the interactions between leukocytes and endothelial cells might therefore block the intracellular signalling pathways that are induced by selectin binding. In some disease contexts, selectin-mediated cellular trafficking might contribute to tumour-cell adhesion in metastasis and leukocyte infiltration in autoimmune disease. Indeed, blocking selectin binding or inhibiting the synthesis of selectin ligands can attenuate the pathogenesis of inflammatory diseases and decrease the tumorigenicity of cancer cells51–55. In other contexts, the induction of selectin ligands can be beneficial. Glycan remodelling of the mesenchymal stem-cell surface in vitro to express selectin ligands increased cell tropism for the bone marrow after transplantation and resulted in increased bone formation56. Pharmacological alteration of the production of selectin ligands and modulation of the binding of selectins in vivo are active areas of research and development that could ultimately lead to new treatments for the diseases of the immune system that are linked with leukocyte trafficking and activation.
Adaptive immunity and autoimmune disease
During their maturation, the antigen receptors of B and T cells are post-translationally modified with N- and O-glycan chains. The addition of different N-glycan linkages to the B-cell receptor (BCR) and T-cell receptor (TCR) by various glycosyltransferases in the Golgi apparatus seems to alter the molecular interactions at the cell surface that regulate receptor trafficking, association with other glycoproteins on the cell surface, signal transduction and receptor internalization by endocytosis. Moreover, glycans and endogenous multivalent lectins that bind to them might, in some cases, be engaged in a precise spatial and geometrically predictable array on the cell membrane (often termed a lattice) that could control glycoprotein function (see later).
TCR signalling
Glycosylation is involved in the cellular mechanisms that control the threshold of TCR activation, and can therefore have profound effects on the function of the adaptive immune system. The GnT5 glycosyltransferase is encoded by Mgat5 (mannoside acetylglucosaminyl transferase 5) and initiates formation of the β1,6 N-glycan-branch structure on various glycoproteins, including the TCR. This N-glycan branch commonly includes polylactosamine, which consists of N-acetyllactosamine disaccharide repeats and is a ligand for various lectins of the galectin family57. In studies of mice that lack GnT5 function, TCR clustering is significantly increased, which coincides with a decreased threshold of T-cell activation (FIG. 3a). GnT5-deficient mice have increased delayed-type hypersensitivity responses, increased susceptibility to experimental autoimmune encephalomyelitis (EAE) and increased glomerulonephritis that is associated with immune-complex deposition58. The phenotype that is conferred by GnT5 deficiency also includes a TCR trafficking abnormality, and the resulting hyperimmune response might be a contributing factor in some human autoimmune diseases59.
Figure 3. Glycans in T-cell-receptor activation.

a. Co-clustering of αβ T-cell receptors (TCRs) decreases in the presence of N-glycan branching on the TCR through lectin binding involving one or more of the galectins. Loss of GnT5 glycosyltransferase activity, which mediates N-glycan branching, and therefore lectin binding, increases the propensity for TCR and co-receptor co-clustering and decreases the threshold of TCR activation. b. Lectin binding might also control TCR signalling in cytotoxic T cells by inhibiting the association of the TCR with the CD8 co-receptor.
Endogenous lectins have been implicated in the formation of homotypic and heterotypic TCR interactions that modulate immunological signalling. Galectin-3 is chemically crosslinked with the TCR on naive resting T cells in a GnT5-dependent manner, and disruption of this interaction with glycan-ligand mimetics of galectin-3 also results in increased TCR clustering and a decreased threshold of activation58. By contrast, in post-activated mature cytotoxic T cells, a decrease in effector activity, such as occurs during anergy, can be reversed by ex vivo treatment with glycan-ligand mimetics of galectin-3. Decreased effector activity is associated with decreased co-localization of the TCR and CD8, and decreased binding of the T cells to HLA–peptide tetramers60 (FIG. 3b). Studies of galectin deficiency in mice also indicate that galectin-1 modulates the threshold of TCR signalling during thymocyte development61. Interestingly, treatment of cells or mice with high concentrations of GlcNAc increases GnT5-mediated N-glycan branching and inhibits TCR activation and autoimmune responses in mouse models of EAE and type 1 diabetes62. This is thought to be the result of a higher concentration of the cellular donor UDP (uridine 5′-diphosphate)–GlcNAc in the Golgi apparatus, which might increase the capacity of GnT5 and other closely related glycosyltransferases to produce GlcNAc linkages that contribute to N-glycan-branch formation.
BCR signalling
The BCR forms complexes with immunoregulatory glycoproteins that modulate the threshold of BCR activation, including the lectin CD22 (also known as Siglec-2), a member of the Siglec (sialic-acid-binding immunoglobulin-like lectin) family63 that is expressed exclusively on B cells. The structure of CD22 includes an extracellular lectin domain at the amino terminus of several immunoglobulin-like domains, and an intracellular immunoreceptor tyrosine-based inhibitory motif repeat, which has an inhibitory role as it recruits the phosphatase SHP1 (SRC-homology-2-domain-containing protein tyrosine phosphatase 1) and thereby dampens BCR signalling64. Synthesis of the glycan ligands for CD22 requires the ST6Gal1 sialyltransferase, which adds sialic acid in an α2,6 linkage to the underlying galactose residue of the N-acetyllactosamine disaccharide65. Although this glycan ligand for CD22 is present on many glycoproteins, in addition to CD22 itself, CD22 tends to bind to the glycan ligands of neighbouring CD22 molecules, thereby forming homotypic multimers 66. In the absence of ligands for CD22 owing to ST6Gal1 deficiency, BCR activation and humoral immunity are markedly decreased, and this coincides with increased co-localization of the BCR with CD22, constitutive recruitment of SHP1 to CD22, decreased protein phosphotyrosine signalling, increased BCR trafficking to clathrin-enriched microdomains and increased BCR endocytosis65,67,68. The absence of both CD22 and ST6Gal1 restored normal BCR signalling, microdomain association and rate of endocytosis. Measurements of intracellular signal transduction in response to BCR stimulation in the absence of the ST6Gal1-dependent ligands of CD22 show dominance of negative signalling that suppresses B-cell activation. This negative signalling complex prevents the development of autoimmune disease and markedly improves survival in LYN-tyrosine-kinase-deficient mice68. These findings are consistent with a model of BCR function in which Siglec-dependent binding of CD22 to the BCR dictates the threshold of immunological activation (FIG. 4).
Figure 4. Glycans in B-cell-receptor activation.

The threshold of B-cell receptor (BCR) activation is modulated by ST6Gal1 and CD22, a member of the Siglec (sialic-acid-binding immunoglobulin-like lectin) family that has a negative role in BCR signalling. CD22 Siglec binding to its glycan ligand promotes homotypic CD22–CD22 interactions, thereby decreasing the frequency of interaction of CD22 with the IgM BCR on mature naive B cells. Loss of CD22 Siglec ligands owing to ST6Gal1 sialyltransferase deficiency markedly increases the co-localization of CD22 with the BCR, thereby increasing the threshold of immune signalling and promoting endocytosis of the BCR in a CD22-dependent manner.
CD22 can bind ligands that are presented in trans and this might modulate glycoprotein complex assembly and its localization at the B-cell surface177. Moreover, the induced co-localization of CD22 with the BCR in cis seems to occur in a Siglec-independent manner that might involve protein–protein interactions that are mediated by the other extracellular (immunoglobulin-like) domains of CD22. Such a mechanism might explain how binding of dendritic cells to CD22 in a Siglec-independent manner inhibits B-cell proliferation69. The biology of CD22 is complex and there is potential for many Siglec-independent functions, some of which could be influenced by the Siglec-binding activity of CD22.
The glycan-dependent immune modulation that has been defined for ST6Gal1 in B cells and GnT5 in T cells indicates that crucial glycoprotein-selective functions can be modulated by single glycosyltransferase genes that contribute to the cellular mechanisms of antigen-receptor activation and the pathogenesis of autoimmune disease.
Antibody function
The secreted immunoglobulins that are produced following B-cell activation and immunoglobulin class switching are glycoproteins with characteristic glycan linkages70. The glycans of IgG are important for binding to Fc receptors and for other effector functions, including binding to complement component 1q (C1q) and complement activation71. Alterations of endogenous IgG glycosylation have been reported in various immune diseases, including rheumatoid arthritis and IgA nephropathy. In patients with rheumatoid arthritis, the N-glycans that are attached to IgG often lack terminal galactose and sialic-acid linkages and therefore bear exposed N-acetylglucosamine72. This terminal linkage is common to non-mammalian vertebrate species as well as various invertebrates and lower organisms, and is a ligand for some endogenous mammalian immune receptors and lectins that might promote inflammatory responses73,74. In patients with IgA nephropathy, the O-glycans that are attached to IgA are similarly truncated to expose N-acetylglucosamine, which correlates with antibody deposition in the inflamed kidney75. Deficiency of the β1,4GalT1 glycosyltransferase (one of several galactosyltransferases) recapitulates the pathology of IgA nephropathy in mice76. By contrast, the extent of IgG sialylation correlates with decreased inflammation following Fc receptor binding77. This seems to be due to the α2,6 sialic acids on IgG that are contributed by ST6Gal1 and are linked to the anti-inflammatory effects of intravenous IgG when used as a therapy for autoimmune diseases78.
Cytokine receptors
The core 1,6 fucose linkage is found on most N-glycans owing to the activity of Fut8 fucosyltransferase, and deficiency of this enzyme results in an emphysema-like disease in mice. Remarkably, this syndrome was linked to impaired binding of the receptor for transforming growth factor β1 (TGFβ1) to its ligand, and could be attenuated by increasing the concentration of TGFβ1 (REFS 79,80). An inherited or sporadic gene mutation that introduces new glycosylation sites into a protein can also produce receptors with ligand-binding defects81. In this study, an N-glycosylation site that was found at a new position in the protein sequence of the interferon-γ receptor 2 (IFNγR2) resulted in the formation of an N-glycan at this location that disrupted the ability of the receptor to bind its ligand. Such pathological changes to protein glycosylation represent another mechanism by which glycosylation can contribute to protein dysfunction and disease aetiology. It seems that glycosylation in the Golgi apparatus can in some cases directly affect the binding of immunomodulatory receptors to cognate ligand.
Innate immunity and autoimmune disease
Glycans might have been used to sense the presence of non-self since the beginning of multicellular life. Glycomic differences in phylogeny and between species can allow for this type of molecular discrimination between self and non-self, and thus glycan structures are activators of the innate immune system and pathogenic triggers for autoimmune diseases. Unlike new-world primates and other vertebrates, old-word primates lack the α1,3 galactosidase (α1,3Gal) galactose-terminated glycan branch linkage owing to a germline mutation of the α1,3 galactosyltransferase. This results in a substantial fraction of circulating immunoglobulins in old-world primates (including humans) that bind the α1,3Gal epitope and contribute to the hyperacute rejection of xenotransplants from other mammals82. Another emerging example concerns endogenous antibodies that are specific for the sialic-acid variant N-glycolylneuraminic acid (NeuGc). Humans seem to be unique in their lack of the enzyme that converts N-acetylneuraminic acid (NeuAc) to NeuGc, and the consumption of NeuGc in the diet could lead to its incorporation into endogenous glycoproteins, resulting in the formation of NeuGc-specific antibodies83. Antibodies specific for glycans are also induced during human Tn syndrome, which is a haematological stem-cell disorder that is associated with thrombocytopaenia and leukocytopaenia and is caused by a mutation of COSMC (also known as C1GALT1C1). This gene normally encodes a protein with chaperone function that is required for β1,3 galactosyltransferase activity in O-glycan biosynthesis84. Mutation of COSMC leads to a truncated O-glycan structure on haematopoietic cells and immunoglobulins (known as the Tn antigen) that functions as an epitope for antibody production and cell lysis and might contribute to IgA nephropathy and Henoch–Schönlein purpura84.
The aberrant synthesis of endogenous glycans can result in the exposure of cryptic epitopes that are perceived by the immune system as non-self and induce an inflammatory condition in the absence of infection that is linked to the pathogenesis of autoimmune disease. This explains how α-mannosidase-II (αM-II; also known as MAN2A1) deficiency causes an autoimmune syndrome in mice that is remarkably similar to systemic lupus erythematosus. Disease pathogenesis in αM-II deficiency is non-haematopoietic in origin (unlike other autoimmune disease models), is independent of both the adaptive immune system and C3 complement activation, can be mitigated by intravenous IgG administration and is linked to chronic activation of the innate immune system85. The block in N-glycan maturation in the Golgi that is caused by αM-II deficiency increases the expression of phylogenetically primitive hybrid N-glycans at the cell surface and among extracellular glycoproteins. The exposed mannose linkages of these primitive hybrid N-glycans on multiple glycoproteins are ligands for endogenous mannose-binding lectin receptors of the innate immune system, and this interaction results in the chronic induction of a pro-inflammatory response that can recruit and activate macrophages. The requirement for complex N-glycan branching to safeguard against this immunological recognition of cryptic mannose ligands reveals a distinct mechanism in the pathogenesis of autoimmune disease that can be termed epitope modification or neo-ligand formation. Mechanisms that impair N-glycan branching could therefore contribute to the pathogenesis of various autoimmune syndromes of presently unknown origin.
The mechanisms that establish and maintain immunological tolerance also involve lectin–glycan ligand interactions that suppress immune-cell activation. For example, galectin-1 is becoming increasingly important in the study of T-cell and dendritic-cell biology and in the pathogenesis of immunological diseases86. Galectins are involved in attenuating graft-versus-host disease, collagen-induced arthritis, type 1 diabetes and T-cell-mediated tumour rejection18,86–91. Galectin-1 seems to bridge innate and adaptive immunity in part by regulating the Fc receptor- and MHC-dependent immune functions of monocytes and macrophages through an extracellular-signal-regulated kinase (ERK) signalling pathway91. The immunomodulatory activity of galectin-1 extends to its involvement in fetal–maternal tolerance during pregnancy. Deficiency of galectin-1 increases the loss of fetuses that have been conceived by the mating of allogeneic mice. In addition, treatment of galectin-1-deficient mice with exogenous galectin-1 restores normal fecundity, and this coincides with the induction of tolerogenic dendritic cells and the proliferation of interleukin-10 (IL-10)-producing regulatory T cells92.
Normal and abnormal catabolism of glycans can also provide signals of immunological importance. Hyaluronan is a glycan that consists of a disaccharide repeat that modulates innate immunity by interacting with TLRs. This glycan is synthesized as a linear chain unattached to either a protein or a lipid and its size seems to be important. In response to tissue injury, endogenous hyaluronidases cleave extracellular hyaluronan into small fragments that induce inflammatory gene expression by binding TLR2 and TLR4 (REFS 93,94). Some pathogens express hyaluronidase as a virulence factor that may cleave hyaluronan into fragments that do not bind TLRs and thereby subvert the immune response. The enzymatic regulation of hyaluronan synthesis and degradation is increasingly recognized as having an important role in innate immunity and the response to infection95.
Immune-cell differentiation
Glycoproteins, such as antigen receptors and MHC molecules, are important regulators of immune-cell differentiation and must be properly folded to function. Without correct folding and maturation in the ER and Golgi apparatus, the expression of improperly folded and dysfunctional glycoproteins on the cell surface and in extracellular compartments can cause defects in the signalling pathways that promote immune-cell differentiation. In addition, acquisition of secondary determinants of the differentiated immune-cell phenotype can also be controlled by glycan-dependent mechanisms. For example, glycosaminoglycan sulphation by the sulphotransferase NDST2 (N-deacetylase/N-sulphotransferase (heparan glucosaminyl) 2) promotes the development of mast-cell secretory granules, which are essential for the function of these immune cells96,97.
T-cell ontogeny and endogenous glycans
The range and structures of peptides that are presented by MHC molecules could be influenced by glycan linkages on native glycoproteins and thus could contribute to the immunological definition of self98. Although peptides that are produced in the cell for presentation on MHC class I molecules are typically deglycosylated during proteolysis in the proteasome, some glycan linkages persist on various peptides and are recognized by cytotoxic T cells99. Peptides that arise from external sources can retain a broader range of glycan linkages, which might influence peptide proteolysis and loading on MHC class II molecules for binding by αβ TCRs. For example, some TCRs from T cells that have been isolated from mice with collagen-induced arthritis recognize an MHC class II-associated peptide of type II collagen that contains galactose linked to hydroxylysine residues of collagen101. Fate decisions in thymic T-cell development can be modulated by galectin-1, which promotes negative selection through ERK signalling100. Altered glycosylation in glycosyltransferase-deficient animals, for example, may influence the formation of the TCR repertoire and thereby have an effect in immunity. The repertoire of T cells in an intact mammal, which is determined by self-antigen presentation, might therefore reflect, in part, the portfolio of endogenous glycopeptides and their presentation by MHC molecules. This possibility could also involve intracellular O-GlcNAc glycan linkages that are attached to many cytoplasmic and nuclear proteins102. Similarly, glycolipids might be involved in development of the natural killer T (NKT)-cell lineage; the presentation of an endogenous glycolipid structure by the CD1 molecule might promote the development of a functional NKT-cell population103.
T-cell repertoire development
Expression of the glycoproteins CD4 and CD8 as TCR co-receptors confers MHC class II and class I restriction of TCR activation, respectively, during T-cell development in the thymus. The sialylation state of glycans of the CD8–α β TCR heterodimer affects co-receptor binding to MHC class I molecules. A low level of O-glycoprotein sialylation in CD4+CD8+ thymocytes is associated with binding to MHC class I tetramers, whereas the increased abundance of sialic-acid linkages on differentiated CD8+ thymic T cells decreases the binding avidity of CD8 for MHC class I molecules, thereby decreasing T-cell adhesion to MHC class I tetramers104–106. This decrease in binding to MHC class I molecules is in part mediated by the sialic acids that are produced by the ST3Gal1 sialyltransferase, as deficiency of this enzyme in mice increases the binding of MHC tetramers to single-positive CD8+ thymic T cells and alters the repertoire of the TCR β-chain variable region (Vβ)105. This mechanism seems to involve a change in the CD8–αβ TCR heterodimer conformation that promotes MHC class I binding in the presence of unsialylated core 1 O-glycans, which lack the steric hindrance and negative charge that are associated with sialic acid105,107. However, another study that used a different MHC–peptide complex showed no significant change in tetramer binding of ST3Gal1-deficient CD8+ thymic T cells108. The reasons for this discrepancy are not clear, but they might relate to the intrinsic binding affinities of the TCR transgenes and MHC tetramer probes that were used in each study. Nevertheless, the expression and deficiency of ST3Gal1 alter the TCR Vβ repertoire in an opposing manner, which indicates that ST3Gal1 has a role in the MHC-restricted thymic differentiation of CD8+ T cells105,109.
Notch ligands and immune-cell development
The binding of Notch ligands to Notch receptors provides another example of how glycosyltransferases can influence intracellular signal transduction and thereby govern the development and function of immune cells. Notch signalling, which controls cell-fate determination in many organisms, dictates lineage commitment in the mammalian lymphoid system and has recently been shown to be involved in myelopoiesis110. The closely related Fringe glycosyltransferases (Manic, Radical and Lunatic in the mammalian genome) were discovered as genes that establish cell-boundary fields in embryogenesis and were later found to intersect with the functions of Notch receptors in early development111–115. The Fringe glycosyltransferases are β1,3 N-acetylglucosaminyltransferases that operate on the O-linked fucose glycans. These glycans are found in the extracellular epidermal growth factor (EGF)-like repeats of Notch receptors in the secretory pathway and control cell-surface Notch signalling116,117. The O-fucose linkage that is the substrate of the Fringe glycosyltransferases is first generated by Pofut1 (protein O-fucosyltransferase). The glycan products of the Fringe glycosyltransferases are then substrates of β1,3 galacto-syltransferases. These glycans provide a physiological mechanism to modulate Notch binding and activation by the extracellular glycoprotein ligands Jagged/Serrate and Delta118–123. In addition to generating the O-fucose linkage that is the substrate of the Fringe glycosyltransferases, Pofut1 provides a chaperone activity that is independent of its glycosyltransferase activity and promotes Notch maturation124. More recently, Notch signalling has been shown to be also influenced by the Rumi glycosyltransferase, which generates O-glucose linkages on the EGF-like repeats of Notch125,126.
Signalling through the Notch axis has profound effects on lymphocyte development and activation in mammals. For example, a constitutively active form of Notch can promote the development of CD8+ T cells in the absence of MHC class I molecules127. The effect of Fringe glycosyltransferases on lymphoid-cell fate includes the suppression of B-cell development from lymphoid progenitors in the thymus128. However, Fringe activity promotes increased Notch binding to Delta-like ligands, which can lead to a block in T-cell development. This might be due to an effect on the access of progenitor cells to intrathymic niches or a direct effect on the induction of T-cell differentiation signals129,130. Moreover, Notch activation leads to the formation of marginal-zone B cells in the spleen and promotes immunoglobulin secretion by a signalling mechanism that involves Notch binding to the Delta-like 1 ligand131–133. Dendritic- and myeloid-cell differentiation also seem to be modulated by Notch signalling in response to the binding of Notch ligands, and the activation of NK-cell cytotoxicity is promoted by the Notch ligand Jagged2 (REFS 134,135). Immune responses can also be inhibited by Notch binding to Fringe-dependent glycan ligands; for example, Delta-like-1 ligand seems to inhibit Notch activation, which skews the outcome in airway inflammation from a T helper 2 (TH2)- to TH1-cell response136. The functional relationships between Notch receptors and their glycan ligands are only partly understood, but already a remarkable complexity in glycan-mediated and Notch-dependent immune-cell differentiation is indicated.
TH-cell polarization
Activated CD4+ effector T cells can differentiate into distinct subsets (TH1, TH2, TH17 and regulatory T cells), each of which produces a unique profile of cytokines that differentially govern immune responses to various stimuli. It seems that mammalian glycosylation has a role in the emergence and phenotypic profiles of these TH-cell subsets, in part by its effect on the production of ligands of various lectins. The Delta-like 1 and Delta-like 4 glycan ligands of Notch promote TH1-cell differentiation of CD4+ T cells in part by suppressing IL-4-mediated signalling, which would otherwise induce TH2-cell differentiation and promote TH2-cell immune responses137,138. By contrast, the GnT5 glycosyltransferase promotes TH2-cell immune responses by inducing differentiation signals that inhibit IFNγ production139. The induced deficiency of galectin-1 restores the TH1–TH2-cell balance of T-cell infiltrates in classical Hodgkin’s lymphoma, apparently by modulating the secretion of TH2-cell cytokines and the proliferation of regulatory T cells140. Differential glycosylation of TH-cell subsets has been linked with their susceptibility to death by galectin-induced apoptosis, which provides an explanation for the increased TH1- and TH17-cell responses that have been observed in galectin-1-deficient mice141. A related mechanism might operate during parasitic infection by which helminth glycans skew CD4+ T-cell immune responses towards the TH2-cell phenotype142.
Apoptosis
Cell death by apoptosis is an important mechanism of immune-system homeostasis during lymphoid development and in the clearance of effector cells after immune activation143. Mammalian glycosylation contributes to this process by generating ligands for endogenous lectins, including galectins57,144,145. These lectins bind to glycans that contain the disaccharide lactose and to polylactosamine, which are present in some cellular glycoproteins and glycolipids.
The T-cell lineage is sensitive to both the endogenous expression and exogenous administration of galectin, and most studies have examined the effects of galectin-1 and galectin-3. Exogenously administered galectin-1 was first reported to induce apoptosis in thymocytes, T-cell lines and activated T cells146. The in vitro methodology used was thought to contribute in part to this result, as a subsequent study reported that activated T cells do not normally undergo apoptosis in response to galectin-1, but become apoptotic after administration of galectin-3 (REFS 147,148). In multiple cell types, including T cells and neutrophils, binding of galectins -1, -2, -3, -4 or -8 to cellular glycans was shown to induce phosphatidyl-serine exposure on the outer leaflet of the plasma membrane, which is associated with cellular apoptosis with phagocytic uptake in vivo, and normal cell viability was retained in vitro147–150. Galectins bind to glycan ligands that are present on various glycoproteins, such as CD2, CD3, CD7 and CD45, thereby forming glycoprotein complexes that induce intracellular signalling cascades and inhibit IFNγ and IL-2 production and IL-2-mediated signalling148,151–154. Among the glycan ligands of the galectins, the polylactosamine moieties that are found in O-glycans are sometimes present on the core 2 branch. T-lymphoma cells that are deficient in core 2 O-glycans as a result of GCNT1 mutation or haploinsufficiency are resistant to galectin-1-induced apoptosis155. In addition, some galectins seem to oppose the functions of others. Galectin-3 binds different glycans and glycoproteins to those that are recognized by galectin-1, and galectin-3 can inhibit galectin-1- but not galectin-9-mediated apoptosis of some human T-cell lines. By contrast, galectin-9 induces T-cell apoptosis that can be inhibited by B-cell lymphoma 2 (BCL-2) (REFS 156–158).
The ST3Gal1 sialyltransferase seems to control the largest and most synchronous apoptotic response in adult mammals, that of eliminating the increase in cytotoxic CD8+ T cells that results from an immune response to infection by various microorganisms and viruses. More than 90% of the clonally expanded CD8+ T-cell population must be rapidly cleared by apoptosis during the resolution of the immune response so that the body is not overcome by lymphocytosis. ST3Gal1 is involved in this process by linking sialic acid in an α2,3 linkage to the core 1 O-glycan of one or more O-glycoproteins. Attenuation of ST3Gal1 function by a post-translational mechanism occurs during the resolution phase of the immune response, and the resulting unsialylated core 1 O-glycan structure is linked to the onset of post-activated CD8+ T-cell apoptosis by a caspase-dependent mechanism that seems to dominate the effects of the intracellular apoptotic modulators BCL-2 and BIM109,159. Remodelling of the glycome in activated CD8+ T cells by downmodulation of ST3Gal1 function induces a signalling pathway that ensures apoptotic cell death in the absence of TCR stimulation and of factors that induce memory-cell differentiation. The lectin (or lectins) that induces post-activated CD8+ T-cell apoptosis in vivo, and the glycoprotein counter-receptor (or receptors) that transmits the apoptotic signal following binding to the unsialylated core 1 O-glycan ligand, are yet to be identified.
Summary and future directions
A rapid expansion of our knowledge of the immunological roles of mammalian glycosylation has resulted from interdisciplinary studies that combine biochemical, enzymatic, structural and genetic techniques. Establishing the relationships between glycan linkages, the enzymes that produce them, and the glycoproteins that are modulated by them remains at the forefront of research (TABLE 1). Studies that bridge immunology and glycobiology, and which link the enzymes of glycosylation with lectin recognition, will continue to provide new insights into the immune system and will reveal new approaches to therapeutic intervention in disease. Studies of glycosylation by the ST3Gal4 sialyltransferase, for example, recently revealed that the Ashwell–Morell receptor of hepatocytes — the main lectin expressed by hepatocytes — mitigates the lethal coagulopathy of sepsis160, exposing an unexpected pathogen–host relationship that might reflect an ancient arm of the innate immune system.
Table 1.
Mammalian glycan structures that are linked to immune functions
| Glycan* | Enzymes‡ | Function | Recognition§ | |
|---|---|---|---|---|
| sLex | 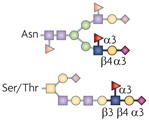 |
ST3Gal4, ST3Gal6, β1,4GalT1, Fut4 and Fut7 | Trafficking, adhesion and signalling | E- and P-selectin |
| 6-sulpho-sLex | 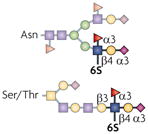 |
ST3Gal4, GlcNAc6ST1, GlcNAc6ST2, Fut4 and Fut7 | Lymphocyte homing | L-selectin |
| Core 1 O-glycan | ST3Gal1 | CD8+ T-cell apoptosis | Unknown | |
| Terminal GlcNAc | 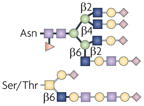 |
GnTs | IgA nephropathy and complement activation | Integrins |
| N-acetyl-lactosamine and polylactosamine | 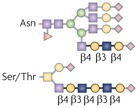 |
β1,4GalTs and β1,3GnTs | T-cell-mediated tumour rejection, fetal–maternal tolerance and DC function | Galectins |
| β1,6 N-glycan branch | 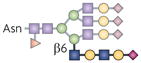 |
GnT5 | TCR activation threshold | Galectin-3 |
| Sia6LacNAc | 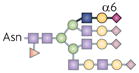 |
ST6Gal1 | BCR activation threshold | CD22 |
| Hybrid N-glycan | 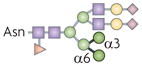 |
αM-II | Deficiency causes SLE-like syndrome in mice | Mannose-binding lectin (or lectins) |
| O-fucose glycan | β1,3GlcNAcTs and Pofut1 | Lymphocyte development and activation, myeloid and DC development, and NK-cell toxicity | Notch | |
| O-glucose glycan | O-glucosyltransferase (Rumi) | Notch signalling | Notch | |
| Hyaluronan | Hyaluronan synthases | Innate immunity, response to infection | TLR2 and TLR4 | |
| N-glycan | 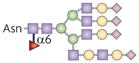 |
Fut8 | TGFβ1 receptor ligand binding and signalling | TGFβ1 |
Glycan structure is indicated using the symbol nomenclature for monosaccharides; see FIG. 1 for further information.
Glycosyltransferase and glycosidase enzymes that are known to be responsible for the presence of the indicated glycan linkages, whether owing to their regulated expression or lack of expression.
Recognition proteins that include classically defined lectins, which function by binding to the highlighted glycan structures. αM-II, α-mannosidase-II; β1,4GalT, β1,4-galactosyltransferase; β1,3GnT, β1,3-glucosaminyltransferase; BCR, B-cell receptor; DC, dendritic cell; Fut, fucosyltransferase; GCNT1, core 2 β1,4-glucosaminyltransferase 1; GlcNAc, β-N-acetylglucosamine; GlcNAc6ST, glycan sulphotransferase; GnT, N-acetylglucosaminyltransferase; NK, natural killer; Pofut1, protein O-fucosyltransferase; SLE, systemic lupus erythematosus; sLex, sialyl-Lewis-X; ST3Gal, α2,3-sialyltransferase, TCR, T-cell receptor; TGFβ1, transforming growth factor β1; TLR, Toll-like receptor.
The mechanisms by which glycans and lectins contribute to immunity are only now beginning to be understood. Among the potential mechanisms, the nature of multivalent lectin–ligand binding as observed in vitro indicates that higher-order structures might exist among glycoproteins and lectins at the cell surface161. Further study could resolve whether galectins normally operate in vivo to construct a repetitive and spatially precise ultrastructure (a lattice) that thereby modulates the presentation, interaction, function and turnover of glycoproteins161–166. There is increasing evidence that mammalian glycosylation controls the rate of receptor endocytosis as a means to modulate cellular responses through the interaction of lectins and their ligands167–169. It seems possible that what is presently considered basal protein-turnover activity at the cell surface is instead a highly orchestrated process that controls glycoprotein fate and function. Furthermore, it remains probable that more lectins without canonical lectin-like domains exist, which are identifiable as proteins that bind their targets and function in a glycan-dependent manner. It follows that glycan-dependent regulation of glycoprotein and glycolipid interactions might contribute in many contexts and in presently unknown ways to the mechanisms that govern the immune responses of cells to various extracellular stimuli.
The evolutionary success of the selectins and their glycan ligands in regulating immune function by controlling cell–cell adhesion and signalling makes it unlikely that they are the only lectin–ligand system that has evolved to direct immune-cell trafficking. Other glycan structures, such as Lewis X and Lewis Y, for example, have recently been found to participate in the cell–cell recognition that modulates dendritic-cell adhesion and signalling170,171. Moreover, hyaluronan has been recently found to modulate neutrophil adhesion in liver sinusoids172. The extent to which glycosylation contributes to immune-cell recognition is uncertain, but the regulated changes that occur in glycan formation during cell maturation and activation are profound and may alter the production of various lec-tin ligands that might engage various molecules and cell types173. Even the glycosyltransferases that participate in selectin-ligand synthesis have yet to be fully identified, and the regulatory events that control the formation of selectin ligands are incompletely understood. In addition, the mechanisms by which selectin ligands differentially affect the immune response, including their ability to modulate intracellular signalling, are just beginning to be studied.
Glycosylation in mammals constructs a glycome that can be distinguished phylogenetically and immunologically from those of lower and divergent organisms, and encompasses glycan linkages with profound immunoregulatory functions. Improved experimental techniques that can rapidly define glycan structure and can image distinct cellular glycoprotein interactions are increasingly relevant. Antibodies that specifically bind the synthetic and degradative enzymes of glycosylation are for the most part not available today, which makes it difficult to detect the molecular interactions that constitute the regulatory machinery of cellular glyco-sylation. Determining the elements of the mammalian glycome and the mechanisms by which the glycome is constructed and regulated will provide integrative and profound immunological insights into the biological roles of mammalian glycosylation.
Acknowledgments
We apologize to the colleagues and researchers who have made contributions that were not referenced owing to space limitations. J.D.M. is supported by the Howard Hughes Medical Institute and grants from the National Institutes of Health, USA (HL57345, HL78784 and GM62116).
- Glycome
The biological repertoire of glycan structures
- Experimental autoimmune encephalomyelitis
An experimental model of multiple sclerosis. Autoimmune disease is induced in animals by immunization with myelin or peptides derived from myelin. The animals develop a paralytic disease with inflammation and demyelination in the brain and spinal cord
- Immunoreceptor tyrosine-based inhibitory motif
(ITIM). A short amino-acid sequence (the consensus sequence of which is V/IxYxxV/L, in which x denotes any amino acid) that is found in the cytoplasmic tail of inhibitory receptors. ITIMs are thought to mediate inhibitory signalling by recruiting phosphatases such as sHP1 (sRC-homology-2-domain-containing protein tyrosine phosphatase 1)
- Clathrin-enriched microdomain
A region of the plasma membrane that is associated with clathrin-dependent endocytic mechanisms of protein trafficking and turnover
- IgA nephropathy
An inflammatory disease of the kidney that is associated with the glomerular deposition of IgA
- Old-world primates
A grouping of primates, which includes humans, that are native to Africa and Asia
- Henoch–Schönlein purpura
An inflammatory disease that involves the blood vessels and causes vasculitis
- Hodgkin’s lymphoma
A type of lymphoma that is characterized by the presence of Reed–sternberg cells (derived from B cells) and that spreads between the lymph nodes
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
αM-II | CD22 | COSMC | E-selectin | galectin-1 | GCNT1 | L-selectin | Mgat5 | Notch | P-selectin | PSGL1 | ST3Gal4 | ST3Gal6
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
rheumatoid arthritis | systemic lupus erythematosus | Tn syndrome
FURTHER INFORMATION
Jamey D. Marth’s homepage: http://www.hhmi.org/research/investigators/marth_bio.html
References
- 1.Paulson JC, Blixt O, Collins BE. Sweet spots in functional glycomics. Nature Chem Biol. 2006;2:238–248. doi: 10.1038/nchembio785. This publication provides an introduction to the definition and concept of the glycome and describes new approaches to research in this field. [DOI] [PubMed] [Google Scholar]
- 2.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. This article reviews the process of glycosylation and the mechanisms by which glycans contribute to molecular interactions that govern health and disease. [DOI] [PubMed] [Google Scholar]
- 3.Raman R, Raguram S, Venkataraman G, Paulson JC, Sasisekharan R. Glycomics: an integrated systems approach to structure–function relationships of glycans. Nature Methods. 2005;2:817–824. doi: 10.1038/nmeth807. [DOI] [PubMed] [Google Scholar]
- 4.Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. This article reviews the developmental biology of glycosylation and the mechanisms by which glycans control ontogeny. [DOI] [PubMed] [Google Scholar]
- 5.Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:732–742. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varki A. Nothing in glycobiology makes sense, except in the light of evolution. Cell. 2006;126:841–845. doi: 10.1016/j.cell.2006.08.022. This review provides a perspective on how glycan function arose and continues to change in the evolution of organisms. [DOI] [PubMed] [Google Scholar]
- 7.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 8.Lowe JB, Marth JD. A genetic approach to mammalian glycan function. Annu Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- 9.Comelli EM, et al. A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology. 2005;16:117–131. doi: 10.1093/glycob/cwj048. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, et al. Mutation of the COG complex subunit COG7 causes a lethal congenital disorder. Nature Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 11.Hart GW. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu Rev Biochem. 1997;66:315–335. doi: 10.1146/annurev.biochem.66.1.315. [DOI] [PubMed] [Google Scholar]
- 12.Dias WB, Hart GW. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol Biosyst. 2007;3:766–772. doi: 10.1039/b704905f. [DOI] [PubMed] [Google Scholar]
- 13.Sharon N. Lectins: carbohydrate-specific reagents and biological recognition molecules. J Biol Chem. 2007;282:2753–2764. doi: 10.1074/jbc.X600004200. This publication describes the origins and activities of lectins as proteinaceous receptors that bind glycans. [DOI] [PubMed] [Google Scholar]
- 14.Taylor ME, Drickamer K. Paradigms for glycan-binding receptors in cell adhesion. Curr Opin Cell Biol. 2007;19:572–577. doi: 10.1016/j.ceb.2007.09.004. This article describes the various lectins and their binding activities in adhesive functions between cells. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein IJ. Lectin structure-activity: the story is never over. J Agric Food Chem. 2002;50:6583–6585. doi: 10.1021/jf0201879. This publication describes the extent of origins and activities of lectins. [DOI] [PubMed] [Google Scholar]
- 16.Gagneux P, Varki A. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology. 1999;9:747–755. doi: 10.1093/glycob/9.8.747. [DOI] [PubMed] [Google Scholar]
- 17.Medzhitov R, Janeway JC. The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8:452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- 18.van Kooyk Y, Rabinovich GA. Protein–glycan interactions in the control of innate and adaptive immune responses. Nature Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 19.Baum LG. Developing a taste for sweets. Immunity. 2002;16:5–8. doi: 10.1016/s1074-7613(02)00265-0. [DOI] [PubMed] [Google Scholar]
- 20.Lowe JB. Glycosylation, immunity, and autoimmunity. Cell. 2001;104:809–812. doi: 10.1016/s0092-8674(01)00277-x. [DOI] [PubMed] [Google Scholar]
- 21.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2374. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 22.Lowe JB. Glycosyltransferases and glycan structures contributing to the adhesive activities of L-, E- and P-selectin counter-receptors. Biochem Soc Symp. 2002;69:33–45. doi: 10.1042/bss0690033. [DOI] [PubMed] [Google Scholar]
- 23.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 24.McEver RP. Leukocyte interactions mediated by selectins. Thromb Haemost. 1991;66:80–87. [PubMed] [Google Scholar]
- 25.Lasky LA. Selectins: interpreters of cell-specific carbohydrate information during inflammation. Science. 1992;258:964–969. doi: 10.1126/science.1439808. [DOI] [PubMed] [Google Scholar]
- 26.Ley K, Kansas GS. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nature Rev Immunol. 2004;4:325–335. doi: 10.1038/nri1351. [DOI] [PubMed] [Google Scholar]
- 27.Rossi FMV, et al. Recruitment of adult thymic progenitors is regulated by P-selectin and its ligand PSGL-1. Nature Immunol. 2005;6:626–634. doi: 10.1038/ni1203. [DOI] [PubMed] [Google Scholar]
- 28.McEver R. Selectin–carbohydrate interactions during inflammation and metastasis. Glycoconj J. 1997;14:585–591. doi: 10.1023/a:1018584425879. [DOI] [PubMed] [Google Scholar]
- 29.McEver RP, Cummings RD. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:S97–103. [PubMed] [Google Scholar]
- 30.Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26:477–489. doi: 10.1016/j.immuni.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zisoulis DG, Kansas GS. H-Ras and phosphoinositide 3-kinase co-operate to induce α(1,3)-fucosyltransferase VII expression in Jurkat T cells. J Biol Chem. 2004;279:39495–39504. doi: 10.1074/jbc.M407904200. [DOI] [PubMed] [Google Scholar]
- 32.Underhill GH, et al. A crucial role for T-bet in selectin ligand expression in T helper 1 (Th1) cells. Blood. 2005;106:3867–3873. doi: 10.1182/blood-2005-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang R, et al. Transcriptional regulation of the N-acetylglucosaminyltransferase V gene in human bile duct carcinoma cells (HuCC-T1) is mediated by Ets-1. J Biol Chem. 1996;271:26706–26712. doi: 10.1074/jbc.271.43.26706. [DOI] [PubMed] [Google Scholar]
- 34.Maly P, et al. The α(1, 3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–653. doi: 10.1016/s0092-8674(00)80137-3. [DOI] [PubMed] [Google Scholar]
- 35.Homeister JW, et al. The α(1, 3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity. 2001;15:115–126. doi: 10.1016/s1074-7613(01)00166-2. [DOI] [PubMed] [Google Scholar]
- 36.Smith PL, et al. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellies LG, et al. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3425. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 38.Sperandio M, et al. α2,3-sialyltransferase-IV is essential for L-selectin ligand function in inflammation. Eur J Immunol. 2006;36:3207–3215. doi: 10.1002/eji.200636157. [DOI] [PubMed] [Google Scholar]
- 39.Frommhold, et al. Sialyltransferase ST3Gal-IV controls CXCR2-mediated firm leukocyte arrest during inflammation. J Exp Med. 2008;205:1435–1446. doi: 10.1084/jem.20070846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellies LG, et al. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- 41.Sperandio M, et al. Differential requirements for core2 glucosaminyltransferase for endothelial L-selectin ligand function in vivo. J Immunol. 2001;16:7, 2268–2274. doi: 10.4049/jimmunol.167.4.2268. [DOI] [PubMed] [Google Scholar]
- 42.Gauguet JM, Rosen SD, Marth JD, von Andrian UH. Core 2 branching β1,6-N-acetyl- glucosaminyltransferase and high endothelial cell N-acetylglucosamine-6-sulfotransferase exert differential control over B- and T-lymphocyte homing to peripheral lymph nodes. Blood. 2004;104:4104–4112. doi: 10.1182/blood-2004-05-1986. [DOI] [PubMed] [Google Scholar]
- 43.Yeh JC, et al. Novel sulfated lymphocyte homing receptors and their control by a core1 extension β1, 3-N-acetylglucosaminyltransferase. Cell. 2001;105:957–969. doi: 10.1016/s0092-8674(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 44.Mitoma J, et al. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nature Immunol. 2007;8:409–418. doi: 10.1038/ni1442. [DOI] [PubMed] [Google Scholar]
- 45.Kawashima H, et al. N-acetylglucosamine-6-O-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nature Immunol. 2005;6:1096–1164. doi: 10.1038/ni1259. [DOI] [PubMed] [Google Scholar]
- 46.Uchimura K, et al. A major class of L-selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nature Immunol. 2005;6:1105–1113. doi: 10.1038/ni1258. [DOI] [PubMed] [Google Scholar]
- 47.Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem. 1997;272:28750–28756. doi: 10.1074/jbc.272.45.28750. [DOI] [PubMed] [Google Scholar]
- 48.Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates β2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. 2000;164:4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 49.Urzainqui A, et al. ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity. 2002;17:401–412. doi: 10.1016/s1074-7613(02)00420-x. [DOI] [PubMed] [Google Scholar]
- 50.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced αLβ2 integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26:773–783. doi: 10.1016/j.immuni.2007.04.011. This study links selectin binding to signal transduction by tyrosine kinases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci USA. 1998;95:9325–9380. doi: 10.1073/pnas.95.16.9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Fukuda M. Cell type-specific roles of carbohydrates in tumor metastasis. Methods Enzymol. 2006;416:371–380. doi: 10.1016/S0076-6879(06)16024-3. [DOI] [PubMed] [Google Scholar]
- 53.Romano SJ. Selectin antagonists: therapeutic potential in asthma and COPD. Treat Respir Med. 2005;4:85–94. doi: 10.2165/00151829-200504020-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uchimura K, Rosen SD. Sulfated L-selectin ligands as a therapeutic target in chronic inflammation. Trends Immunol. 2006;27:559–565. doi: 10.1016/j.it.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Woollard KJ, Chin-Dusting J. Therapeutic targeting of P-selectin in atherosclerosis. Inflamm Allergy Drug Targets. 2007;6:69–74. doi: 10.2174/187152807780077345. [DOI] [PubMed] [Google Scholar]
- 56.Sackstein R, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nature Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 57.Rapoport EM, Kurmyshkina OV, Bovin NV. Mammalian galectins: structure, carbohydrate specificity, and functions. Biochemistry (Mosc) 2008;73:393–405. doi: 10.1134/s0006297908040032. [DOI] [PubMed] [Google Scholar]
- 58.Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- 59.Lee SU, et al. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J Biol Chem. 2007;282:33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- 60.Demotte N, et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414–424. doi: 10.1016/j.immuni.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 61.Liu SD, et al. Endogenous galectin-1 enforces class I-restricted TCR functional fate decisions in thymocytes. Blood. 2008;112:120–130. doi: 10.1182/blood-2007-09-114181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grigorian A, et al. Control of T cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J Biol Chem. 2007;282:20027–20035. doi: 10.1074/jbc.M701890200. [DOI] [PubMed] [Google Scholar]
- 63.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nature Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. This review covers the structure and activities of the Siglec family of lectins and their immunological functions. [DOI] [PubMed] [Google Scholar]
- 64.Tedder TF, Tuscano J, Sato S, Kehrl JH. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev Immunol. 1997;15:481–504. doi: 10.1146/annurev.immunol.15.1.481. [DOI] [PubMed] [Google Scholar]
- 65.Hennet T, Chui D, Paulson JC, Marth JD. Immune regulation by the ST6Gal sialyltransferase. Proc Natl Acad Sci USA. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han S, Collins BE, Bengtson P, Paulson JC. Homo-multimeric complexes of CD22 in B cells revealed by protein–glycan cross-linking. Nature Chem Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 67.Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nature Immunol. 2006;7:199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 68.Grewal PK, et al. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santos L, et al. Dendritic cell-dependent inhibition of B cell proliferation requires CD22. J Immunol. 2008;180:4561–4569. doi: 10.4049/jimmunol.180.7.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kobata A. The N-linked sugar chains of human immunoglobulin G: their unique pattern, and their functional roles. Biochim Biophys Acta. 2008;1780:472–478. doi: 10.1016/j.bbagen.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 71.Wright A, Morrison SL. Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotech. 1997;15:26–32. doi: 10.1016/S0167-7799(96)10062-7. [DOI] [PubMed] [Google Scholar]
- 72.Parekh RB, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature. 1985;316:452–457. doi: 10.1038/316452a0. [DOI] [PubMed] [Google Scholar]
- 73.Hoffmeister KM, et al. Glycosylation restores survival of chilled blood platelets. Science. 2003;301:1531–1534. doi: 10.1126/science.1085322. [DOI] [PubMed] [Google Scholar]
- 74.Jones AJS, et al. Selective clearance of glycoforms of a complex glycoprotein pharmaceutical caused by terminal N-acetylglucosamine is similar in humans and cynomolgus monkeys. Glycobiology. 2007;17:529–540. doi: 10.1093/glycob/cwm017. [DOI] [PubMed] [Google Scholar]
- 75.Hiki Y, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 76.Nishie T, et al. Development of immunoglobulin A nephropathy-like disease in β-1,4-galactosyl-transferase-I-deficient mice. Am J Pathol. 2007;170:447–456. doi: 10.2353/ajpath.2007.060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 78.Anthony RM, et al. Racapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, et al. Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad Sci USA. 2005;102:15791–15794. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X, et al. Phenotype changes of fut8 knockout mouse: core fucosylation is crucial for the function of growth factor receptor(s) Methods Enzymol. 2006;417:11–22. doi: 10.1016/S0076-6879(06)17002-0. [DOI] [PubMed] [Google Scholar]
- 81.Vogt G, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nature Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 82.Macher BA, Galili U. The Galα1, 3Galβ1, 4GlcNAc-R. (α-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta. 2007;1780:75–88. doi: 10.1016/j.bbagen.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Padler-Karavani V, et al. Diversity in specificity, abundance and composition of anti-Neu5Gc antibodies in normal humans: potential implications for disease. Glycobiology. 2008 Jul 31; doi: 10.1093/glycob/cwn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ju T, Cummings RD. Protein glycosylation: chaperone mutation in Tn syndrome. Nature . 2005;437:1252. doi: 10.1038/4371252a. [DOI] [PubMed] [Google Scholar]
- 85.Green RS, et al. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity. 2007;27:308–320. doi: 10.1016/j.immuni.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 86.Bianco GA, Toscano MA, Ilarregui JM, Rabinovich GA. Impact of protein–glycan interactions in the regulation of autoimmunity and chronic inflammation. Autoimmunity Rev. 2006;5:349–356. doi: 10.1016/j.autrev.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 87.Baum LG, et al. Amelioration of graft versus host disease by galectin-1. Clin Immunol. 2003;109:295–307. doi: 10.1016/j.clim.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 88.Rubinstein N, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: a potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–251. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 89.Perone MJ, et al. Dendritic cells expressing transgenic galectin-1 delay onset of autoimmune diabetes in mice. J Immunol. 2006;177:5278–5289. doi: 10.4049/jimmunol.177.8.5278. [DOI] [PubMed] [Google Scholar]
- 90.Seki M, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Barrionuevo P, et al. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol. 2007;178:436–445. doi: 10.4049/jimmunol.178.1.436. [DOI] [PubMed] [Google Scholar]
- 92.Blois SM, et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nature Med. 2007;13:1450–1457. doi: 10.1038/nm1680. [DOI] [PubMed] [Google Scholar]
- 93.Taylor KR, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44 and MD-2. J Biol Chem. 2007;282:18265–18275. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 94.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Develop Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 95.Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921–1943. doi: 10.1016/j.lfs.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 96.Forsberg E, et al. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 1999;400:773–776. doi: 10.1038/23488. [DOI] [PubMed] [Google Scholar]
- 97.Humphries DE, et al. Heparin is essential for the storage of specific granule proteases in mast cells. Nature. 1999;400:769–772. doi: 10.1038/23481. [DOI] [PubMed] [Google Scholar]
- 98.Carbone FR, Gleeson PA. Carbohydrates and antigen recognition by T cells. Glycobiology. 1997;7:725–730. doi: 10.1093/glycob/7.6.725-d. [DOI] [PubMed] [Google Scholar]
- 99.Zhao XJ, Cheung NK. GD2 oligosaccharide: target for cytotoxic T lymphocytes. J Exp Med. 1995;182:67–74. doi: 10.1084/jem.182.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu SD, et al. Endogenous galectin-1 enforces class I-restricted TCR functional fate decisions in thymocytes. Blood. 2008;112:120–130. doi: 10.1182/blood-2007-09-114181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corthay A, et al. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur J Immunol. 1998;28:2580–2590. doi: 10.1002/(SICI)1521-4141(199808)28:08<2580::AID-IMMU2580>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 102.Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–2378. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 103.Zajonc DM, Kronenberg M. CD1 mediated T cell recognition of glycolipids. Curr Opin Struct Biol. 2007;17:521–529. doi: 10.1016/j.sbi.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Daniels MA, et al. CD8 binding to MHC class I molecules is influenced by T cell maturation and glycosylation. Immunity. 2001;15:1051–1061. doi: 10.1016/s1074-7613(01)00252-7. [DOI] [PubMed] [Google Scholar]
- 105.Moody AM, et al. Developmentally regulated glycosylation of the CD8αβ coreceptor stalk modulates ligand binding. Cell. 2001;107:501–512. doi: 10.1016/s0092-8674(01)00577-3. [DOI] [PubMed] [Google Scholar]
- 106.Kao C, Daniels MA, Jameson SC. Loss of CD8 and TCR binding to class I MHC ligands following T cell activation. Int Immunol. 2005;17:1607–1617. doi: 10.1093/intimm/dxh340. [DOI] [PubMed] [Google Scholar]
- 107.Moody AM, et al. Sialic acid capping of CD8β core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–7246. doi: 10.1074/jbc.M210468200. [DOI] [PubMed] [Google Scholar]
- 108.Kao C, Sandau MM, Daniels MA, Jameson SC. The sialyltransferase ST3Gal-I is not required for regulation of CD8-class I MHC binding during T cell development. J Immunol. 2006;176:7421–7430. doi: 10.4049/jimmunol.176.12.7421. [DOI] [PubMed] [Google Scholar]
- 109.Van Dyken SJ, Green RS, Marth JD. Structural and mechanistic features of protein O glycosylation linked to CD8+ T-cell apoptosis. Mol Cell Biol. 2007;27:1096–1111. doi: 10.1128/MCB.01750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou L, et al. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 2008;112:308–319. doi: 10.1182/blood-2007-11-115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Irvine KD, Wieschaus E. Fringe, a boundary-specific signaling molecule, mediates interactions between dorsal and ventral cells during Drosophila wing development. Cell. 1994;79:595–606. doi: 10.1016/0092-8674(94)90545-2. [DOI] [PubMed] [Google Scholar]
- 112.Wu JY, Wen L, Zhang WJ, Rao Y. The secreted product of Xenopus gene lunatic fringe, a vertebrate signaling molecule. Science. 1996;273:355–358. doi: 10.1126/science.273.5273.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goode S, Perrimon N. Brainiac and fringe are similar pioneer proteins that impart specificity to notch signaling during Drosophila development. Cold Spring Harb Symp Quant Biol. 1997;62:177–184. [PubMed] [Google Scholar]
- 114.Johnston SH, et al. A family of mammalian Fringe genes implicated in boundary determination and the Notch pathway. Development. 1997;124:2245–2254. doi: 10.1242/dev.124.11.2245. [DOI] [PubMed] [Google Scholar]
- 115.Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates Notch–ligand interactions. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- 116.Brückner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch–Delta interactions. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 117.Moloney DJ, et al. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 118.Chen J, Moloney DJ, Stanley P. Fringe modulation of Jagged1-induced Notch signaling requires the action of β4galactosyltransferase-1. Proc Natl Acad Sci USA. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y, et al. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem. 2001;276:40338–40345. doi: 10.1074/jbc.M107849200. [DOI] [PubMed] [Google Scholar]
- 120.Okajima T, Irvine KD. Regulation of Notch signaling by O-linked fucose. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- 121.Okajima T, Xu A, Irvine KD. Modulation of Notch-ligand binding by protein O-fucosyltransferase 1 and Fringe. J Biol Chem. 2003;278:42340–42345. doi: 10.1074/jbc.M308687200. [DOI] [PubMed] [Google Scholar]
- 122.Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci USA. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang LT, et al. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell. 2005;16:927–942. doi: 10.1091/mbc.E04-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes Notch receptor folding. Science. 2005;307:1599–1603. doi: 10.1126/science.1108995. [DOI] [PubMed] [Google Scholar]
- 125.Acar M, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247–258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stanley P. Glucose: a novel regulator of Notch signaling. ACS Chem Biol. 2008;3:210–213. doi: 10.1021/cb800073x. [DOI] [PubMed] [Google Scholar]
- 127.Robey E, et al. An activated form of Notch influences the choice between CD4 and CD8 T cell lineages. Cell. 1996;87:483–492. doi: 10.1016/s0092-8674(00)81368-9. [DOI] [PubMed] [Google Scholar]
- 128.Koch U, et al. Subversion of the T/B lineage decision in the thymus by lunatic fringe-mediated inhibition of Notch-1. Immunity. 2001;15:225–234. doi: 10.1016/s1074-7613(01)00189-3. [DOI] [PubMed] [Google Scholar]
- 129.Visan I, et al. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nature Immunol. 2006;7:634–643. doi: 10.1038/ni1345. [DOI] [PubMed] [Google Scholar]
- 130.Jenkinson EJ, Jenkinson WE, Rossi SW, Anderson G. The thymus and T-cell commitment: the right niche for Notch. Nature Rev Immunol. 2006;6:551–555. doi: 10.1038/nri1883. [DOI] [PubMed] [Google Scholar]
- 131.Tanagaki K, et al. Notch–RBP-J signaling in involved in cell fate determination of marginal zone B cells. Nature Immunol. 2002;3:443–450. doi: 10.1038/ni793. [DOI] [PubMed] [Google Scholar]
- 132.Hozumi K, et al. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nature Immunol. 2004;5:638–644. doi: 10.1038/ni1075. [DOI] [PubMed] [Google Scholar]
- 133.Santos MA, et al. Notch1 engagement by Delta-like-1 promotes differentiation of B lymphocytes to antibody-secreting cells. Proc Natl Acad Sci USA. 2007;104:15454–15459. doi: 10.1073/pnas.0702891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheng P, Gabrilovich D. Notch signaling in differentiation and function of dendritic cells. Immunol Res. 2008;41:1–14. doi: 10.1007/s12026-007-8011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kijima, et al. Dendritic cell-mediated NK cell activation is controlled by Jagged2–Notch interaction. Proc Natl Acad Sci USA. 2008;105:7010–7015. doi: 10.1073/pnas.0709919105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Okamoto, et al. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Exp Med. 2008;205:1087–1097. doi: 10.1084/jem.20072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schaller MA, et al. Notch ligand Delta-like 4 regulates disease pathogenesis during respiratory viral infections by modulating Th2 cytokines. J Exp Med. 2007;204:2925–2934. doi: 10.1084/jem.20070661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun J, Krawczyk CJ, Pearce EJ. Suppression of Th2 cell development by Notch ligands Delta1 and Delta4. J Immunol. 2008;180:1655–1661. doi: 10.4049/jimmunol.180.3.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morgan R, et al. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol. 2004;173:7200–7208. doi: 10.4049/jimmunol.173.12.7200. [DOI] [PubMed] [Google Scholar]
- 140.Juszczynski P, et al. The AP1-dependent secretion of galectin-1 by Reed–Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci USA. 2007;104:13134–13139. doi: 10.1073/pnas.0706017104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Toscano MA, et al. Differential glycosylation of TH1, TH2, and TH-17 effector cells selectively regulates susceptibility to cell death. Nature Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 142.Thomas PG, Harn DA. Immune biasing by helminth glycans. Cell Microbiol. 2004;6:13–22. doi: 10.1046/j.1462-5822.2003.00337.x. [DOI] [PubMed] [Google Scholar]
- 143.Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nature Immunol. 2003;4:410–415. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- 144.Hernandez JD, Baum LG. Ah, sweet mystery of death! Galectins and control of cell fate. Glycobiology. 2002;12:127R–136R. doi: 10.1093/glycob/cwf081. This publication reviews galectins in immune-cell apoptosis. [DOI] [PubMed] [Google Scholar]
- 145.Hsu DK, Yang RY, Liu FT. Galectins in apoptosis. Methods Enzymol. 2006;417:256–273. doi: 10.1016/S0076-6879(06)17018-4. [DOI] [PubMed] [Google Scholar]
- 146.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 147.Stowell SR, et al. Human galectin-1, -2, and -4 induce surface expression of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood. 2008;109:219–227. doi: 10.1182/blood-2006-03-007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stowell SR, et al. Differential roles of galectin-1 and galectin-3 in regulating leukcoyte viability and cytokine secretion. J Immunol. 2008;180:3091–3102. doi: 10.4049/jimmunol.180.5.3091. [DOI] [PubMed] [Google Scholar]
- 149.Stowell SR, et al. Dimeric galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the carboxyl terminal domain. J Biol Chem. 2008;283:20547–20559. doi: 10.1074/jbc.M802495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dias-Baruffi M, et al. Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. J Biol Chem. 2003;278:41282–41293. doi: 10.1074/jbc.M306624200. [DOI] [PubMed] [Google Scholar]
- 151.Vespa GN, et al. Galectin-1 specifically modulates TCR signals to enhance TCR apoptosis but inhibits IL-2 production and proliferation. J Immunol. 1999;162:799–806. [PubMed] [Google Scholar]
- 152.Pace KE, Hahn HP, Pang M, Nguyen JT, Baum LG. CD7 delivers a pro-apoptotic signal during galectin-1-induced T cell death. J Immunol. 2000;165:2331–2334. doi: 10.4049/jimmunol.165.5.2331. [DOI] [PubMed] [Google Scholar]
- 153.Waizel H, Blach M, Hirabayashi J, Kasai KI, Brock J. Involvement of CD2 and CD3 in galectin-1 induced signaling in human Jurkat T-cells. Glycobiology. 2000;10:131–140. doi: 10.1093/glycob/10.2.131. [DOI] [PubMed] [Google Scholar]
- 154.Fuertes MB, et al. Regulated expression of galectin-1 during T-cell activation involves Lck and Fyn kinases and signaling through MEK1/ERK, p38 MAP kinase and p70S6 kinase. Mol Cell Biochem. 2004;267:177–185. doi: 10.1023/b:mcbi.0000049376.50242.7f. [DOI] [PubMed] [Google Scholar]
- 155.Cabrera PV, et al. Haploinsufficiency of C2GnT-I glycosyltransferase renders T lymphoma cells resistant to cell death. Blood. 2006;108:2399–2406. doi: 10.1182/blood-2006-04-018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hahn HP, et al. Galectin-1 induces nuclear translocation of endonuclease G in caspase- and cytochrome c-independent T cell death. Cell Death Diff. 2004;11:1277–1286. doi: 10.1038/sj.cdd.4401485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stillman BN, et al. Galectin-2 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 158.Bi S, Earl LA, Jacobs L, Baum LG. Structural features of galectin-9 and galectin-1 that determine distinct T cell death pathways. J Biol Chem. 2008;283:12248–12258. doi: 10.1074/jbc.M800523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Priatel JJ, et al. The ST3Gal-I sialyltransferase controls CD8+ T cell homeostasis by modulating O-glycan biosynthesis. Immunity. 2000;12:273–283. doi: 10.1016/s1074-7613(00)80180-6. [DOI] [PubMed] [Google Scholar]
- 160.Grewal PK, et al. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nature Med. 2008;14:648–655. doi: 10.1038/nm1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays, and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616–623. doi: 10.1016/s0959-440x(02)00364-0. This article reviews the hypothesis that galectins generate a spatially precise, repetitive structure (lattice) that controls glycoprotein trafficking and function on the cell surface. [DOI] [PubMed] [Google Scholar]
- 162.Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. 2007;282:1374–1383. doi: 10.1074/jbc.M604506200. [DOI] [PubMed] [Google Scholar]
- 163.Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin–glycoprotein lattices. Curr Opin Struct Biol. 2007;17:513–520. doi: 10.1016/j.sbi.2007.09.002. This publication reviews findings regarding the galectins and their potential to form various lattices that can define functional units and membrane domains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hernandez JD, et al. Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J Immunol. 2006;177:5328–5336. doi: 10.4049/jimmunol.177.8.5328. [DOI] [PubMed] [Google Scholar]
- 165.Chen IJ, Chen HL, Demetriou M. Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling. J Biol Chem. 2007;282:35361–35372. doi: 10.1074/jbc.M706923200. [DOI] [PubMed] [Google Scholar]
- 166.Lajoie P, et al. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol. 2007;179:341–356. doi: 10.1083/jcb.200611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Partridge EA, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 168.Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Marth JD. Dietary and genetic control of glucose transporter-2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 169.Lau KS, et al. Complex N-glycan number and degree of branching co-operate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 170.van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y. Neutrophils medicate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med . 2005;20:1, 1281–1292. doi: 10.1084/jem.20041276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Garcia-Vallejo JJ, et al. DC-SIGN mediates adhesion and rolling of dendritic cells on primary human umbilical vein endothelial cells through Lewis(Y) antigen expressed on ICAM-2. Mol Immunol. 2008;45:2359–2369. doi: 10.1016/j.molimm.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 172.McDonald B, et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med. 2008;205:915–927. doi: 10.1084/jem.20071765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bax M, et al. Dendritic cell maturation results in pronounced changes in glycan expression affecting recognition by siglecs and galectins. J Immunol. 2007;179:8216–8224. doi: 10.4049/jimmunol.179.12.8216. [DOI] [PubMed] [Google Scholar]
- 174.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci USA. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Freeze HH. Genetic defects in the human glycome. Nature Rev Genet. 2006;7:537–551. doi: 10.1038/nrg1894. A review of the human genomic data that are known to contribute to genetic disease by altered glycosylation. [DOI] [PubMed] [Google Scholar]
- 176.Brooks SA, et al. Altered glycosylation of proteins in cancer: what is the potential for new anti-tumour strategies. Anticancer Agents Med Chem. 2008;8:2–21. doi: 10.2174/187152008783330860. [DOI] [PubMed] [Google Scholar]
- 177.Collins BT, et al. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc Natl Acad, Sci USA. 2004;282:6104–6109. doi: 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]