

Abstract
Mucin antigen 1 (MUC1) is overexpressed on various human adenocarcinomas and haematological malignancies and has long been used as a target antigen for cancer immunotherapy. Most of the preclinical and clinical studies using MUC1 have used the tandem repeat region of MUC1, which could be presented by only a limited set of major histocompatibility complex haplotypes. Here, we evaluated N-terminal region (2–147 amino acids) of MUC1 (MUC1-N) for dendritic cell (DC)-based cancer immunotherapy. We used Esherichia coli-derived MUC1-N that was fused to the protein transduction domain of human immunodeficiency virus Tat protein for three reasons. First, mature DCs do not phagocytose soluble protein antigens. Secondly, tumour cells express underglycosylated MUC1, which can generate epitopes repertoire that differs from normal cells, which express hyperglycosylated MUC1. Finally, aberrantly glycosylated MUC1 has been known to impair DC function. In our study, Tat-MUC1-N-loaded DCs induced type 1 T cell responses as well as cytotoxic T lymphocytes efficiently. Furthermore, they could break tolerance in the transgenic breast tumour mouse model, where MUC1-positive breast cancers grow spontaneously. Compared with DCs pulsed with unconjugated MUC1-N, DCs loaded with Tat-conjugated MUC1-N could delay tumour growth more effectively in the transgenic tumour model as well as in the tumour injection model. These results suggest that the recombinant N-terminal part of MUC1, which may provide a diverse epitope repertoire, could be utilized as an effective tumour antigen for DC-based cancer immunotherapy.
Keywords: cancer immunotherapy, DC, HIV Tat, mucin antigen 1, protein transduction domain
Introduction
MUC1 is expressed normally on the apical surface of most simple glandular epithelial cells and consists of an N-terminal non-tandem repeat (NTR, 124 amino acids) including signal sequence, variable number of tandem repeats (VNTR, consisting of 20 amino acids) and a C-terminal 228 amino acids. Because the VNTR is enriched with serine and threonine residues that are attached by O-linked glycans, MUC1 is hyperglycosylated in normal cells [1]. In various cancers, such as breast, ovary, lung, pancreas and prostate tumours, MUC1 is increased more than 100-fold and aberrantly underglycosylated [2]. These quantitative and qualitative changes in MUC1 expression in cancers render it immunogenic. A number of reports have identified anti-MUC1 immune responses in preclinical studies and in cancer patients after active immunization with MUC1 [2,3]. However, MUC1-specific immune responses in those trials have rarely translated into clinical efficacy [3], and there are still many hurdles that must be overcome to elicit efficient and protective immune responses and eradicate cancers.
Dendritic cells (DCs) are used as potent therapeutic vaccines against human cancers, because DCs initiate and regulate tumour-specific immune responses [4,5]. Although early clinical trials with ex vivo-generated DCs that were pulsed with MUC1 have provided a proof of principle, the efficacy has been inadequate to apply to clinical settings [5,6]. To improve the efficacy of DC vaccination various strategies have been developed, such as the generation of specific DC subsets, efficient antigen loading, efficacious delivery of DCs to regional lymph nodes (LNs), enhanced survival of DCs and activation of DCs [4,5,7].
First of all, a sufficient amount of MUC1 must be loaded into DCs. Because activated DCs cannot phagocytose exogenous antigens efficiently, antigen-loading strategies should be designed carefully [5,7]. Recombinant proteins, peptides, viral vectors, RNA, immune complexes and killed tumour cell lysates have been used in this capacity [5], among which the use of the protein transduction domain (PTD) has been given much attention, because it is safer than viral vectors yet equally effective in loading MUC1[8,9]. It has been hypothesized that the intracellular delivery of tumour-associated antigens (TAAs) into mature DCs by a PTD, such as the human immunodeficiency virus (HIV) Tat peptide, allows DCs to process and present the internalized antigens to T cells by major histocompatibility complex (MHC) class I and II molecules efficiently [10,11]. DCs that are pulsed with Tat-TAA have been demonstrated to induce antigen-specific CD4 T cells effectively as well as cytotoxic T lymphocytes (CTLs) [11,12].
In addition to loading antigen into DCs, the epitope repertoire that is presented by DCs may be another determinant of DC-based immunotherapy using MUC1 as TAA [13]. Two factors may determine the efficacy of current MUC1-based immunotherapy. First, the VNTR, which consists of 20-amino acid repeats and has been used intensively in multiple studies [3,14–16], generates a limited range of epitopes and certain MHC haplotypes may not be able to present them efficiently [17]. Even though the immunogenicity of an extracellular non-VNTR region and cytoplasmic tail of MUC1 has been evaluated in several studies [18–21], the efficacy of DC-based vaccine utilizing N-terminal region of MUC-1 (MUC1-N) has as yet been poorly characterized. Secondly, glycosylation of MUC1 influences the epitope repertoire generated during the antigen processing steps through proteasomal and endosomal degradation [22]. It has been reported previously that three different forms of MUC1, ranging from glycosylated and underglycosylated protein to unglycosylated synthetic peptide, were able to elicit MUC1-specific, class I-restricted CTL responses [23]. The efficiency of processing and the resulting strength in CTL activity correlate inversely with the degree of glycosylation.
For these reasons, we have tried to develop strategies using unglycosylated recombinant MUC1-N, which is purified from Escherichia coli and includes NTR and a VNTR of MUC1, to generate more diverse epitope subsets that can be presented on various MHC haplotypes [18]. In addition, we also investigated the efficacy of a DC vaccine that uses a Tat-fused MUC1-N as TAA. We found that the Tat-MUC1-N-based DC vaccine elicited MUC1-specific cellular immune responses and anti-tumour immunity in both a tumour-injection model and a transgenic mouse model that develops spontaneous MUC1-positive tumours.
Materials and methods
Mice and cell lines
C57BL/10NAGCSnAi-[KO] Rag2 (H-2b) mice were obtained from Taconic Farms, Inc. (Hudson, NY, USA). C57BL/6 (H-2b) mice were purchased from the Center for Animal Resource Development, Seoul National University (SNU) College of Medicine (Seoul, Korea). Polyomavirus middle-T oncogene (PyMT) and human-MUC1 transgenic mice [24] were kindly provided by Dr S. Gendler (Mayo Clinic, Scottsdale, AZ, USA). The MUC1/PyMT double transgenic mice (MMT) were generated by crossing female MUC1 transgenic mice with male PyMT transgenic mice. MMT mice developed spontaneous MUC1-expressing mammary carcinomas with 100% penetrance at 8–15 weeks of age, as described [24]. Only female MMT mice were used for the experiments. The animal experiments were performed after approval by the SNU animal welfare committee (permission ID: SNU-080115-7). EL-4 (H-2b) and MC57G (H-2b), mouse tumour cell lines, were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Both EL4 and MC57G were transfected individually with a plasmid that encoded full-length human MUC1 that contained 22 tandem repeats, which was kindly provided by Dr Olivera Finn (University of Pittsburgh, Pittsburgh, PA, USA), to generate cell lines that stably expressed human MUC1. A pcDNA3·1 (Invitrogen, Grand Island, NY, USA)-transfected cell line was generated for MUC1-negative control cells. EL4 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Welgene Inc., Daegu, Korea) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 2 mM l-glutamine, penicillin (100 units/ml), streptomycin (100 µg/ml) and gentamicin (50 µg/ml) (Life Technologies, Gaithersburg, MD, USA). MC57G cells were cultured in Eagle's minimum essential medium (EMEM; ATCC) containing 10% FBS, 2 mM l-glutamine and antibiotics. Both cell lines, stably expressing MUC1, were maintained in the presence of G418 (400 µg/ml; Stratagene, Santa Clara, CA, USA).
Construction of recombinant MUC1 expression vectors
Human MUC1 cDNA containing 22 tandem repeats (TR) was used as a template for the polymerase chain reaction (PCR) amplification of the N-terminal of MUC1, corresponding to nucleotides 4–441. PCR was performed using the forward primer 5′-CGGAATTCCGACACCGGGCACCCAG-3′ (MUC14–18, EcoRI site underlined) and the reverse primer 5′-CCCTCGAGGGCCGGCCTGGTGT-3′ (MUC1428–441, XhoI site underlined). The PCR products were digested with EcoRI and XhoI (Takara, Shiga, Japan) and gel-purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Purified PCR products were cloned into EcoRI/XhoI-digested pET-23a (Novagen, Darmstadt, Germany) or pET-23a that contained 11 amino acids of the Tat PTD (YGRKKRRQRRR) at the NheI/EcoRI sites to generate pET-MUC1-N and pET-Tat-MUC1-N, respectively. E. coli DH5α (Real Biotech Corp., Taipei, Taiwan) were transformed with the resulting constructs by heat shock and selected on LB agar plates containing 100 µg/ml of ampicillin (Sigma, St Louis, MO, USA).
Expression and purification of recombinant MUC1 proteins
To express and purify recombinant MUC1 proteins, E. coli BL21 star (DE3) strains (Novagen, Madison, WI, USA) were transformed with pET-MUC1-N or pET-Tat-MUC1-N. Bacteria were then grown in Luria–Bertani broth containing ampicillin (100 µg/ml). Protein expression was induced by isopropyl β-D-thiogalactoside (IPTG) (Duchefa, Zwijndrecht, the Netherlands) at a final concentration of 0·4 mM for 4 h at 37°C. Bacterial pellets were resuspended in lysis buffer (2% Tween-20, 10 mM imidazole, 50 mM sodium phosphate, 0·3 M NaCl, pH 7·4), followed by sonication on ice for 15 min. Sonicated lysates were centrifuged at 16 000 g for 20 min at 4°C and subjected to HisPur cobalt resin affinity chromatography (Pierce, Woburn, MA, USA). His-tagged proteins that were bound to the resin were eluted with elution buffer (150 mM imidazole, 50 mM sodium phosphate, 0·3 M NaCl, pH 7·4). Finally, the identity and purity of purified proteins were assessed by Western blot and Coomassie blue staining, respectively. Purified proteins were treated with endotoxin removal columns (Pierce, Woburn, MA, USA) before being added to DCs. Endotoxin contamination of the purified recombinant proteins was determined using the QCl-1000® End-Point Chromogenic Endotoxin Detection kit (Lonza, Basel, Switzerland).
Western blot
Whole bacterial cell lysates and purified proteins were separated on 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked with 5% non-fat milk (BD, San Jose, CA, USA) in Tris-buffered saline solution (TBST) [20 mM Tris-Cl (pH 7·6), 100 mM NaCl and 0·05% Tween-20], incubated with mouse anti-human MUC1 monoclonal antibody (clone VU4H5; Santa Cruz, CA, USA) or mouse anti-His monoclonal antibody (clone 27E8; Cell Signaling, Danvers, MA, USA) at 4°C overnight, and washed three times with TBST. The membrane was incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Santa Cruz) at room temperature (RT) for 1 h. After washing, immunoreactive bands were detected using enhanced chemiluminescence (ECL) (Amersham, Piscataway, NJ, USA) and LAS-4000 (Fujifilm, Miami Beach, FL, USA). To confirm MUC1 expression in the transfected tumour cell lines, cells were lysed with NP40 lysis buffer (150 mM NaCl, 1% NP40, 50 mM Tris, pH 7·4) and analysed by Western blot using anti-human MUC1 monoclonal antibody (clone VU4H5; Santa Cruz).
In vitro generation of bone marrow (BM)-derived DCs
DCs were generated from BM of 6–10-week-old Rag2 knock-out mice. BM cells were flushed out of the femurs and tibias with serum-free CellGro medium (CellGenix, Freiburg, Germany). The single cell suspension was then filtered through a nylon cell strainer (70-mm Nylon mesh; BD), washed twice with complete CellGro medium [CellGro supplemented with recombinant mouse granulocyte–macrophage colony-stimulating factor (GM–CSF) (0·75 ng/ml) and mouse interleukin (IL)-4 (1·5 ng/ml, PeproTech, Rocky Hill, NJ, USA), penicillin (100 units/ml), streptomycin (100 µg/ml), gentamicin (50 µg/ml), l-glutamine (2 mM) and β-mercaptoethanol (ME) (50 nM, Invitrogen)], and seeded at a concentration of 1 × 106 cells per well in a 24-well plate in a final volume of 2 ml of complete CellGro medium. Half the medium was replaced every other day with an equal volume of complete CellGro medium for 6 days.
Confocal laser microscopy
At day 6, DCs were pulsed with MUC1-N or Tat-MUC1-N proteins (50 µg/ml) for 1 h, washed twice in phosphate-buffered saline (PBS), transferred onto poly-L-lysine-coated microscope slides (Menzel-Glaser, Braunschweig, Germany) and fixed with 4% paraformaldehyde solution at RT for 10 min. Fixed DCs were then permeabilized with 0·25% Triton-X100, blocked with Superblock [a mixture of 10% rat, hamster and mouse sera each, with 10 µg/ml 2·4G2 monoclonal antibody (Invitrogen)] at RT for 20 min, and stained with fluorescein isothiocyanate (FITC)-conjugated anti-human MUC1 (clone HMPV; BD Pharmingen, San Diego, CA, USA) at RT for 1 h. Nuclei were stained with 0·5 µg/ml 4,6-diamidino-2-phenylindole (DAPI) (Molecular Probe, Eugene, OR, USA) at RT for 5 min. Cells were analysed on a FluoView1000 confocal laser microscope (Olympus, Nagano, Japan).
Flow cytometric analysis
At day 6 of DC cultures, purified MUC1-N or Tat-MUC1-N proteins were added at 50 µg/ml and incubated for 1 h. After stimulation with LPS (300 ng/ml) for 18 h, DCs were washed with ice-cold fluorescence activated cell sorter (FACS) buffer [PBS containing 1% bovine serum albumin (BSA) (AMResco, Solon, OH, USA) and 1 mM ethylenediamine tetraacetic acid (EDTA) (Sigma)] and blocked on ice for 30 min with Superblock. DCs were stained with antibodies and 7-aminoactinomycin D (7AAD, BD Pharmingen) for FACS analysis using FITC-conjugated antiI-Ab (clone AF6-120·1), allophycocyanin (APC)-conjugated anti-CD11c (clone HL3), phycoerythrin (PE)-conjugated anti-CD86 (clone GL1), FITC-conjugated anti-CD80 (clone 16-10A1) and PE-conjugated anti-CD40 (clone 3/23). All antibodies were purchased from BD Pharmingen. Cells were analysed on a FACS Canto II flow cytometer (BD Biosciences, Mountain View, CA, USA). To analyse protein transduction efficiency, MUC1-N and Tat-MUC1-N were labelled with Alexa Fluor 594 using the Alexa Fluor® 594 protein labelling kit (Invitrogen) according to the manufacturer's instructions. DCs were pulsed with either free Alexa Fluor 594 or Alexa Fluor 594-labelled proteins (50 µg/ml) for 30 min and washed three times with FACS buffer to quantify intracellular MUC1 by FACS.
Lymphocyte proliferation assay
B6 mice (6–8 weeks old) were injected subcutaneously (s.c.) at the base of the tail at day 0 with 1 × 106 untransduced DCs or DCs that were transduced with MUC1-N or Tat-MUC1-N. At day 10 after vaccination, lymph node (LN) cells were harvested and incubated with varying concentrations of MUC1-N proteins in 96-well round-bottomed microtitre plates (2 × 105 cells/well) for 2 days. Cells were labelled with [3H]-methylthymidine (1 µCi) (Amersham) for an additional 18 h. The cells were harvested onto fibreglass filters, and cell-associated radioactivity was measured by Micro Beta TriLux (PerkinElmer, Waltham, MS, USA).
Evaluation of cytokine secretion
LN cells, obtained as above, were cultured in the presence or absence of MUC1-N (50 µg/ml) proteins for 48 h. Culture supernatants were analyzed by T helper type 1/2 (Th1/2) cytometric bead array (BD Biosciences, San Diego, CA, USA). The results were analysed according to the manufacturer's instructions using a FACS Canto II flow cytometer (BD). Each sample was analysed in triplicate.
Cytotoxicity assay
B6 mice (6–8 weeks old) were immunized twice at weekly intervals with MUC1-N- or Tat-MUC1-N-transduced DCs (1 × 106). At day 7 after final immunization, the cytotoxicity of splenocytes to target cells was assessed by P-JAM test, as described [25]. Briefly, total splenocytes that were harvested from each immunized mouse were restimulated with 10 µg/ml MUC1-N protein for 3 days and used as effector cells. Target cells (MC57G cells transduced with pcDNA3·1 or MC57G cells transduced with pcDNA-MUC1) were incubated with effector cells for 4 h at various effector : target ratios. After incubation, the effector cells and dead targets were washed away four times with PBS. Live target cells, attached to the bottom of the culture plates, were labelled with [3H]-methylthymidine (Amersham) in 200 ml medium at a final concentration of 5 µCi/ml for 3 h. Incorporation of [3H]-methylthymidine was quantified by Micro Beta TriLux (Wallac, Turku, Finland) after harvesting. The percentage of specific lysis = [counts per minute (cpm)targets − (cpmtargets+killers − cpmkillers)]/cpmtargets × 100 [25].
Tumour growth
For tumour rejection experiments, 6-week-old B6 mice were injected with EL4-pcDNA-MUC1 cells (1 × 105) into the right flank at day 0. From day 1 after tumour challenge, the mice received vaccines. Mice were vaccinated subcutaneously four times at weekly intervals with DCs (1 × 106) that were pulsed with MUC1-N or Tat-MUC1-N protein (50 µg/ml). Mice that were immunized with PBS or DCs served as controls. Tumour volume was calculated using the following formula: tumour volume (mm3) = (A × B2)/2, where A is the long diameter and B is the short diameter [26]. On every other day, we calculated the average tumour volume and standard error of the mean (s.e.m.) from all tumour masses of the mice in each group. The percentage of survival in each group was also recorded after tumour inoculation. To assess the efficacy of DCs (Tat-MUC1-N) in a more physiological system, 6-week-old MMT mice were vaccinated subcutaneously with 1 × 106 DCs. The immunization was repeated five additional times at biweekly intervals. The mice were observed for up to 22 weeks. The tumour volume was calculated using the formula that is described above.
Statistical analysis
Statistical analyses were performed using the Student's t-test with SigmaPlot software (Jandel, San Rafael, CA, USA). The data are presented as mean ± standard error (s.e.) and were considered statistically significant at P < 0·05.
Results
Expression of recombinant MUC1
Nucleotides 4–441 (438 base pairs) of human MUC1 were PCR-amplified and subsequently cloned in-frame to generate pET-MUC1-N and pET-Tat-MUC1-N (Fig. 1a). The recombinant proteins were expressed in E. coli BL21 that were transformed with pET-MUC1-N or pET-Tat-MUC1-N after induction with IPTG and purified by HisPur cobalt resin affinity chromatography (Fig. 1b). Recombinant human MUC1 was expressed readily in E. coli as a soluble 18-kDa protein in the presence or absence of Tat. From a 1-litre culture, we obtained 2·0 ± 0·5 mg and 1·0 ± 0·2 mg of purified recombinant MUC1-N and Tat-MUC1-N, respectively. The purified proteins were verified by Western blot using anti-His antibody and anti-human MUC1 antibody (Fig. 1b). Coomassie blue staining of the purified proteins revealed that they were > 95% pure. Endotoxin contamination of the purified recombinant proteins was < 0·1 EU/ml in all protein samples after passage through an endotoxin removal column.
Fig. 1.
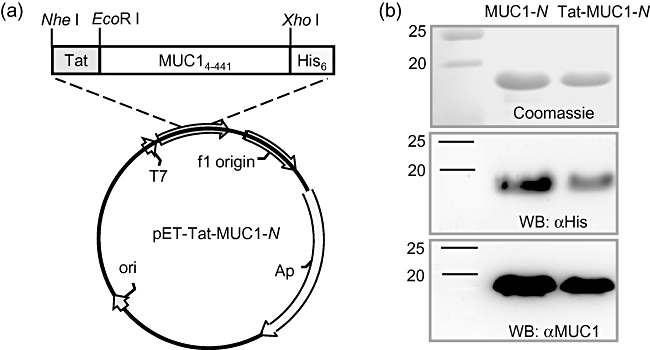
Expression and purification of Tat-N-terminal region mucin antigen 1 (MUC1)-N fusion proteins. (a) Schematic of pET-Tat-MUC1-N vector, which carries Tat-MUC1-N upstream of the 6× His tag. (b) Purification of MUC1-N and Tat-MUC1-N proteins. To express recombinant MUC1 proteins, BL21 Star (DE3) cells were transformed with pET-MUC1-N or pET-Tat-MUC1-N. Recombinant MUC1 proteins were purified from the Escherichia coli lysates and stained with Coomassie blue (upper panel) and analysed by immunoblotting with anti-His (middle panel) or anti-human MUC1 (lower panel).
Transduction of DCs with recombinant proteins
To determine the transduction efficiency of purified MUC1 fusion proteins into DCs, MUC1-N or Tat-MUC1-N fusion protein, labelled with Alexa594, was incubated with DCs for 30 min, and cells were analysed by FACS analysis. As shown in Fig. 2a, the percentage of Alexa594-positive DCs was enhanced markedly at 30 min after transduction when incubated with Tat-MUC1-N (72·1%) compared with those incubated with MUC1-N (39·0%). The mean fluorescence intensity (MFI) of Tat-MUC1-N-transduced DCs (53·1) was higher than that of MUC1-N-transduced DCs (23·1). Efficient delivery of Tat-MUC1-N was also confirmed by confocal microscopy (Fig. 2b). Intracellular MUC1 levels were greater in DC (Tat-MUC1-N) than in DC (MUC1-N).
Fig. 2.
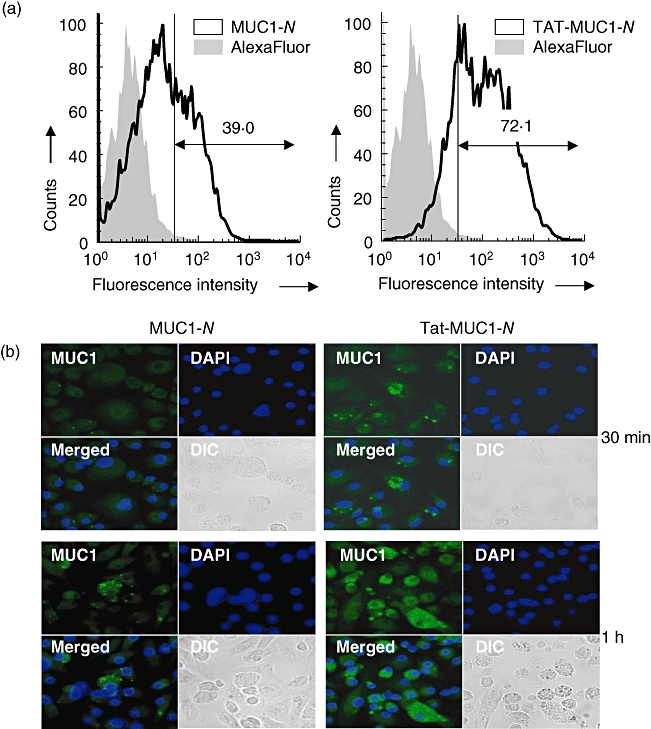
Transduction of recombinant N-terminal region mucin antigen 1 (MUC1) into dendritic cells (DCs). (a) Flow cytometric analysis of MUC1 in DCs transduced with N-terminal region MUC1 (MUC1-N) or Tat-MUC1-N. DCs were incubated with either free AlexaFluor 594 (grey area) or AlexaFluor 594-labelled proteins (solid line) for 30 min. (b) Confocal microscopic analysis of DCs transduced with MUC1-N or Tat-MUC1-N. After incubation of DCs with recombinant MUC1 proteins for 30 min or 1 h, intracellular MUC1 was stained with anti-human MUC1-fluorescein isothiocyanate (green). Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (blue). Two pseudocoloured images (MUC1 + DAPI) were merged (merged), and differential interference contrast (DIC) images are shown.
DC maturation after recombinant MUC1 transduction
Because MUC1 affects the maturation and function of DCs [27], we examined whether incubating DCs with recombinant MUC1 influences DC maturation by analysing the expression of co-stimulatory molecules on DCs. DCs were transduced with recombinant MUC1 for 1 h and stimulated with lipopolysaccharide (LPS) for an additional 18 h. DCs were stained with 7AAD and antibodies against CD40, CD80, CD86 and I-Ab. Live DCs (7AADneg/CD11cpos) were analysed by FACS analysis. DCs that were transduced with MUC1-N or Tat-MUC1-N up-regulated MHC I and co-stimulatory molecules to levels that were similar to control DCs (Fig. 3), suggesting that transduction of DCs with recombinant MUC1 does not affect DC maturation.
Fig. 3.
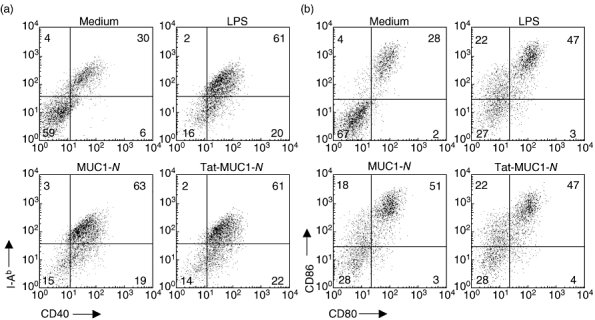
Expression of co-stimulatory molecules and I-Ab of dendritic cells (DCs) pulsed with recombinant mucin antigen 1 (MUC1). Because it has been known that MUC1 could impair DC function, we analysed co-stimulatory molecule expression on DCs after they were pulsed with purified N-terminal region MUC1 (MUC1-N) or Tat-MUC1-N proteins. After DCs were incubated with lipopolysaccharide for another 18 h after protein pulsing, cells were stained with antibodies reactive with I-Ab, CD40, CD86, CD80 and CD11c. Live DCs (7AADneg/CD11cpos) were analysed for the surface molecule expression using a flow cytometer.
Priming lymphocytes in vivo
To assess whether DCs pulsed with MUC1-N or Tat-MUC1-N prime MUC1-specific lymphocytes, lymphocytes were harvested from lymph nodes of mice 10 days after vaccination with DC (MUC1-N) or DC (Tat-MUC1-N) (1 × 106 cells) and restimulated in vitro with various amounts of MUC1-N antigen. As shown in Fig. 4, lymphocytes from mice that were immunized with DC (Tat-MUC1-N) showed increased proliferation in response to MUC1-N in a dose-dependent manner. This proliferation was significantly higher compared with mice that were immunized with DC (MUC1-N) (P < 0·05). Lymphocytes from mice that were immunized with control DCs did not proliferate in response to MUC1. These results suggest that DC (Tat-MUC1-N) prime MUC1-specific lymphocytes more efficiently than DC (MUC1-N) in vivo.
Fig. 4.
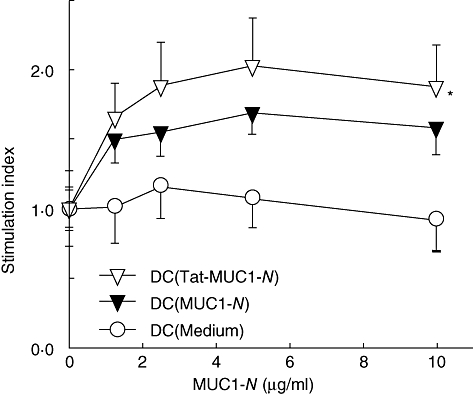
Proliferation of lymph node (LN) cells from mice immunized with dendritic cell (DC) vaccines. DCs (1 × 106 cells) pulsed with N-terminal region mucin antigen 1 (MUC1-N) or Tat-MUC1-N proteins were injected into the base of tails of B6 mice (three mice per group). LN cells were harvested from inguinal and peri-aortic lymph nodes 10 days later. LN cells were co-cultured with the indicated amounts of MUC1-N proteins for 2 days and labelled with [3H]-methylthymidine (1 mCi) for an additional 18 h. Proliferation of LN cells was determined by [3H]-methylthymidine uptake. Stimulation index = counts per minute (cpm)test/cpmmedium, presented as mean ± standard error of the mean. *P < 0·05 when compared with DC (MUC1-N).
Cytokine production of lymphocytes
Cytokine secretion in lymph node cells from immunized mice was measured at 48 h after MUC1-N restimulation in vitro. As shown in Fig. 5, vaccination of mice with DC (Tat-MUC1-N) enhanced interferon (IFN)-γ (Fig. 5a) and tumour necrosis factor (TNF)-α production (Fig. 5b) in response to MUC1-N antigen compared with DC (MUC1-N)-vaccinated mice (P < 0·05 for IFN-γ and P = 0·08 for TNF-α); cytokines were not induced significantly in the absence of MUC1.
Fig. 5.
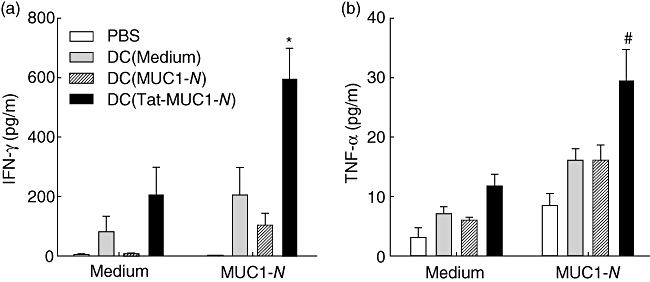
Interferon (IFN)-γ (a) and tumour necrosis factor (TNF)-α (b) secretion of lymph node (LN) cells in response to mucin antigen 1 (MUC1). LN cells were harvested as in Fig. 4 and cultured in the presence [N-terminal region MUC1 (MUC1-N), 50 µg/ml] or absence of MUC1-N proteins (medium) for 48 h. The levels of cytokines in the culture supernatants were measured by cytometric bead array. Each sample was analysed in triplicate. Results are presented as mean ± standard error of the mean. *P < 0·05 or #P = 0·08 when compared with DC (MUC1-N).
When we examined other cytokines, such as the Th2 cytokines IL-4 and IL-5, there were no significant differences between the groups (data not shown). Only basal levels of Th2 cytokines were detected in the immunized and mock-immunized mice. These results indicate that immunization with DC (Tat-MUC1-N) induce MUC1-specific Th1 responses more efficiently compared with DC (MUC1-N) vaccination.
Induction of MUC1-specific cytotoxic splenocytes in vivo
To assess MUC1-specific cytotoxic cells after immunization with DC (Tat-MUC1-N), a P-JAM test was performed [25]. As shown in Fig. 6, splenocytes from mice that were immunized with DC (Tat-MUC1-N) lysed MC57G-pcDNA-MUC1 target cells more efficiently than those from DC (MUC1-N)-immunized mice (P < 0·05). Splenocytes from control DC- or PBS-immunized mice had barely detectable CTL responses against MC57G-pcDNA-MUC1 target cells (Fig. 6a). The cytotoxicity of splenocytes against MUC1-negative MC57G-pcDNA3·1 target cells was not observed in any group (Fig. 6b).
Fig. 6.
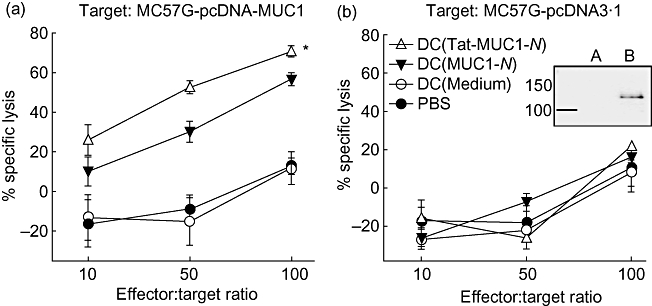
Induction of mucin antigen 1 (MUC1)-specific cytotoxic T lymphocytes (CTLs). Dendritic cells (DCs) (1 × 106 cells) pulsed with either N-terminal region MUC1 (MUC1-N) or Tat-MUC1-N protein were injected twice at weekly intervals into B6 mice (three mice per group). At 7 days after the second immunization, splenocytes were harvested, restimulated with MUC1-N proteins for 3 days and used as effector cells. Effector cells were incubated with MC57G-pcDNA-MUC1 (a) or MC57G-pcDNA3·1 (b). MUC1 expression of target cells was confirmed by Western blot with anti-MUC1 antibody (boxed figure: MC57G-pcDNA3·1 (a) and MC57G-pcDNA-MUC1 (b). After incubation for 4 h, the effector cells and killed target cells were washed off. The target cells that remained in the plates were labelled with [3H]-methylthymidine (5 µCi/ml) for 3 h. [3H]-Methylthymidine uptake was quantified on a microbetacounter. Percentage of specific lysis = [counts per minute (cpm)targets − (cpmtargets+effectors − cpmkillers)]/cpmtargets × 100. Data are presented as mean ± standard error of the mean. *P < 0·05 when compared with DC (MUC1-N).
MUC1 expression in target cells was confirmed by Western blot with anti-MUC1 antibody [Fig. 6b, boxed figure: MC57G-pcDNA3·1(a) and MC57G-pcDNA-MUC1(b)]. These results suggest that immunization of mice with DC (Tat-MUC1-N) induces potent CTL response specifically against MUC1-expressing tumours.
Tumour growth and survival
We examined the anti-tumour immune response that was elicited by vaccination with DC (Tat-MUC1-N) using two different mouse tumour models. First, we inoculated mice with EL4-pcDNA-MUC1 cells and vaccinated them four times with DC vaccines or PBS. MUC1-positive tumours grew steadily in all groups (Fig. 7a). Mice that were immunized with DC (Tat-MUC1-N), however, showed significantly delayed tumour growth compared with mice that were immunized with DC (MUC1-N), controls DCs or PBS (Fig. 7a, P < 0·05).
Fig. 7.
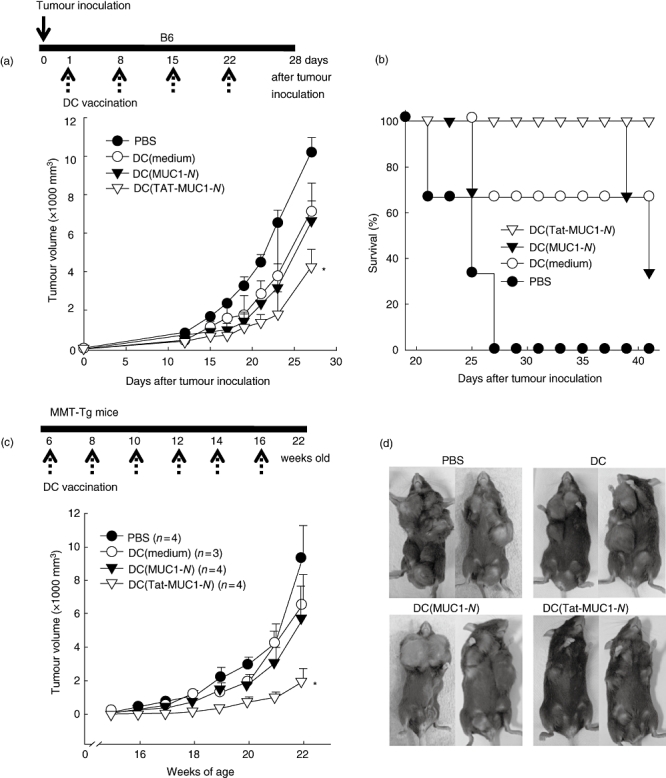
Tumour growth and survival of mice immunized with dendritic cell (DC) vaccines. Tumour growth (a) and survival (b) of B6 mice (three mice per group) were monitored after inoculation with EL4-pcDNA-mucin antigen 1 (MUC1) tumour cells (1 × 105). On the day after tumour cell inoculation, mice were immunized with DC [Tat-N-terminal region MUC1 (MUC1-N), DC (MUC1-N)], control DCs or phosphate-buffered saline (PBS) four times subcutaneously at the base of the tail at weekly intervals as shown in the upper diagram. (c) Spontaneous tumour growth of MUC1/PyMT double transgenic mice after vaccination with DCs. The first vaccine was given to 6-week-old MUC1/PyMT double transgenic mice with 1 × 106 DCs. A total of six vaccinations were given every other week, as shown in diagram. Numbers in parentheses denote number of mice in each group. (d) Representative tumours are shown when the mice were 22 weeks old to compare the volumes and numbers of masses. Data are presented as the mean ± standard error of the mean. *P < 0·05 when compared with DC (MUC1-N).
Furthermore, 100% of DC (Tat-MUC1-N)-immunized mice survived longer than 40 days after tumour inoculation. In contrast, 66% of DC (MUC1-N)-immunized mice and 33% of control DC-immunized mice died 40 days after tumour inoculation. Notably, all mock-immunized mice were dead 27 days after tumour inoculation (Fig. 7b).
In the second model, MUC1/PyMT double transgenic mice (MMT mice), which grow breast tumours spontaneously, were immunized with DCs. We immunized 6-week-old mice and repeated immunization five times at biweekly intervals. All MMT mice that were treated with PBS, control DCs or DC (MUC1-N) developed multiple large mammary tumours at as early as 16 weeks of age. In contrast, immunization of MMT mice with DC (Tat-MUC1-N) delayed significantly the development of spontaneous mammary tumours (Fig. 7c, P < 0·05).
As shown in Fig. 7d, at 22 weeks of age PBS- and control DC-treated mice developed more than six large tumours (2140 ± 355 mm3). DC (MUC1-N)-immunized mice developed two or three large tumours (2047 ± 500 mm3), in addition to two to four small tumours (121 ± 50 mm3). Mice that were vaccinated with DC (Tat-MUC1-N), however, developed only several small tumours (531 ± 187 mm3). At 22 weeks of age, the number of tumours in each mouse that was immunized with DC (Tat-MUC1-N) was 4·3 ± 0·5, which was not significantly different from DC (MUC1-N)-immunized mice (5·3 ± 0·9, P > 0·05). This figure, however, was significantly lower than that of PBS- (6·3 ± 0·3) and DC-immunized (6·3 ± 0·3) mice. These results suggest strongly that DC (Tat-MUC1-N) effect greater immunity against MUC1-positive tumours than DC (MUC1-N) in both the tumour-injection and MMT TG mouse models.
Discussion
Vaccination with MUC1-loaded DCs is becoming a major immunotherapeutic strategy for the treatment of MUC1-positive tumours due to the high immunogenicity of MUC1 [2,3]. Despite the desirable immune responses that are elicited in mice, however, the clinical effects of this approach vary widely depending on the formulation of the DC vaccines, and the clinical responses have not been satisfactory [3,28–32]. Among those trials, the most promising result was reported in a study by Kontani et al.[33], which used DCs pulsed with MUC1 antigen or tumour lysates. After vaccination, the survival of MUC1-positive patients was prolonged significantly compared with MUC1-negative patients (mean survival: 16·75 versus 3·80 months) [33]. In other clinical trials, however, anti-tumour effects have been observed in less than 20% of enrolled patients [3]. None the less, these clinical outcomes clearly support the possible use of DC-based immunotherapy for MUC1-positive cancers.
In considering MUC1 as a target antigen for cancer immunotherapy, several safety concerns of MUC1 and glycosylation-dependent efficacy need to be assessed carefully, because MUC1 is glycosylated aberrantly in malignant cells. First, MUC1 can be deleterious to cells on introduction to patients during immunotherapy. For example, MUC1 can function as an oncoprotein by blocking death receptor-mediated apoptosis [34] and by activating nuclear factor kappa B (NF-κB) signalling, leading to constitutive, enhanced cell growth [35]. In non-malignant epithelial cells, the cytoplasmic domain of MUC1 interacts with caspase-8 and Fas-associated death domain in response to death receptor stimulation [34]. The cytoplasmic domain of MUC1 competes with caspase-8 in binding to Fas-associated death domain protein (FADD) and blocks the recruitment of caspase-8 to the death-inducing signalling complex. For this reason, the cytoplasmic domain of MUC1 must be removed for use in immunotherapy to discard possible oncogenic potential. The overexpression of MUC1 in malignant cells also accelerates tumour progression by enhancing the breakdown of cell–cell and cell–matrix contacts, facilitating migration and metastasis [36]. Aberrantly glycosylated MUC1, which sheds during cancer transformation, can also impair the differentiation and function of DCs by altering the balance in IL-12/IL-10 production [27].
The efficacy of recombinant MUC1 in inducing immunity might be influenced by the extent of glycosylation on MUC1. Heavy glycosylation of MUC1 causes steric hindrance by introducing bulky carbohydrate chains [37,38]. O-linked glycans protect proteins from proteolytic digestion, thereby causing those proteins to be processed ineffectively [22]. Furthermore, their binding to MHC molecules can be influenced by glycans [38]. Similar steric hindrances can develop during complex formation with T cell receptors (TCRs) [38]. The crystal structure of MHC class I/glycopeptide complexes shows that glycans can be accommodated by the TCR [39,40]. Not all peptide-attached glycans, however, can elicit a T cell response [41]. It appears that αβT cells do not recognize large and highly complex glycan structures [38]. The central CDR3 region of the TCR may not accommodate very large glycans, suggesting that epitopes of hyperglycoslyated MUC1 cannot be presented by MHC molecules in healthy cells. Thus, it is not surprising that the natural glycoforms of MUC1 are poor immunogens [23]. Therefore, glycosylated recombinant MUC1 from a eukaryotic expression system might reduce the efficacy of DC-based immunotherapies. The inefficacy of glycosylated MUC1 in preclinical settings also can explain the limited effects of DC-based therapies that use MUC1-positive tumour cell lysates or fusion strategies in cancer patients [3]. In this respect, use of recombinant proteins that are purified from bacteria, and thus are unglycosylated, may be a reasonable approach to enhance the immunogenic potential of DCs.
By using recombinant N-terminal region of MUC1 (amino acids 2–147 that include NTR and one VNTR of MUC1), we expected that a broader range of MUC1 epitopes could be presented by the diverse sets of MHC haplotypes [37] than by using VNTR only, which comprises only tandem 20-amino acids repeats. It has been reported that the NTR region of MUC1 contains CTL epitopes (M1·2) for HLA-A and induces CTL responses as strong as those that are generated against VNTR [18,42]. Several MUC1 epitopes also have been mapped outside the VNTR that comply with the peptide-binding motif for HLA-A 0201 and that form stable MHC-peptide complexes, as assessed by in vitro assays [18]. In A2/Kb transgenic mice, three peptides from the NTR region elicited peptide-specific CTL responses, which protected these mice against challenges with MUC1 A2/Kb-expressing tumour cells. These peptides therefore represent naturally processed MUC1-derived CTL epitopes that map to the NTR region, suggesting that the N-terminal region spanning NTR and VNTR could be better candidates than peptides containing only VNTR for patients whose MHC haplotypes are heterogeneous.
In this study, the conjugation of MUC1-N with HIV Tat, a well-known PTD, enhanced the immunogenicity of MUC1, possibly by facilitating the delivery of the MUC1-N into the cytosol of DCs. When we used Tat-fused carcinoembryonic antigen, the maximum uptake was achieved within 30 min [43]. Furthermore, the maximum uptake of antigens by DCs was greater when Tat-fusion proteins were used compared with unconjugated proteins, even when we incubated DCs with unconjugated antigens for longer periods. We observed diffuse staining of Tat-MUC1-N in the cytoplasm of DCs by confocal microscopy, whereas punctate MUC1-N staining was detected within DCs, suggesting that MUC1-N was delivered by endocytosis. This approach might allow exogenous proteins to be channelled into the MHC class I and class II presentation pathways and to be highly effective in inducing anti-MUC1 responses [7,44]. Indeed, we observed stronger CTL responses, preferential type 1 T cell responses, slower tumour growth and increased survival in mice that were vaccinated with DC (Tat-MUC1-N), compared with those from DC (MUC1-N)-immunized mice. In particular, delayed tumour growth in the transgenic tumour mouse model indicates clearly that DC (Tat-MUC1-N) can break tolerance in vivo and elicit sufficient anti-MUC1 immunity in mice [45]. A more detailed analysis of anti-cancer immune mechanisms by DC (Tat-Muc1-N), however, will improve clinical efficacy. For example, antibody-dependent cell-mediated immunity, immunity by NK cells and altered distribution or functional derepression of regulatory T cells or myeloid-derived suppressor cells need to be tested.
Acknowledgments
This work was supported by a grant of the Korea Healthcare technology R&D Project, the Ministry of Health and Welfare, Republic of Korea (grant A062260) and the National Research Foundation of Korea through the Pioneer Research Center Program funded by the Ministry of Education, Science and Technology (M10711160001-08M1116-00110).
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Hanisch FG. O-glycosylation of the mucin type. Biol Chem. 2001;382:143–9. doi: 10.1515/BC.2001.022. [DOI] [PubMed] [Google Scholar]
- 2.Tang CK, Apostolopoulos V. Strategies used for MUC1 immunotherapy: preclinical studies. Expert Rev Vaccines. 2008;7:951–62. doi: 10.1586/14760584.7.7.951. [DOI] [PubMed] [Google Scholar]
- 3.Tang CK, Katsara M, Apostolopoulos V. Strategies used for MUC1 immunotherapy: human clinical studies. Expert Rev Vaccines. 2008;7:963–75. doi: 10.1586/14760584.7.7.963. [DOI] [PubMed] [Google Scholar]
- 4.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–83. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Palucka AK, Ueno H, Fay JW, Banchereau J. Taming cancer by inducing immunity via dendritic cells. Immunol Rev. 2007;220:129–50. doi: 10.1111/j.1600-065X.2007.00575.x. [DOI] [PubMed] [Google Scholar]
- 6.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 7.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–8. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 9.Shibagaki N, Udey MC. Dendritic cells transduced with protein antigens induce cytotoxic lymphocytes and elicit antitumor immunity. J Immunol. 2002;168:2393–401. doi: 10.4049/jimmunol.168.5.2393. [DOI] [PubMed] [Google Scholar]
- 10.Wang HY, Fu T, Wang G, et al. Induction of CD4(+) T cell-dependent antitumor immunity by TAT-mediated tumor antigen delivery into dendritic cells. J Clin Invest. 2002;109:1463–70. doi: 10.1172/JCI15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viehl CT, Tanaka Y, Chen T, et al. Tat mammaglobin fusion protein transduced dendritic cells stimulate mammaglobin-specific CD4 and CD8 T cells. Breast Cancer Res Treat. 2005;91:271–8. doi: 10.1007/s10549-005-0450-4. [DOI] [PubMed] [Google Scholar]
- 12.Shibagaki N, Udey MC. Dendritic cells transduced with TAT protein transduction domain-containing tyrosinase-related protein 2 vaccinate against murine melanoma. Eur J Immunol. 2003;33:850–60. doi: 10.1002/eji.200323709. [DOI] [PubMed] [Google Scholar]
- 13.Day EB, Zeng W, Doherty PC, Jackson DC, Kedzierska K, Turner SJ. The context of epitope presentation can influence functional quality of recalled influenza A virus-specific memory CD8+ T cells. J Immunol. 2007;179:2187–94. doi: 10.4049/jimmunol.179.4.2187. [DOI] [PubMed] [Google Scholar]
- 14.Ciborowski P, Finn OJ. Non-glycosylated tandem repeats of MUC1 facilitate attachment of breast tumor cells to normal human lung tissue and immobilized extracellular matrix proteins (ECM) in vitro: potential role in metastasis. Clin Exp Metastasis. 2002;19:339–45. doi: 10.1023/a:1015590515957. [DOI] [PubMed] [Google Scholar]
- 15.Barratt-Boyes SM, Vlad A, Finn OJ. Immunization of chimpanzees with tumor antigen MUC1 mucin tandem repeat peptide elicits both helper and cytotoxic T-cell responses. Clin Cancer Res. 1999;5:1918–24. [PubMed] [Google Scholar]
- 16.Yamamoto K, Ueno T, Kawaoka T, et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005;25:3575–9. [PubMed] [Google Scholar]
- 17.Domenech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithelial tumor antigen mucin. J Immunol. 1995;155:4766–74. [PubMed] [Google Scholar]
- 18.Pietersz GA, Li W, Osinski C, Apostolopoulos V, McKenzie IF. Definition of MHC-restricted CTL epitopes from non-variable number of tandem repeat sequence of MUC1. Vaccine. 2000;18:2059–71. doi: 10.1016/s0264-410x(99)00515-0. [DOI] [PubMed] [Google Scholar]
- 19.Kohlgraf KG, Gawron AJ, Higashi M, et al. Tumor-specific immunity in MUC1.Tg mice induced by immunization with peptide vaccines from the cytoplasmic tail of CD227 (MUC1) Cancer Immunol Immunother. 2004;53:1068–84. doi: 10.1007/s00262-004-0557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heukamp LC, van der Burg SH, Drijfhout JW, Melief CJ, Taylor-Papadimitriou J, Offringa R. Identification of three non-VNTR MUC1-derived HLA-A*0201-restricted T-cell epitopes that induce protective anti-tumor immunity in HLA-A2/K(b)-transgenic mice. Int J Cancer. 2001;91:385–92. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1051>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 21.Apostolopoulos V, Pouniotis DS, van Maanen PJ, et al. Delivery of tumor associated antigens to antigen presenting cells using penetratin induces potent immune responses. Vaccine. 2006;24:3191–202. doi: 10.1016/j.vaccine.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Ninkovic T, Hanisch FG. O-glycosylated human MUC1 repeats are processed in vitro by immunoproteasomes. J Immunol. 2007;179:2380–8. doi: 10.4049/jimmunol.179.4.2380. [DOI] [PubMed] [Google Scholar]
- 23.Hiltbold EM, Alter MD, Ciborowski P, Finn OJ. Presentation of MUC1 tumor antigen by class I MHC and CTL function correlate with the glycosylation state of the protein taken up by dendritic cells. Cell Immunol. 1999;194:143–9. doi: 10.1006/cimm.1999.1512. [DOI] [PubMed] [Google Scholar]
- 24.Chen D, Xia J, Tanaka Y, et al. Immunotherapy of spontaneous mammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology. 2003;109:300–7. doi: 10.1046/j.1365-2567.2003.01656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Usharauli D, Perez-Diez A, Matzinger P. The JAM test and its daughter P-JAM: simple tests of DNA fragmentation to measure cell death and stasis. Nat Protoc. 2006;1:672–82. doi: 10.1038/nprot.2006.107. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar K, Bose A, Chakraborty K, et al. Neem leaf glycoprotein helps to generate carcinoembryonic antigen specific anti-tumor immune responses utilizing macrophage-mediated antigen presentation. Vaccine. 2008;26:4352–62. doi: 10.1016/j.vaccine.2008.06.048. [DOI] [PubMed] [Google Scholar]
- 27.Rughetti A, Pellicciotta I, Biffoni M, et al. Recombinant tumor-associated MUC1 glycoprotein impairs the differentiation and function of dendritic cells. J Immunol. 2005;174:7764–72. doi: 10.4049/jimmunol.174.12.7764. [DOI] [PubMed] [Google Scholar]
- 28.Maraskovsky E, Sjolander S, Drane DP, et al. NY-ESO-1 protein formulated in ISCOMATRIX adjuvant is a potent anticancer vaccine inducing both humoral and CD8+ T-cell-mediated immunity and protection against NY-ESO-1+ tumors. Clin Cancer Res. 2004;10:2879–90. doi: 10.1158/1078-0432.ccr-03-0245. [DOI] [PubMed] [Google Scholar]
- 29.Rieser C, Ramoner R, Holtl L, et al. Mature dendritic cells induce T-helper type-1-dominant immune responses in patients with metastatic renal cell carcinoma. Urol Int. 1999;63:151–9. doi: 10.1159/000030438. [DOI] [PubMed] [Google Scholar]
- 30.Morse MA, Nair SK, Mosca PJ, et al. Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Invest. 2003;21:341–9. doi: 10.1081/cnv-120018224. [DOI] [PubMed] [Google Scholar]
- 31.Homma S, Matai K, Irie M, Ohno T, Kufe D, Toda G. Immunotherapy using fusions of autologous dendritic cells and tumor cells showed effective clinical response in a patient with advanced gastric carcinoma. J Gastroenterol. 2003;38:989–94. doi: 10.1007/s00535-002-1183-3. [DOI] [PubMed] [Google Scholar]
- 32.O'Rourke MG, Johnson M, Lanagan C, et al. Durable complete clinical responses in a phase I/II trial using an autologous melanoma cell/dendritic cell vaccine. Cancer Immunol Immunother. 2003;52:387–95. doi: 10.1007/s00262-003-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kontani K, Taguchi O, Ozaki Y, et al. Dendritic cell vaccine immunotherapy of cancer targeting MUC1 mucin. Int J Mol Med. 2003;12:493–502. [PubMed] [Google Scholar]
- 34.Agata N, Ahmad R, Kawano T, Raina D, Kharbanda S, Kufe D. MUC1 oncoprotein blocks death receptor-mediated apoptosis by inhibiting recruitment of caspase-8. Cancer Res. 2008;68:6136–44. doi: 10.1158/0008-5472.CAN-08-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad R, Raina D, Trivedi V, et al. MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat Cell Biol. 2007;9:1419–27. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carraway KL, III, Funes M, Workman HC, Sweeney C. Contribution of membrane mucins to tumor progression through modulation of cellular growth signaling pathways. Curr Top Dev Biol. 2007;78:1–22. doi: 10.1016/S0070-2153(06)78001-2. [DOI] [PubMed] [Google Scholar]
- 37.Hanisch FG. Design of a MUC1-based cancer vaccine. Biochem Soc Trans. 2005;33:705–8. doi: 10.1042/BST0330705. [DOI] [PubMed] [Google Scholar]
- 38.Werdelin O, Meldal M, Jensen T. Processing of glycans on glycoprotein and glycopeptide antigens in antigen-presenting cells. Proc Natl Acad Sci USA. 2002;99:9611–13. doi: 10.1073/pnas.152345899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glithero A, Tormo J, Haurum JS, et al. Crystal structures of two H-2Db/glycopeptide complexes suggest a molecular basis for CTL cross-reactivity. Immunity. 1999;10:63–74. doi: 10.1016/s1074-7613(00)80007-2. [DOI] [PubMed] [Google Scholar]
- 40.Speir JA, Abdel-Motal UM, Jondal M, Wilson IA. Crystal structure of an MHC class I presented glycopeptide that generates carbohydrate-specific CTL. Immunity. 1999;10:51–61. doi: 10.1016/s1074-7613(00)80006-0. [DOI] [PubMed] [Google Scholar]
- 41.Galli-Stampino L, Meinjohanns E, Frische K, et al. T-cell recognition of tumor-associated carbohydrates: the nature of the glycan moiety plays a decisive role in determining glycopeptide immunogenicity. Cancer Res. 1997;57:3214–22. [PubMed] [Google Scholar]
- 42.Heukamp LC, van Hall T, Ossendorp F, et al. Effective immunotherapy of cancer in MUC1-transgenic mice using clonal cytotoxic T lymphocytes directed against an immunodominant MUC1 epitope. J Immunother. 2002;25:46–56. doi: 10.1097/00002371-200201000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Bae MY, Cho NH, Seong SY. Protective anti-tumor immune responses by murine dendritic cells pulsed with recombinant Tat-carcinoembryonic antigen derived from Escherichia coli. Clin Exp Immunol. 2009;157:128–38. doi: 10.1111/j.1365-2249.2009.03943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morse MA, Lyerly HK, Gilboa E, Thomas E, Nair SK. Optimization of the sequence of antigen loading and CD40-ligand-induced maturation of dendritic cells. Cancer Res. 1998;58:2965–8. [PubMed] [Google Scholar]
- 45.Mukherjee P, Madsen CS, Ginardi AR, et al. Mucin 1-specific immunotherapy in a mouse model of spontaneous breast cancer. J Immunother. 2003;26:47–62. doi: 10.1097/00002371-200301000-00006. [DOI] [PubMed] [Google Scholar]