

Abstract
Early onset disease is frequently examined in genetic studies because it is presumed to contain a more severe subset of patients under a higher influence of genetic effects. In light of the dramatic success of Crohn’s disease (CD) gene discovery efforts, we aimed to characterize the contribution of established common risk variants to pediatric CD. Using 35 confirmed CD risk alleles, we genotyped 384 parent-child trios (mean age of onset 11.7 years) along with 321 healthy controls. We performed association tests on the independent pediatric cohort and compared results to those previously published(1). We also computed a weighted CD genetic risk score for each affected person. Six variants not previously validated in children (at 5q33, 1q24, 7p12, 12q12, 8q24 and 1q32) were significantly associated with pediatric CD (P<0.03). We detected no significant association between risk score and age at onset through age 30. This analysis illustrates that the genetic effect of established CD risk variants is similar in early and later onset CD. These results motivate joint analyses of genome-wide association data in early and late onset cohorts and suggest that, rather than established risk variants, independent variants or environmental exposures should be sought as modulators of age of onset.
Keywords: Crohn Disease/*genetics, genetic predisposition to disease, polymorphism, single nucleotide, Age of Onset, Child
Introduction
Studying early-onset presentations of complex disease is appealing to geneticists because of the expectation that these efforts will increase the probability of finding novel risk variants. Implicit in this strategy is the assumption that these patients represent a more severe, more genetically influenced group of affected individuals. Some studies have identified specific genes that predispose to early onset—others, aided by the enrichment in gene burden, have discovered general risk variants. The discovery by linkage and fine mapping of BRCA1 on chromosome 17q21 in early onset, familial breast cancer(2, 3), for example, provides encouragement that there is a genetic basis to common age of onset phenotypes(4–6). The yields of this approach have been particularly illustrative in early-onset forms of Alzheimer’s disease(7), Huntington’s disease(4) and myocardial infarction(8).
CD has provided a highlight of the recent efforts to implicate genetic variation in complex disease pathogenesis. Genome-wide association studies (GWAS) and a subsequent meta-analysis performed in thousands of predominantly adult onset CD cases have led to confirmation of more than 30 risk alleles explaining approximately 20% of the genetic variance in CD(1, 9–11). Through these recent efforts it has now become possible to study the collective influence of many risk variants in CD pathogenesis.
Phenotypic heterogeneity between adult and pediatric onset CD is well documented but the causal mechanisms underlying these differences are unclear. Different anatomical distributions, responses to medical therapy, and prognoses(12–17) suggest a physiologic basis for these observations. This diversity is likely heritable, suggested by increased familial aggregation(12, 18, 19), higher concordance in disease location(20), and genotype-phenotype correlations(21–27) seen in early onset disease.
Whether genetic variation can explain observed differences between pediatric and adult onset CD is largely unexplored. Several CD susceptibility alleles are confirmed as common to both adult and pediatric populations(27–31) but the majority have not been explored in children. Characterizing the role of DNA variation in influencing earlier onset CD has important implications for drug development, diagnostic testing, and risk stratification.
Although unlikely, pediatric CD may represent a distinct disease entity from later onset disease, with unique genetic risk factors—as is the case with early onset Alzheimer’s disease and breast cancer. In the other extreme, pediatric CD and later onset disease could have identical genetic architecture, but with earlier onset patients inheriting a larger dose of genetic risk factors. An intermediate hypothesis is that environmental exposures, genetic variation outside the CD causal pathway, or rare variants in common risk loci modulate the age at which disease presents.
We aimed to test the hypothesis that the timing of CD might be influenced by the overall burden of common genetic risk or through the action of common variation at individual risk alleles.
Materials and Methods
Study Subjects
Pediatric CD cohort (PED)
Through ongoing IRB-approved genetic studies at Children’s Hospital Boston (CHB) and Milwaukee Children’s Hospital (MCH), we collected detailed phenotypic and demographic information on 384 parent-parent-affected trios with a child age 19 or younger at the time of diagnosis; 189 trios were enrolled at CHB and 195 at MCH. These samples were not involved in any previous linkage or genome-wide association analysis contributing to the discovery of the risk loci being examined. We had previously collected peripheral blood and extracted genomic DNA on all individuals. Physicians at the home institutions made the diagnosis of CD, which was based on a combination of clinical, radiographic, and gross endoscopic findings, as well as on a review of intestinal mucosal biopsies. Patients with CD in any area of the intestine were eligible for analysis. We excluded from the cohort patients with indeterminate colitis.
Pediatric Ulcerative Colitis (UC) Cohort
We collected phenotype and genotype information from 70 patients with pediatric UC to compare risk scores with the PED cohort. These patients, diagnosed under age 20, were enrolled at CHB in the same protocol in which PED was enrolled.
Meta-analysis replication cohort (REP)
In order to study the age of onset across children and adults, we used previously genotyped data from 1295 parent-parent-affected offspring trios with onset of disease at any age. This sample had been obtained for replicating CD risk loci identified from the recent meta-analysis of CD GWAS. Details about this dataset are explained elsewhere(1).
Controls (CON)
321 healthy adult controls from the Boston area were genotyped to calculate a control group risk score. This cohort had not been typed previously for any other CD analysis.
The above cohorts and sample sizes reflect the Caucasian-only subset of the data. Non-Caucasians were removed prior to analysis because of the known population differences in CD genetic architecture.
Phenotype ascertainment
Age of onset was defined as the age at which the patient was initially diagnosed with CD. Pediatric onset disease was defined as a CD diagnosis before age 20. A genetic risk score was calculated to describe the overall burden of CD risk alleles for each individual. Using recently published ORs at all confirmed CD risk variants(1), we multiplied the number of risk alleles at each locus (0, 1, or 2) by the log(OR) of that risk allele. We took the average score across all successfully genotyped loci as a summary score for each individual. The scores were normalized to reflect a Z distribution. Since the five independent risk alleles across CARD15 and IL23R have relatively large effect sizes, we only scored individuals with 100% genotyping at all of these loci.
Genotyping
Using the polymerase chain reaction (PCR) based Sequenom genotyping platform (San Diego CA), primers were designed to amplify and genotype all 35 confirmed independent CD risk alleles or a nearby proxy SNP with an r2=1. These 35 independently acting alleles represent 32 different loci across the genome.
Quality Control (QC)
The dataset was filtered to exclude SNPs with a Hardy-Weinberg p-value <0.001 and a call rate > 95%. Individuals with <80% genotyping were excluded.
Data Analysis and Statistics
Clinical and genotype data from CHB samples were read into R, PLINK, and Haploview statistical packages for analysis. Because quality control measures disrupted some intact trios, we evaluated a small subset of affected patients in case-control analysis using the CON dataset as the control group. Family based association testing employed the transmission disequilibrium test (TDT). Association tests employed a chi-squared test with one degree of freedom.
Using observed and expected allele counts and variance estimates for both TDT samples and case-control samples, we combined the association tests into a single meta Z score for each SNP. OR’s were calculated separately for TDT and for case-control samples and were then combined using a stratified approach(32). Alleles were deemed ‘significantly associated’ with pediatric CD if the p value was <=0.03. The rationale for this threshold is that if all 34 tested SNPs were found to be significantly associated with pediatric CD, we would expect one of these associations to be below the threshold significance by chance alone (i.e. a false positive).
Risk scores were compared using the Student’s t-test. Age of onset was compared to risk score by computing the Pearson correlation coefficient. Power analysis was performed using an online genetic power calculator(33).
Ethical Considerations
The home institution’s institutional review board (IRB) approved the recruitment of patients and families at CHB and MCH. Patients and/or parents gave written, informed consent before enrollment. We de-identified patient information before samples were analyzed by the authors. Ethical considerations in patient recruitment for the de-identified REP dataset, are described elsewhere(1).
Results
Clinical Characteristics
The PED cohort had a higher proportion of children with colonic, ileo-colonic, and peri-anal CD than the children in the REP cohort (Table 1a and 1b). Since PED is entirely a hospital-based cohort, it is possible that these individuals had more severe and/or extensive disease. Despite these clinical differences, their mean genetic risk scores were similar (see below).
Table 1.
| Table 1a. Baseline demographic and clinical data in the CD and control cohorts studied. Shown in separate columns are PED, the pediatric onset component of REP (REP=<19), the older onset component of REP (REP (>19), the full REP cohort, and CON. Children in PED have a higher proportion of L2, L3, and peri-anal disease than the similarly aged REP<=19. | |||||
|---|---|---|---|---|---|
| Patient Cohorts | |||||
| PED | REP (=<19) | REP (>19) | REP(ALL)* | CON | |
| Number of Affected | 384 | 578 | 527 | 1125 | 312 |
| Males (%) | 235 (61.2) | 315 (54.5) | 201 (38.1) | 528 (46.9) | 117 (41.2) |
| Caucasian (%) | 372 (96.9) | 578 (100.0) | 547 (100.0) | 1125 (100.0) | 312 (100.0) |
| Mean Age of Onset (%) | 11.7 | 13.4 | 28.8 | 20.8 | NA |
| Perianal Disease (%) | 149 (41.9) | 172 (32.3) | 162 (30.7) | 338 (30.0) | NA |
| Montreal Classification Disease Location | |||||
| L1—Ileal (%) | 49 (13.0) | 178 (34.2) | 167 (31.7) | 350 (35.5) | NA |
| L2—Colon Only (%) | 81 (21.5) | 57 (11.0) | 85 (16.1) | 146 (14.8) | NA |
| L3—Ileo-Colonic (%) | 241 (63.9) | 279 (53.7) | 205 (38.9) | 489 (49.6) | NA |
| Not Recorded(%) | 7 (1.8) | 58 (10.0) | 70 (13.3) | 139 (12.4) | NA |
| Table 1b. Baseline demographic information and clinical data in UC patients studied. | |
|---|---|
| Number | 71 |
| Males (%) | 36 (50.7) |
| Caucasian (%) | 68 (95.8) |
| Mean age of onset | 11.4 |
| Montreal Classification disease location | |
| E1—ulcerative proctitis | 0 |
| E2—left-sided UC | 16 (22.5) |
| E3—pancolitis | 52 (73.2) |
| Missing data | 3 (4.2) |
Includes unknown age of onset patients in the calculations
Validation of the Set of CD Risk Alleles
To determine if predominantly adult-identified risk variants are valid and act with similar effect sizes in children, we genotyped the PED cohort at all 35 independent CD risk alleles. All SNPs passed QC filters except for rs17622378, representing the IBD5 locus at 5q31, which was excluded from the analysis. Of the remaining 34 SNPs, 15 were associated with pediatric onset CD with our predetermined significance threshold of P<0.03 (Table 2). This observation is extremely unlikely to occur by chance (P=1.5 × 10−14). For six of these alleles, this is the first validation in pediatric CD. Of note, all independently acting CARD15, IL23R, and ATG16L1 alleles replicated in our cohort. Although association with pediatric CD is well established for lesions in these important pathogenic mechanisms, replication in our cohort provides reassurance that our samples are representative of pediatric CD in the larger population.
Table 2.
Summary of CD risk allele validation in the PED sample. Grouped by meta-Z statistics, 15 independently acting risk alleles meet conservative significance thresholds. In six of these alleles, represented in bold, this is the first report of association with pediatric CD. P-values are one-tailed. CI are 95% confidence intervals around the combined odds ratio. Risk alleles are defined relative to the + strand of the reference and were obtained from previously published results(1).
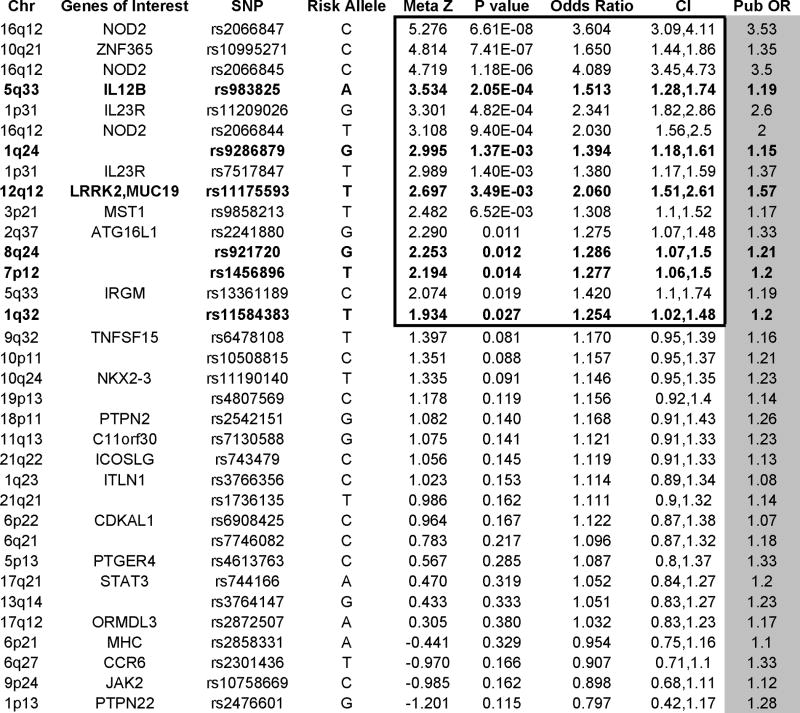 |
In 28 of 34 SNPs tested, the OR 95% confidence bounds include the previously established values (1). Thirty of 34 ORs reflect effect sizes in the same direction as reported ORs. Furthermore, roughly half of the observed OR’s are larger than published values. These data argue for similar genetic determinants with similar effect sizes in pediatric and later onset CD.
Genetic Burden and Age of Onset
Under the assumption that all adult-identified CD risk loci participate in the pathogenesis of pediatric CD, we calculated a genetic risk score for each individual in the PED sample as well as the REP and CON samples. Of the 34 genotyped SNPs, 30 were used in the risk score calculation (or a proxy SNP with an r2>0.89). Four variants (rs7130588, rs10508815, rs2858331and rs2872507) were not sufficiently genotyped in all three samples and were therefore excluded in the risk score calculation. The international meta-analysis reported modest effect sizes for these SNPS (ORs 1.17–1.23).
The risk score distributions from PED and CON overlap to some degree (figure 1), however the mean score was significantly higher in the affected patients than in controls (0.701 -vs- 0 respectively, P=1.74×10−10).
Figure 1.

The distribution of risk scores in the pediatric onset CD cases (PED) and in a control population (CON). The mean score was significantly higher in the affected patients than in controls (0.701 -vs- 0, P=1.74×10−10).
In order to address whether the risk scores in the REP sample were artificially enriched for allelic dosage due to its previous use in replicating the CD meta-analysis findings, we compared the risk scores in the REP children under age 20 at diagnosis to those of the independent PED sample. We found no difference in mean score (0.707 and 0.739 respectively, P=0.635), suggesting that the samples can be combined for further analyses.
Risk score distributions, stratified by the Montreal classification for age at diagnosis(34) reveal a small deflation in risk scores in individuals diagnosed over the age of 40 (figure 2). We detected a very weak negative correlation between age of CD onset and risk score (i.e. lower gene dosage is associated with later CD onset), with a pearson r=−0.088 (P=0.002). Of note, there is a disproportionate amount of outlying data in older onset patients (over age 30). When we analyze all patients diagnosed only up to age 30, there is no longer a detectable correlation between age of onset and risk score.
Figure 2.

The distribution of cumulative risk scores stratified by age of onset. Risk score distributions appear very similar in the under 17 and 17 to 40 group. There is mild deflation in the score in the over 40 group. The risk score distribution in the control group is lower than all affected groups.
We also subanalyzed the very early onset group of patients (diagnosed under age 9) and compared them to older onset children (diagnosed age 9 through 19). There was no difference in the mean risk score between these two groups. Additionally, males and females with CD had similar mean risk scores.
Individual Risk Alleles and Age of Onset
To test for single alleles that might exert a disproportionate influence on the age of onset of CD, we modeled age of onset against all 30 alleles in the risk score acting as independent variables using linear regression. We detected no significant action of a single risk variant on age of onset.
Differences in CD Gene Dosage in Pediatric CD and UC
We undertook an initial analysis of how the collective burden of CD risk alleles differs in pediatric CD and UC patients. We computed risk scores in 70 cases of pediatric UC and compared this distribution to that of the 371 CD patients in the PED cohort (figure 3). We found a significantly deflated risk score in the UC compared with CD patients (0.197 -vs- 0.739, P=4.8 × 10−7). The score in UC patients was similar to controls (P=0.067). Finally, we noted that when repeating the score calculation solely using 10 alleles that are associated with both CD and UC(35), the mean scores in CD and UC were similar (p=0.4), reflecting the loss of discriminating information (i.e. CD-specific alleles) from the analysis. Not surprisingly in this analysis of shared alleles, all IBD patients combined (UC and CD) had higher mean scores than the controls still (p=0.008), reflecting the fact that we still had ten highly significant IBD loci in the analysis.
Figure 3.

The distribution of risk scores for the CD and UC cases from the PED cohort and from controls. The risk score is inflated in CD over UC (0.739 -vs- 0.197, P=4.8×10−7). UC and controls have similar risk scores (P=0.067)
Discussion
This is the first comprehensive analysis of the contribution of common genetic risk to pediatric and adult-onset CD—two heterogeneous presentations of IBD. Previous attempts to compare the genetic influences on age of CD onset examined either individual genes such as NOD2 and IL23R, or a small combination of genetic variants (21–27). Here, we show that the overall burden of confirmed risk alleles, estimated to explain 20% of the genetic variance of CD(1), is at most a very minor factor in the age at which CD presents. The findings argue for heritable, but as of yet undiscovered factors as the important determinants of an early presentation of CD. Rare, penetrant genetic variation and differences in the timing of host-environment interactions are attractive mechanisms that deserve additional attention in light of these findings.
We also report that at common DNA variants, similar loci act with similar effect sizes in early and later onset CD. Through this effort, we confidently validate six such alleles in children for the first time. 15 of 34 tested alleles were significantly associated with pediatric CD by conservative thresholds, reflecting our limited power to detect true associations at all tested alleles. It should be noted that we did not have adequate power to confidently rule out any adult-identified locus as associated with pediatric onset CD.
We further observed that children with UC have a marked deflation in CD risk scores and that their scores are similar to controls. Even without any UC-specific alleles used to calculate the risk score, there was a notable separation in the distributions for UC and CD patients. This raises the possibility that with further refinement—perhaps by accounting for both CD and UC-specific associations in the score--genotype-based approaches could have utility in distinguishing IBD subtypes in clinically ambiguous cases.
This report is timely as the IBD genetics field explores how best to analyze the vast amounts of data generated from genome-wide analyses. Our results provide reassurance that genotype data from children and adults can be combined for more powerful analyses. Deep sequencing of risk loci in search of rare variants that may be active in early onset CD should also be encouraged. It is unlikely that further discovery of common CD risk alleles will increase the ability to detect a major effect of ‘overall genetic load’ in age of onset since current known risk variants explain less than 10% of the variance in this phenotype.
We defined the age of onset phenotype as ‘the age of CD diagnosis by a physician’, which could cause a spurious association between age of onset and risk score. The initial presentation of CD can be insidious, and upon subsequent reflection, patients may recall symptoms for several years preceding their presentation to a physician. Subtle symptoms are more likely to be ignored, resulting in a delay in presentation to a physician. Thus, if highly insidious (but active) disease is associated with both a decrease in CD gene dosage and with the age at which one presents to a physician, there would be an artificial association between genetic dose and our ‘age of onset’ phenotype. This systematic error might be less active in young patients, where parents and pediatricians are more likely to seek prompt referral for any subtle signs or symptoms in their children. In a secondary analysis, we removed all individuals reportedly diagnosed over the age of 30 and we detected no correlation between genetic risk score and age of onset, lending further support for information bias as a source for the weak observed correlation.
Acknowledgments
We thank the IBD patients and families recruited at Children’s Hospital Boston and at Milwaukee Children’s Hospital. We also thank the General Clinical Research Center at Children’s Hospital and the genotyping platform at the Broad Institute for support, supplies, and technical advise. The NIDDK IBD Genetics Consortium, which collected and curated a large dataset for our analysis includes the following principal investigators: John D Rioux, Ramnik J. Xavier, Kent D Taylor, Mark S Silverberg, Philippe Goyette, Alan Huett, Stephan R Targan, A Hillary Steinhart, Jerome I Rotter, Richard H Duerr, Judy H Cho, Mark J Daly, and Steven R Brant.
References
- 1.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature genetics. 2008 doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science (New York, NY. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 3.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (New York, NY. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 4.Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nature genetics. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald ME, Vonsattel JP, Shrinidhi J, et al. Evidence for the GluR6 gene associated with younger onset age of Huntington’s disease. Neurology. 1999;53:1330–1332. doi: 10.1212/wnl.53.6.1330. [DOI] [PubMed] [Google Scholar]
- 6.Li JL, Hayden MR, Almqvist EW, et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. American journal of human genetics. 2003;73:682–687. doi: 10.1086/378133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chartier-Harlin MC, Crawford F, Houlden H, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 8.Kathiresan S. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. The New England journal of medicine. 2008;358:2299–2300. doi: 10.1056/NEJMc0707445. [DOI] [PubMed] [Google Scholar]
- 9.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (New York, NY. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS genetics. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature genetics. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polito JM, 2nd, Childs B, Mellits ED, et al. Crohn’s disease: influence of age at diagnosis on site and clinical type of disease. Gastroenterology. 1996;111:580–586. doi: 10.1053/gast.1996.v111.pm8780560. [DOI] [PubMed] [Google Scholar]
- 13.Paul T, Birnbaum A, Pal DK, et al. Distinct phenotype of early childhood inflammatory bowel disease. J Clin Gastroenterol. 2006;40:583–586. doi: 10.1097/00004836-200608000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Heyman MB, Kirschner BS, Gold BD, et al. Children with early-onset inflammatory bowel disease (IBD): analysis of a pediatric IBD consortium registry. The Journal of pediatrics. 2005;146:35–40. doi: 10.1016/j.jpeds.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 15.Marx G, Seidman EG, Martin SR, et al. Outcome of Crohn’s disease diagnosed before two years of age. The Journal of pediatrics. 2002;140:470–473. doi: 10.1067/mpd.2002.123281. [DOI] [PubMed] [Google Scholar]
- 16.Farmer RG, Michener WM. Prognosis of Crohn’s disease with onset in childhood or adolescence. Digestive diseases and sciences. 1979;24:752–757. doi: 10.1007/BF01317207. [DOI] [PubMed] [Google Scholar]
- 17.Markowitz J, Grancher K, Kohn N, et al. A multicenter trial of 6-mercaptopurine and prednisone in children with newly diagnosed Crohn’s disease. Gastroenterology. 2000;119:895–902. doi: 10.1053/gast.2000.18144. [DOI] [PubMed] [Google Scholar]
- 18.Satsangi J, Grootscholten C, Holt H, et al. Clinical patterns of familial inflammatory bowel disease. Gut. 1996;38:738–741. doi: 10.1136/gut.38.5.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orholm M, Fonager K, Sorensen HT. Risk of ulcerative colitis and Crohn’s disease among offspring of patients with chronic inflammatory bowel disease. The American journal of gastroenterology. 1999;94:3236–3238. doi: 10.1111/j.1572-0241.1999.01526.x. [DOI] [PubMed] [Google Scholar]
- 20.Colombel JF, Grandbastien B, Gower-Rousseau C, et al. Clinical characteristics of Crohn’s disease in 72 families. Gastroenterology. 1996;111:604–607. doi: 10.1053/gast.1996.v111.pm8780563. [DOI] [PubMed] [Google Scholar]
- 21.Leshinsky-Silver E, Karban A, Buzhakor E, et al. Is age of onset of Crohn’s disease governed by mutations in NOD2/caspase recruitment domains 15 and Toll-like receptor 4? Evaluation of a pediatric cohort. Pediatric research. 2005;58:499–504. doi: 10.1203/01.PDR.0000175640.75468.D6. [DOI] [PubMed] [Google Scholar]
- 22.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. American journal of human genetics. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine A, Kugathasan S, Annese V, et al. Pediatric onset Crohn’s colitis is characterized by genotype-dependent age-related susceptibility. Inflammatory bowel diseases. 2007;13:1509–1515. doi: 10.1002/ibd.20244. [DOI] [PubMed] [Google Scholar]
- 24.Shaoul R, Karban A, Weiss B, et al. NOD2/CARD15 mutations and presence of granulomas in pediatric and adult Crohn’s disease. Inflammatory bowel diseases. 2004;10:709–714. doi: 10.1097/00054725-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Levine A, Karban A, Eliakim R, et al. A polymorphism in the TNF-alpha promoter gene is associated with pediatric onset and colonic location of Crohn’s disease. The American journal of gastroenterology. 2005;100:407–413. doi: 10.1111/j.1572-0241.2005.41126.x. [DOI] [PubMed] [Google Scholar]
- 26.Levine A, Shamir R, Wine E, et al. TNF promoter polymorphisms and modulation of growth retardation and disease severity in pediatric Crohn’s disease. The American journal of gastroenterology. 2005;100:1598–1604. doi: 10.1111/j.1572-0241.2005.41737.x. [DOI] [PubMed] [Google Scholar]
- 27.de Ridder L, Weersma RK, Dijkstra G, et al. Genetic susceptibility has a more important role in pediatric-onset Crohn’s disease than in adult-onset Crohn’s disease. Inflammatory bowel diseases. 2007;13:1083–1092. doi: 10.1002/ibd.20171. [DOI] [PubMed] [Google Scholar]
- 28.Dubinsky MC, Wang D, Picornell Y, et al. IL-23 receptor (IL-23R) gene protects against pediatric Crohn’s disease. Inflammatory bowel diseases. 2007;13:511–515. doi: 10.1002/ibd.20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell RK, Drummond HE, Nimmo EE, et al. Genotype-phenotype analysis in childhood-onset Crohn’s disease: NOD2/CARD15 variants consistently predict phenotypic characteristics of severe disease. Inflammatory bowel diseases. 2005;11:955–964. doi: 10.1097/01.mib.0000183423.38037.f3. [DOI] [PubMed] [Google Scholar]
- 30.Biank V, Broeckel U, Kugathasan S. Pediatric inflammatory bowel disease: clinical and molecular genetics. Inflammatory bowel diseases. 2007;13:1430–1438. doi: 10.1002/ibd.20213. [DOI] [PubMed] [Google Scholar]
- 31.Peterson N, Guthery S, Denson L, et al. Genetic variants in the autophagy pathway contribute to paediatric Crohn’s disease. Gut. 2008;57:1336–1337. doi: 10.1136/gut.2008.152207. author reply 1337. [DOI] [PubMed] [Google Scholar]
- 32.Kazeem GR, Farrall M. Integrating case-control and TDT studies. Annals of human genetics. 2005;69:329–335. doi: 10.1046/j.1529-8817.2005.00156.x. [DOI] [PubMed] [Google Scholar]
- 33.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics (Oxford, England) 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 34.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Canadian journal of gastroenterology = Journal canadien de gastroenterologie. 2005;19(Suppl A):5–36. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 35.Anderson CA, Massey DC, Barrett JC, et al. Investigation of Crohn’s Disease Risk Loci in Ulcerative Colitis Further Defines Their Molecular Relationship. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]