

Abstract
Background
Ventricular hypertrophy is a physiological response to pressure overload that if left untreated can ultimately result in ventricular dysfunction, including diastolic dysfunction. The aim of this study was to test the hypothesis that frequency-dependent myofilament desensitization, a physiological response of healthy myocardium, is altered in hypertrophied myocardium.
Methods and Results
New Zealand White rabbits underwent a pulmonary artery banding (PAB) procedure to induce pressure overload. After 10 weeks the animals were euthanized, hearts removed, and suitable trabeculae harvested from the free wall of the right ventricle. Twitch contractions, calibrated bis-fura-2 calcium transients, and myofilament calcium sensitivity (potassium contractures) were measured at frequencies of 1, 2, 3, and 4 Hz. The force frequency response, relaxation frequency response, and calcium frequency relationships were significantly blunted and diastolic tension significantly increased with frequency in the PAB rabbits compared to sham operated animals. Myofilament calcium sensitivity was virtually identical at 1 Hz in the treatment versus sham group (pCa 6.11±0.03 vs. 6.11±0.06) but the frequency-dependent desensitization that takes place in the sham group (ΔpCa 0.14±0.06, p<0.05) was not observed in the PAB animals (ΔpCa 0.02±0.05). Analysis of myofilament protein phosphorylation revealed that the normally observed frequency-dependent phosphorylation of TnI is lost in PAB rabbits.
Conclusions
The frequency-dependent myofilament desensitization is significantly impaired in right ventricular hypertrophy, and contributes to the frequency-dependent elevation of diastolic tension in hypertrophy.
Keywords: Hypertrophy, Calcium Sensitivity, Heart Rate, EC-coupling, Myofilaments
INTRODUCTION
Ventricular hypertrophy can occur as a result of sustained pressure overload on the ventricles arising from hypertension, valvular stenosis, or ventricular dysfunction. The hypertrophic response is thought to be compensatory at first, but in later stages can result in ventricular dysfunction, and eventually pump failure1. The normal myocardial responses to increases in heart rate can begin to change during the transition from compensatory hypertrophy to decompensation. The force frequency relationship (FFR), normally positive in healthy myocardium, is usually severely blunted or even negative in cases of decompensated hypertrophy, and becomes worse as the heart approaches failure2, 3.
Although it is incompletely understood how the force frequency relationship changes with disease, it is clear that alterations in calcium handling play a major role3–5. The role (if any) the myofilaments play in the contractile dysfunction of decompensating ventricular hypertrophy remains unresolved, in particular as it relates to changes in heart rate. Myofilament calcium sensitivity has been reported to be unaltered in left ventricular myocytes from rapid paced dogs6 and in right ventricular trabeculae of pulmonary artery banded cats4. Other reports have shown myofilament calcium sensitivity to be decreased in spontaneously hypertensive (SHHF) rats7, aortic banded rats8, rats with myocardial infarctions8. In addition, protein kinase C (PKC) dependent myofilament phosphorylations have been implicated in heart failure associated decrease in myofilament calcium sensitivity9. Recently, Lamberts et al. showed that myofilament calcium sensitivity is elevated in rats with right ventricular hypertrophy at low but not high heart rates10. Their study implicated that frequency-dependent desensitization was directly implicated in the blunted FFR. Even now when calcium sensitizing drugs such as levosimendan are being recommended for treatment of cardiac dysfunction,11 the issue of how myofilament calcium sensitivity and its response to frequency change in models of cardiac dysfunction remains unresolved. Understanding how myofilament calcium sensitivity changes with hypertrophy is essential towards understanding cardiac dysfunction and developing better therapies.
In this study, we tested the hypothesis that in hypertrophied myocardium, in which relaxation disorders are known to occur, impaired frequency-dependent myofilament desensitization is a potential contributing factor.
METHODS
All surgical procedures described throughout this publication were completed in accordance with the Institutional Animal Care and Use Committee and National Institutes of Health guidelines.
Pulmonary artery banding (PAB)
Male New Zealand White rabbits (2–3 months old, ∼2 kg weight) were premedicated with Acepromazine (1.25 mg/kg) administered subcutaneously approximately 30 minutes prior to anesthetizing with isoflurane (rate of 2.5–5.0% as needed). Animals received 100% oxygen (rate of 400–600 ml/min) through a loose-fitting mask. Rabbits were then placed in dorsal recumbency and surgical anesthesia was confirmed by the absence of the pedal reflex. Chloramphenicol (30 mg/kg) was administered subcutaneously prior to surgery. After opening of the thorax, the pulmonary artery was constricted to 3.2 mm (outer diameter) using a sterile tube as a gauge at the origin of the vessel. Ligatures were performed using monofilament polypropylene suture. The muscle layers and the skin were sutured closed and after recovery rabbits were given a postoperative dose of buprenorphine (0.01 mg/kg) intramuscularly. Chloramphenicol (30 mg/kg) was given subcutaneously 12 hours from the first preoperative dose and again the following morning. The weights and body temperature of each animal were monitored and recorded 7 days post-op. Sham-operated animals were treated and handled identically, with the sole omission of placing the band around the pulmonary artery. Of 32 animals initially included in the study, 16 were banded and 11 were SHAM operated, while 5 died during the procedure. Post-operative death did not occur in any of the banded or SHAM animals. Because not always a suitable muscle could be obtained from a heart, most experiments were performed with a subset of the total number of animals.
Measurement of twitch contractions, calcium transients, and force-pCa
At 10 weeks post banding (and in a subset of animals at 4 weeks), rabbits were anesthetized with 50 mg/kg pentobarbital and injected with 5,000 units/kg heparin. After bilateral thoracotomy, hearts were rapidly washed with a modified Krebs Henseleit solution with the addition of 2 g/L 2,3-butandione monoxime (BDM) to prevent cutting injury12. From the free right ventricular wall, thin uniform trabeculae were carefully dissected and mounted in the set-up as previously described13–15. Prior to dissection of trabeculae, in a subset of hearts, the total hear, and isolated right and left ventricles were weighed, and samples for protein analysis and for histological analysis were taken. Muscles were stimulated at 2 Hz until force reached a steady state. Auto-fluorescence background was measured at 340 and 380 nm, and trabeculae were iontophorically loaded (at 22 °C) with the calcium indicator bis-fura-2 to a level of 4–8 times background as described previously16. After loading of the dye, the muscle was allowed to twitch-contract at 1 Hz for 20 minutes at 37 °C to allow time for the dye to spread. Twitch contractions and fluorescence emissions at excitation wavelengths of 340 nm and 380 nm were collected at 1, 2, 3, and 4 Hz. Next, the force-calcium relationship was determined while the muscle was contracting at 1 or 4 Hz using potassium induced contractures as described previously13, 14. Briefly, while contracting at 1 or 4 Hz, the superfusion solution was switched from normal Krebs Henseleit solution to a modified version with 110 mM potassium and 40 mM sodium. During the ensuing contracture, calcium enters the myocytes 1000 times slower than during the twitch allowing for a pseudo steady state measurement of myofilament calcium sensitivity. The recorded bis-fura-2 emission signals were calibrated and converted to [Ca2+]i by determining the Rmin and Rmax in each muscle. Force-[Ca2+]i data were plotted and a curve was fit using an iterative fitting procedure based on the Hill-equation. pCa50, maximal developed force (Fmax), and the Hill-coefficient (nHill) were calculated for each curve.
Western Blots
The total tissue homogenate was prepared from freshly-frozen right ventricle of sham and PAB rabbits (taken immediately after washing the heart with the Krebs Henseleit solution with BDM), and western blot analysis was carried out as described earlier17. Briefly, tissue homogenates containing equal amount of proteins were resolved by performing sodium do-decyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on different gel concentrations (10% for Seca2a and CASQ, 9% for NCX-1, 15% for ANP, 12% for TnI, 14% for PLB, Ser16PPLB and Thr17PPLB). Signals were detected by Super Signal WestDura substrate (Pierce) and quantified by densitometry. Although BDM is a reversible myosin ATP-ase inhibitor, it has a phosphatase like activity that may impact phosphorylation status. However, BDM is only applied briefly, and in a previous study we were able to detect changes in PLB-phosphorylation18 after an exposure to BDM longer than used in this study, indicating that results obtained even after brief BDM exposure reflect the prevailing phosphorylation status of PLB.
Phosphoprotein Analysis
Phosphoprotein analysis via the Pro-Q Diamond Stain was performed as described previously13. Briefly, PAB and sham trabeculae twitching at either 1 or 4 Hz were doused with liquid nitrogen until frozen and immediately removed from the experimental set-up. The tissue was then homogenized in an SDS protein lysis buffer, and samples were quantitated using Bradford Protein quantitation assay. Appropriate amounts of each sample were added with 2× SDS-loading buffer to achieve 60ng/ul. Samples were heated to 95 degrees Celsius for 8 minutes and cooled to room temperature. 10 µl of each sample were loaded on a 15% SDS-PAGE gel (1.5 mm thick, 4% stacking) along with two molecular weight standards (Bio-Rad Broad Range Marker 600ng, and 300ng in 1× loading buffer). The gel was run for 53 minutes at 170 Volts. The gel was fixed (50% methanol, 7% acetic acid) overnight. After fixing, the gel was stained with Pro-Q Diamond phosphoprotein stain (Invitrogen) following the manufacturers protocol. After destaining, the gel was equilibrated to water and imaged with the Typhoon variable mode scanner (GE Healthcare) with an excitation wavelength of 532nm (610 BP30 emission filter). The gel was washed in water for 1 hour and stained with SYPRO Ruby total protein stain (Invitrogen) overnight following manufactures protocol. After staining, the gel was imaged with the Typhoon variable mode scanner with an excitation wavelength of 488nm excitation (610 BP30 emission filter). Densitometric analysis was performed on bands corresponding to Troponin I, myosin light chain-2, MyBP-C, Troponin T, and Tropomyosin and the ratio of Pro-Q stain intensity to total protein intensity was calculated.
Statistics
Heart weights, myofilament calcium sensitivity curve parameters, and protein expression level data were analyzed using paired or unpaired t-tests (where applicable) with two-tailed p-value of <0.05 being considered significant. Twitch parameters were analyzed with one-way and two-way (repeated measures where applicable) ANOVA with post hoc t-test. Interaction terms between variables was tested for. Data are depicted as mean ± SEM.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
RESULTS
First, to characterize the model of PA banding, we show in Figure 1A cross-sections of a typical PAB heart and a sham heart. After 10 weeks post-banding, hearts with PAB showed marked right ventricular hypertrophy. The left ventricles appear unaltered from the banding procedure, To quantify the extent of hypertrophy, in a subset of animals, we determined the right and left ventricular weights of PAB rabbits and compared this to SHAM operated rabbits, see table 1. Both the RV weight and RV to heart weight ratio were significantly increased, whereas the left ventricular weight was unaltered. In a separate study, we recently showed19 that in this model the right atrium also hypertrophies, likely as an indication of increase filling pressure. Histological analysis (not shown) on fixed-frozen sections confirmed an increase in myocyte thickness: 16.00 ± 0.27 µm in PAB RV versus 13.91 ± 0.19 µm in the LV of the same animals (99 cells/group, P<0.0001). Figure 1B and 1C show a western blot of right ventricular homogenate of 4 PAB rabbits and 4 sham operated rabbits. The myofilament protein troponin-I (TnI) was used as a marker of hypertrophy. The densitometry analysis of each band showed that, the ratio of TnI/GAPDH, and TnI/calsequestrin was significantly greater for the PAB rabbits compared to the sham-operated rabbits (p<0.01). A further protein analysis was conducted to characterize the expression levels of several key calcium handling proteins as well as ANP, a reliable marker of hypertrophy. In figure 2, we show that both in comparison to the left ventricle of the same hearts and compared to the right ventricle of sham rabbits, serca and phospholamban expression levels are lower, while NCX and ANP levels are increased in the PAB RV myocardium. Phospholamban phosphorylation levels at Ser16 and Thr17 are not different between the groups.
Figure 1.
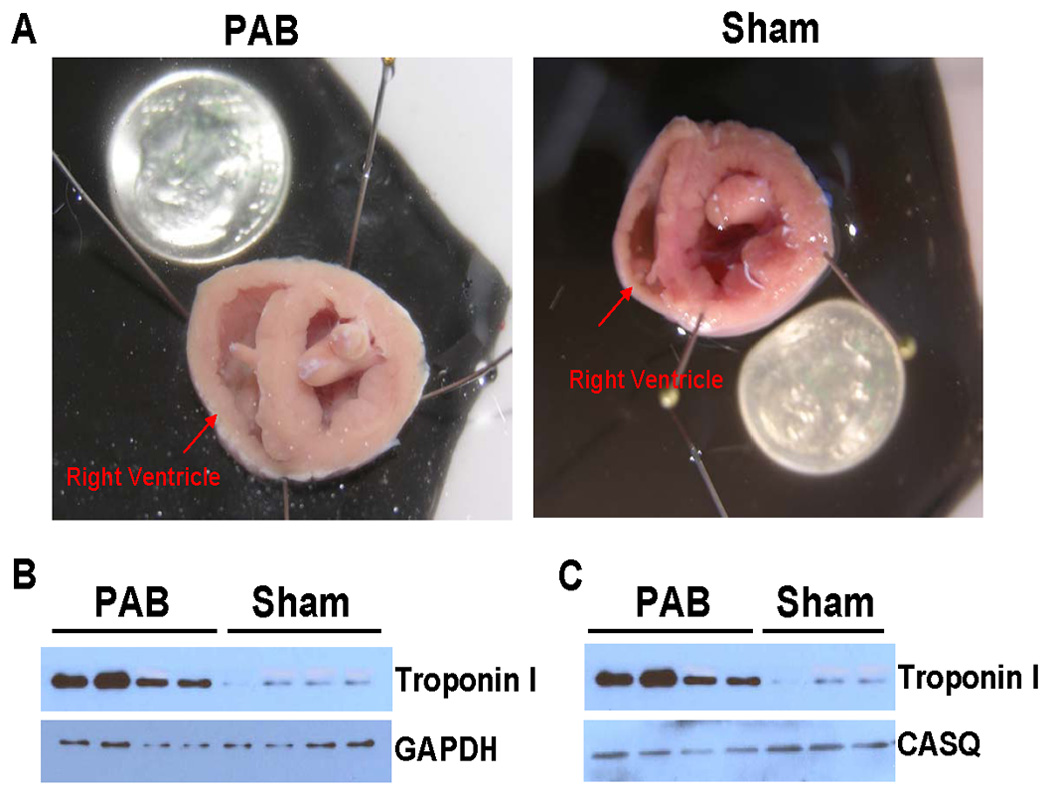
Panel A shows cross sectional slices of hearts (looking towards the apex) from PAB rabbits and sham operated rabbits. The pictures show right ventricular free wall thickening in the PAB heart with little or no change in the left ventricular free wall or interventricular septum. Panel B and C show western blots of homogenized right ventricle from 4 PAB and 4 sham rabbits. Increased TnI levels relative to controls (p<0.01, unpaired t-test) are consistent with the observed anatomical hypertrophy.
Table 1.
Weight comparison of hearts from pulmonary artery banded (PAB) rabbits and sham-operated rabbits. Values are mean ± SEM.
| SHAM | PAB | p-value | ||
|---|---|---|---|---|
| Group size | n=6 | n=9 | ||
| Body weight (BW) | (g) | 3108 ± 148 | 3149 ± 113 | 0.83 |
| Heart weight (HW) | (g) | 8.52 ± 0.53 | 9.45 ± 0.46 | 0.22 |
| HW/BW | (g/kg) | 2.74 ± 0.08 | 3.00 ± 0.10 | 0.08 |
| RV weight | (g) | 1.64 ± 0.09 | 2.50 ± 0.15 | <0.001 |
| LV weight | (g) | 6.16 ± 0.37 | 6.08 ± 0.28 | 0.87 |
| RV/HW | (%) | 19.4 ± 0.7 | 26.4 ± 0.8 | <0.0001 |
Figure 2.
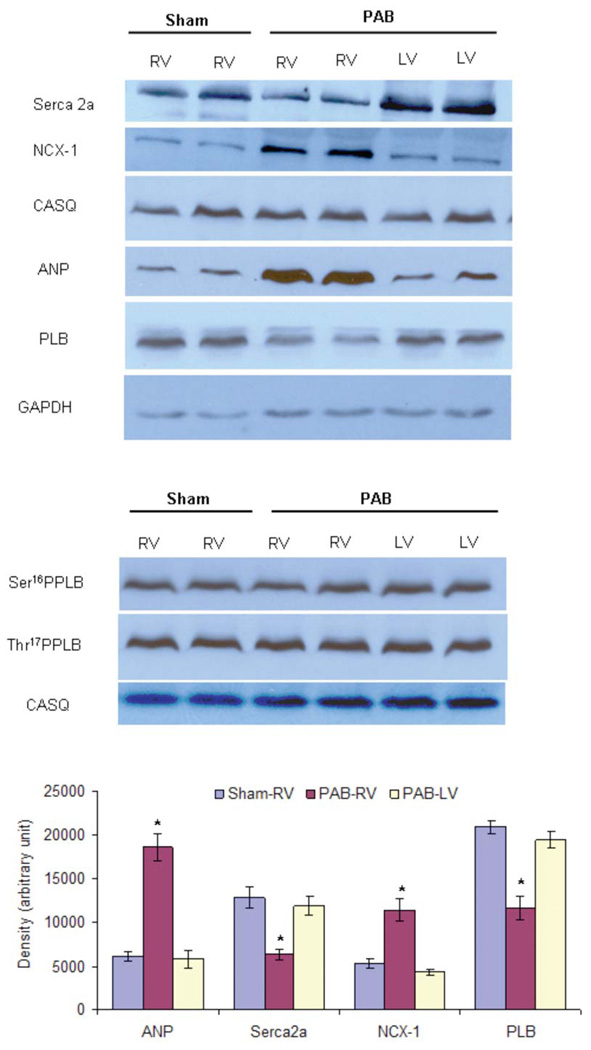
Protein Quantitation in the ventricular tissues of sham-operated and PAB-rabbits. (A, B) Different SDS-PAGE gel concentrations (10% for Seca2a and CASQ, 9% for NCX-1, 15% for ANP, 14% for PLB, Ser16PPLB and Thr17PPLB) were used to resolve total ventricular homogenates and immunoprobed with specific antibodies. A total of 12 µg (panel A) or 20 µg (panel B) of total homogenate was loaded in each well. GAPDH and CASQ were used as loading controls. (C) The expression of SERCA2a and PLB was significantly decreased while expression of ANP and NCX-1 was significantly upregulated in the right ventricle of PAB-rabbits (PAB-RV) in comparison to the left ventricle of PAB-rabbits (PAB-LV) and right ventricle of sham-operated rabbits (Sham-RV). Values given are mean ± SEM from 4 rabbits/group (unpaired t-test). * indicates values significantly (p<0.05) different in comparison to both sham-RV and PAB-LV. PAB, pulmonary artery banding; RV, right ventricle; LV, left ventricle.
In a pilot study, four PAB rabbits were euthanized 4 weeks post surgery to assess right ventricular size and FFR (as a marker of dysfunction). While marked right ventricular hypertrophy was noted as early as 4 weeks post-op (increased wall thickness of the right ventricle), the FFR was still positive through 4 Hz (Supplement figure 1). For the remainder of the study, all animals were kept 10 weeks post surgery to ensure further development of hypertrophy and contractile dysfunction phenotype.
Figure 3 shows representative calcium transients, twitch contractions, and phase plane plots from both sham (A, B, and E) and PAB animals (C, D, and F) 10 weeks post operation. Average twitch contraction and calcium transient data from 11 PAB animals and 9 sham animals 10 weeks post surgery are shown in figure 4. Statistical analysis (two-way repeated measures ANOVA) revealed a significant interaction between the FFR and the two groups indicating the relationship between force and frequency was different between PAB and sham trabeculae. In addition, the 4 Hz group was found to be significantly different from the 1 Hz group in the sham but not in the PAB trabeculae determined by one-way ANOVA (Figure 4A). Analysis of relaxation time (measured as RT50 and RT50 to RT90) revealed different results. The first half of the relaxation curve (RT50) showed normal frequency-dependent acceleration of relaxation (FDAR) in both groups with no significant interaction (Figure 4B). However, the second half of the relaxation curve (RT50 to RT90) displayed significant FDAR in the sham but not the PAB (Figure 4C). Figure 4D shows the resulting effect on diastolic tension as a function of frequency. Diastolic tension significantly decreased with frequency in sham trabeculae. In contrast, diastolic tension in the banded rabbit trabeculae tended to increase or remain unchanged, resulting in a lack of difference in the change of diastolic force from 1–4 Hz. The drop in diastolic force in the sham animals represented an average 20% decrease (one way ANOVA significant difference from 1Hz, p<0.05) while PAB animals exhibited a 4% increase in resting force (not significant). Intracellular calcium measurements revealed that systolic and diastolic calcium rose significantly with frequency within each group (Figure 4E). However, the rise in systolic calcium with frequency was significantly lower in the PAB rabbits compared to sham while the rise in diastolic calcium with frequency was not different between the groups (two way ANOVA). The time of calcium decline from peak to 50% of peak (RT50) accelerated significantly with frequency similarly in sham and PAB trabeculae (Figure 4F).
Figure 3.
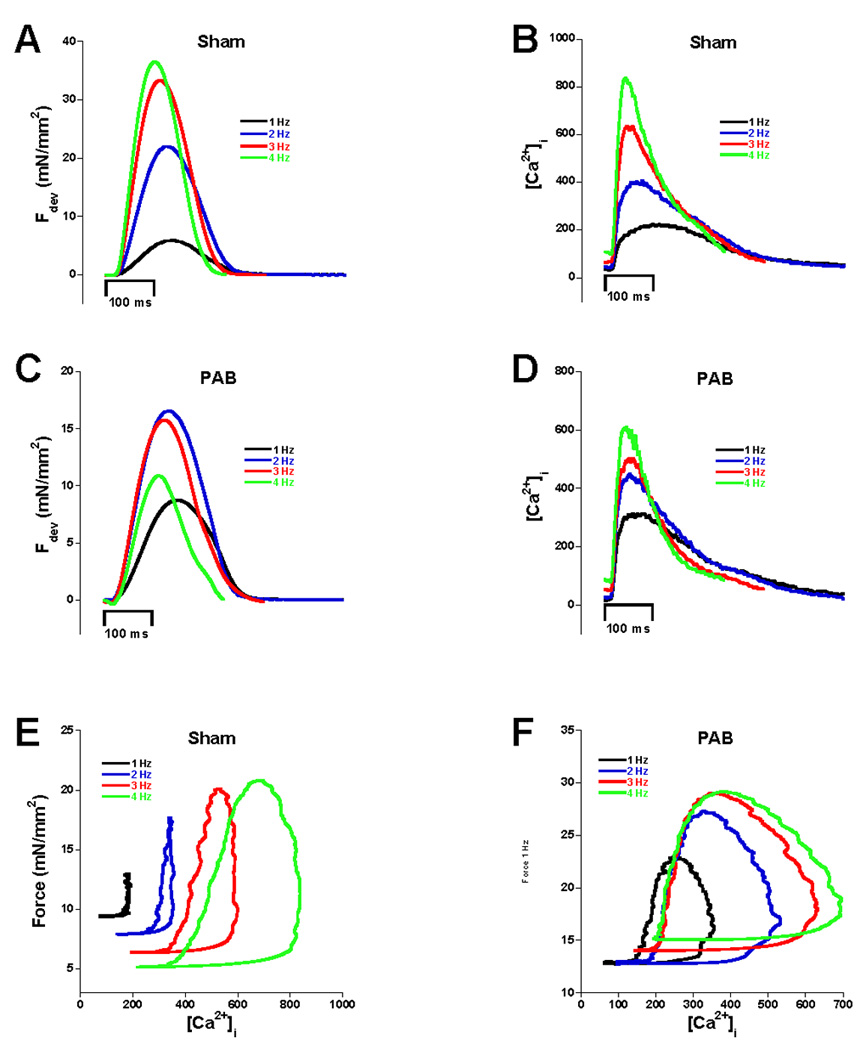
Examples of force twitches and calcium transients. Panels A and B show force tracings and calcium respectively for sham animals. Positive FFR and calcium frequency relationships were observed. PAB animals displayed blunted FFR (C), and a slightly blunted though still positive calcium frequency relationship (D). Phase planes of the relationship of force and calcium during a twitch are shown for sham (E) and PAB (F). Note that the loops shift downward and to the right in the sham example but upward and only slightly to the right in the PAB example.
Figure 4.
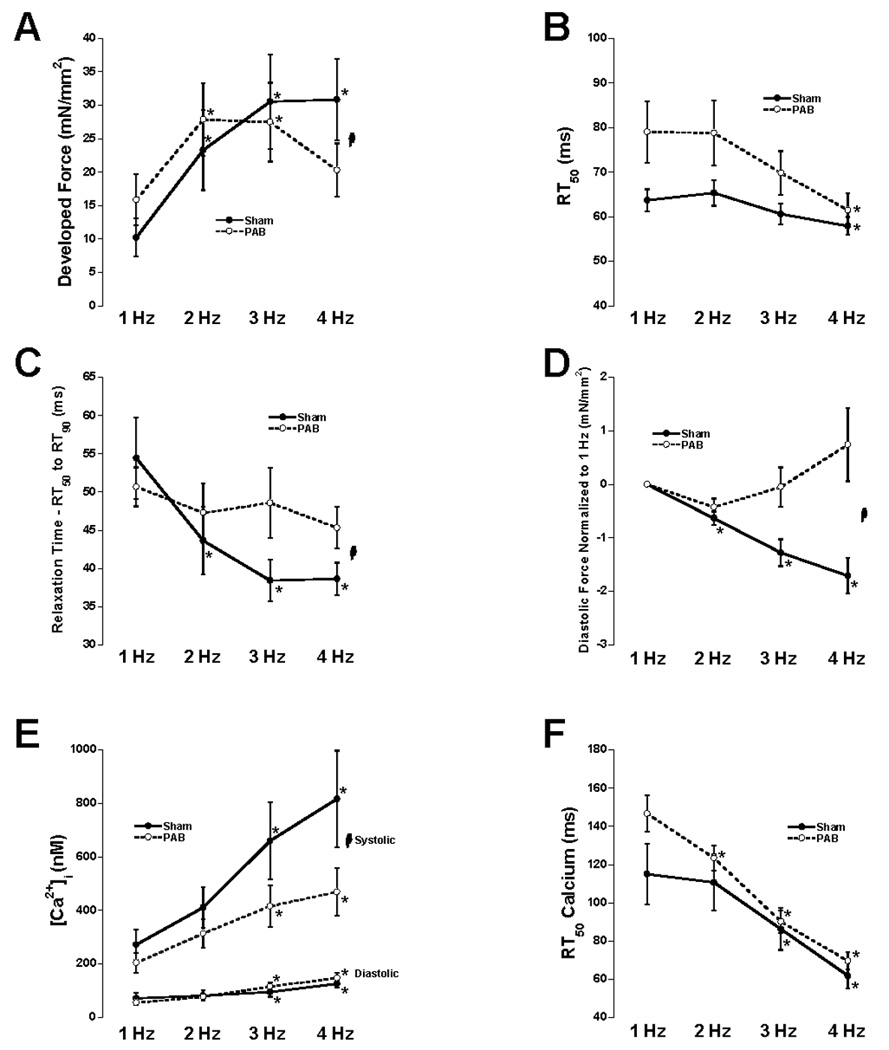
Average data at all 4 frequencies of developed force (A), RT50 of force decline (B), RT50 to RT90 of force decline (C), diastolic tension relative to 1 Hz (D), diastolic and systolic calcium (E), RT50 of intracellular calcium decline (F). Two-way repeated measures ANOVA revealed a significant interaction (indicated with  ) between frequency and the two groups (indicating a difference in the relationship) with respect to developed force, RT50 to RT90, diastolic tension relative to 1 Hz, and systolic calcium. No significant interactions were found between frequency and the two groups with respect to RT50 of force decline, diastolic calcium, or RT50 of calcium decline. Analysis by each group separately by one-way ANOVA revealed a significant difference from 1 Hz in all 7 parameters measured (post-hoc t-test). * denotes significantly different compared to 1 Hz (PAB: n=11, SHAM: n=9).
) between frequency and the two groups (indicating a difference in the relationship) with respect to developed force, RT50 to RT90, diastolic tension relative to 1 Hz, and systolic calcium. No significant interactions were found between frequency and the two groups with respect to RT50 of force decline, diastolic calcium, or RT50 of calcium decline. Analysis by each group separately by one-way ANOVA revealed a significant difference from 1 Hz in all 7 parameters measured (post-hoc t-test). * denotes significantly different compared to 1 Hz (PAB: n=11, SHAM: n=9).
To examine the role of myofilament calcium sensitivity modulations on the observed alterations in relaxation, we employed potassium-induced contractures on twitching sham and PAB trabeculae to construct myofilament calcium sensitivity curves. Figure 5A shows the chart recording of a potassium contracture where force and intracellular calcium were measured simultaneously. The grey area on the graph represents the time where the high potassium solution superfused the trabeculae. Figure 5B and C show representative force-calcium relationships from potassium contractures in sham and PAB animals at 1 and 4 Hz and average pCa50 (Fig 5D) and maximum developed force (Fmax) values (Fig 5E) for all 11 PAB and 9 sham rabbits. We observed a significant decrease in myofilament calcium sensitivity at 4 Hz compared to 1 Hz in the sham animals. This is consistent with previous findings and the decrease in diastolic force in combination with an increase in diastolic calcium. However, the PAB animals exhibited no shift in myofilament calcium sensitivity from 1 to 4 Hz (p>0.5). In addition, we found no significant differences in maximum developed force of any group (Fig 5E) or the nHill coefficient (data not shown).
Figure 5.
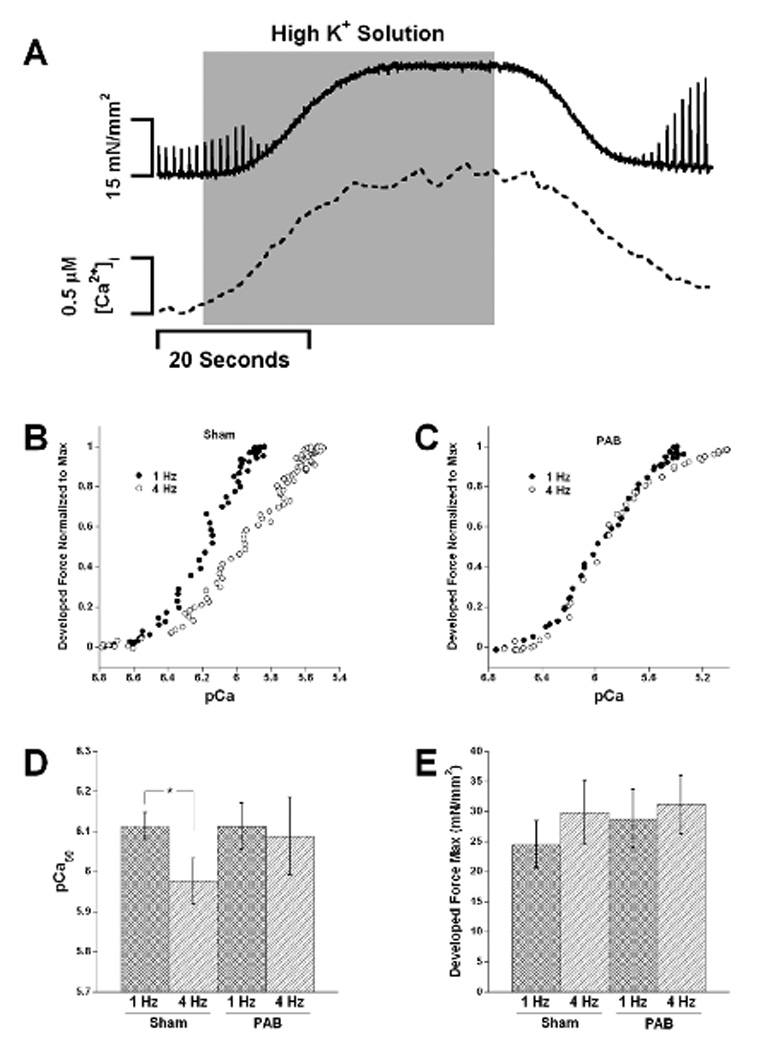
Panel A shows a representative chart recording during a potassium contracture where force and calcium were measured simultaneously. Panels B and C show representative force-calcium relationships (displayed pCa and normalized to max) that were derived from the potassium contractures. We observed a shift in myofilament calcium sensitivity from 1 to 4 Hz in the sham trabeculae and observed a blunted or no shift in sensitivity in the PAB trabeculae. Panel D shows the average pCa50 for 9 sham and 11 PAB animals. The only significant change in pCa50 between frequencies was between 1 and 4 Hz in the sham trabeculae (p<0.05, Two-way repeated measures ANOVA, followed by paired t-test). Panel D shows average max force for all steady state curves at 1 and 4 Hz in sham and PAB trabeculae. No significant difference was found between any of the groups.
To explore a potential correlation between changes in frequency-dependent myofilament desensitization and the changes in mechanical function, we plotted the change in myofilament sensitivity between 1 and 4 Hz versus the change in diastolic tension between 1 and 4 Hz in Figure 6. Despite a considerable spread in both groups, a general trend was observed where muscles exhibiting the greatest change in myofilament calcium sensitivity with frequency (mostly in the sham group) also show the best response in diastolic function (i.e. a decrease in diastolic tension), whereas the muscles with no loss, or even an increase in sensitivity showed a worsening (i.e. increase) in diastolic tension.
Figure 6.
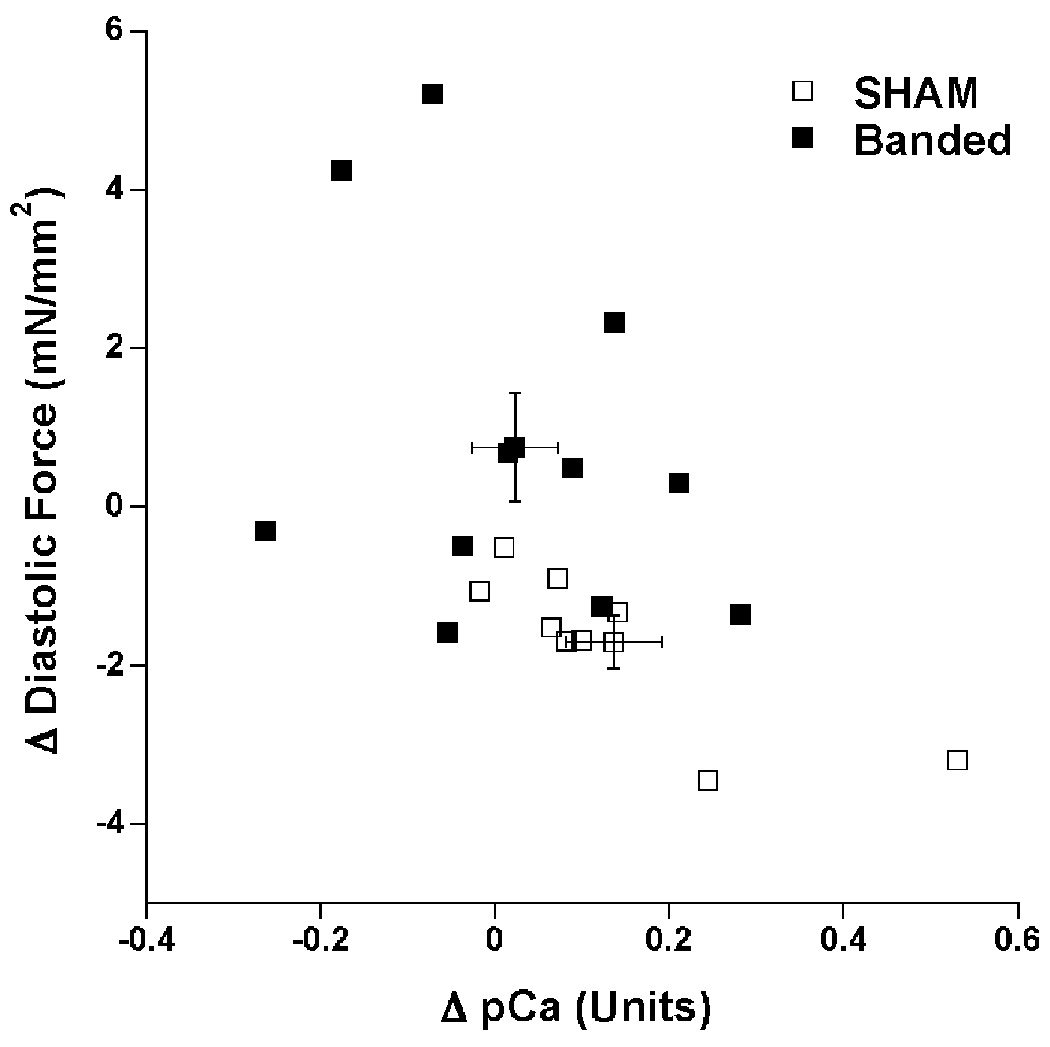
Correlation of change in myofilament calcium sensitivity between 1 and 4 Hz in units of pCa with the change in diastolic tension between 1 and 4 Hz in SHAM and PAB rabbits. Error bars indicate group average values and their standard error. Both the change in diastolic tension and change in pCa were significant between the groups, PAB: n=11, vs. SHAM: n=9, P=0.01 and P=0.02 resp.).
Lastly, we examined whether myofilament protein phosphorylation was altered with frequency. Myofilament protein phosphorylation at TnI and MLC-2 has been previously implicated in shifting myofilament calcium sensitivity13. Accordingly, we examined the phosphorylation status of myofilament proteins TnI, TnT, Tm, MyBP-C, and MLC-2 using the Pro-Q Diamond phosphoprotein stain in trabeculae from sham and PAB hearts twitching at either 1 or 4 Hz. Figure 7 A and B show a gel image stained with Sypro ruby for total protein (Fig 7A) and later with the Pro-Q Diamond stain (Fig 7B). Each gel had at least 1 of the 1 Hz and 2 of the 2 Hz control (SHAM) tissues loaded. For each gel, per protein, the weighed average of these 1 and 4 Hz controls was taken as a value of 1, and the other bands on the same gel were normalized to this value. This normalization now allows density ratios (phosphoprotein/total) for each band to be compared between gels, and lanes on the same gel. We chose this method, as it further reduced noise compared to when data is only normalized to 1 Hz (less bands used in the correction factor leading to less accurate correction factors). In addition, it allows the scaling of all data, and variability of the 1 Hz sham group is so not lost (values would be 1.0 with no error if all is normalized to 1 Hz Sham). Band densitometry revealed that the ratio of phosphoprotein/total protein at the band for TnI was significantly higher at 4 Hz compared to 1 Hz in the sham trabeculae consistent with the frequency dependent myofilament desensitization observed, whereas the change in MyBP-C phosphorylation was borderline significant in the sham trabeculae (P=0.07). In contrast, the PAB trabeculae displayed no difference in phosphorylation status of TnI between 1 and 4 Hz, but the 4 Hz in the PAB group was significantly lower than that of the SHAM group. The bands corresponding with phosphorylation of MLC-2, MyBP-C, TnT, and Tm displayed no significant changes between 1 and 4 Hz in PAB trabeculae.
Figure 7.
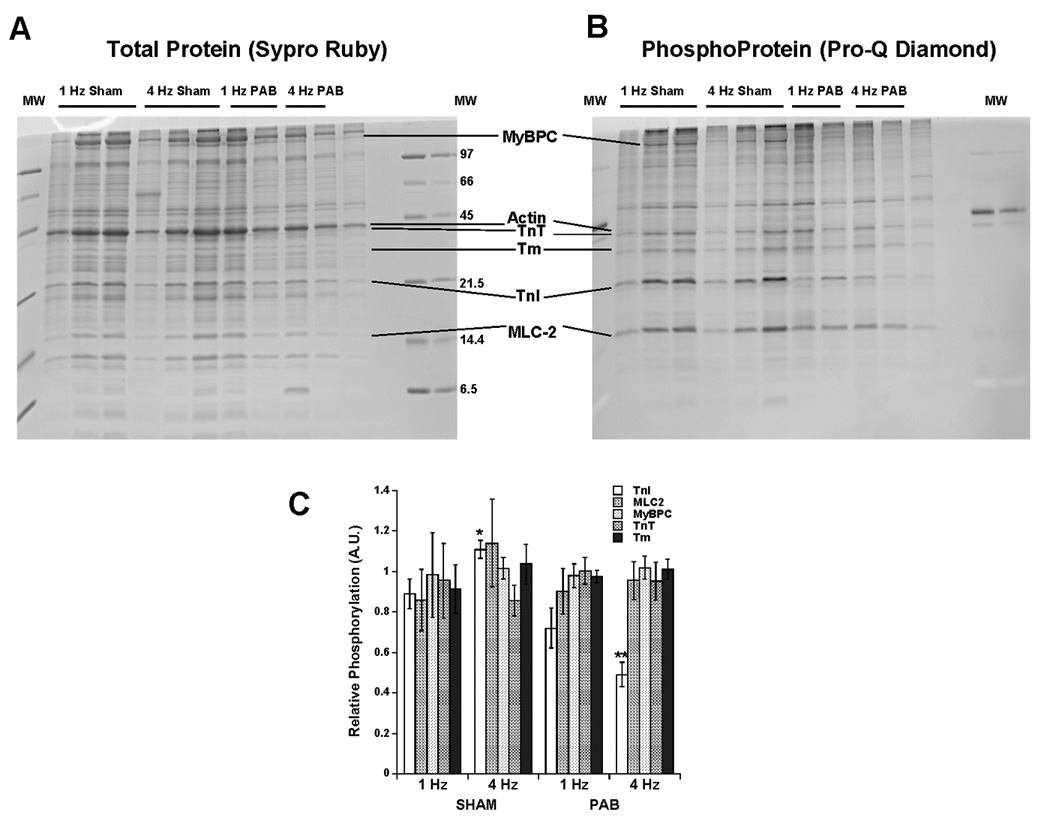
An analysis of myofilament phosphorylation status in sham and PAB trabeculae at 1 and 4 Hz. Panels A and B show a gel stained for total protein (A), and subsequently phosphoprotein (B). Ratio of phosphoprotein/total protein band density is shown in panel C for Troponin-I (TnI), Myosin Light Chain-2 (MLC2), Myosin binding protein-C (MyBPC), Troponin-T (TnT), and Tropomyosin (Tm). A significant increase in TnI phosphorylation was found between 1 and 4 Hz in the sham trabeculae. (n=5–6 muscles/group, un-paired t-test).
DISCUSSION
The key novel finding in this study is the demonstration of a severely blunted or even absent frequency-dependent myofilament calcium desensitization in a model of right ventricular hypertrophy. This finding, in conjunction with the relaxation deficiencies observed at high stimulation rates in the trabeculae isolated from the right ventricles, suggests that frequency-dependent myofilament desensitization may be a mechanism to prevent diastolic dysfunction at higher heart rates, and that this mechanism is impaired in dysfunctional myocardium, contributing to, or even causing diastolic dysfunction.
Pulmonary artery banding (PAB) has been used in adult cats for 11 weeks4, and for 8 weeks in young lambs20 to induce right ventricular hypertrophy. Initially, we assessed contractile parameters in rabbits banded after only 4 weeks post-surgery. We observed a marked hypertrophic response to the banding but at this timepoint did not yet observe a significant deterioration of the FFR. This pilot study indicates that after 4 weeks, the hearts were still in the compensatory hypertrophy stage. We subsequently prolonged the post-op period to 10 weeks, where we found a significantly blunted FFR and calcium frequency response. These assessments suggest that the myocardium had entered the decompensated hypertrophy stage at this point. At this state animals did not show any clinical signs of heart failure (peripheral edema, lethargy, etc.) and thus are not yet in the failing stage, while their right ventricle has increased in weight by 52% on average, with the left ventricular weight being not affected.
Key findings in this study include the blunted frequency-dependent acceleration of relaxation in the late stage of the relaxation trajectory, analogous to increased ventricular filling pressure, as well as a subsequent elevation in diastolic tension with frequency in trabeculae from the PAB group relative to shams. The latter observation is in line with previous findings where PAB in lambs induced diastolic dysfunction measured by an increase in end-diastolic pressure21. Ventricular hypertrophy has also been shown to be a direct precursor to diastolic dysfunction in human patients22. Possible mechanisms proposed for diastolic dysfunction are increased ventricular fibrosis, alterations in myocyte cytoskeleton, slowed myocyte relaxation, and residual myofibrillar cycling23. In this study we showed an elevation in diastolic tension in trabeculae from hypertrophied right ventricles relative to control trabeculae as frequency is increased from 1–4 Hz. Elevation in resting tension with increased frequency can be caused by a number of abnormalities. Slowed relaxation can elevate diastolic force by leaving insufficient time for force decline before the next contraction. Although trabeculae from the PAB animals did not show a difference in FDAR with respect to early relaxation (RT50), there was significant blunting of FDAR in the second half of relaxation (late relaxation: RT50 to RT90). This indicates that the transition from low level contraction to complete relaxation did not accelerated normally with frequency and may contribute (in the whole heart) to inadequate filling at higher heart rates. Elevated diastolic calcium with frequency could also be an effector of elevated resting tension. While diastolic calcium did rise with frequency, it was elevated similarly in sham and banded animals, and therefore is unlikely to be the primary cause of the observed diastolic abnormality. A third possible cause of elevated resting tension is residual cross bridge cycling during rest due to elevated myofilament calcium sensitivity. We have previously shown that myofilament calcium sensitivity decreases with frequency in the healthy myocardium13, while we find here that there is no such change in the trabeculae from PAB rabbits. Frequency-dependent myofilament desensitization appears to be a physiological mechanism to counterbalance sustained elevations in diastolic calcium that occurs in normal myocardium, specifically in larger mammals, when it performs at high pacing rates. The blunting of myofilament desensitization can potentially be seen as a loss of this counterbalance mechanism, allowing for an elevation in diastolic tension with an increase in heart rate. It is also worthy to note that we observed that resting tension decreased with frequency in the sham while there was a slight (but not significant) increase in the PAB. This implies that there may be a small amount of residual cross-bridge binding at the lower frequency in normal myocardium when filling times are sufficient, causing no functional abnormalities. This is consistent with previous findings where left ventricular end-diastolic pressure decreased with an increase in heart rate in humans24. Diastolic calcium rises markedly in rabbits when frequency is increased5, 13, 14 compared to rat where it either does not increase, or only increases by very small amounts10. Therefore, the frequency-dependent myofilament desensitization that occurs in rabbits to potentially counterbalance elevated diastolic calcium levels may not be necessary to prevent diastolic dysfunction in the rat or mouse, and thus alterations in the response may be a phenomenon that is typical of larger mammals. In addition, the force-frequency response in large mammals is much more prominent than in rats and mice, and may contribute to the frequency-dependent differences between models25. Considering the above differences, we believe that these results in intact rabbit trabeculae at body temperature likely closely reflect the human situation, and thus provide a potential novel angle via which to approach potential future therapies for diastolic dysfunction.
Decreased activity of key kinases in hypertrophied myocardium that are not properly activated may be responsible for this lack of myofilament desensitization in PAB myocardium. This is consistent with previous studies that have shown reduced activity of kinases such as PKA and corresponding increases in myofilament calcium sensitivity in myofilaments from failing hearts.26, 27 Our data show that at least one of the molecular events underlying frequency-dependent myofilament calcium desensitization is the phosphorylation of TnI. Although changes in TnI phosphorylation within a group of animals can explain the lack of frequency-dependent changes in sensitivity, we did observed that the absolute values between groups myofilament calcium sensitivity does not directly correspond to TnI phosphorylation level. Thus, it is likely that during the development of hypertrophy, other changes occur that impact on this parameter. In fact, it is still unclear whether in hypertrophied tissue myofilament calcium sensitivity is affected, and if so, in what direction. Previous methods of determining calcium sensitivity often are preformed in demembraned, or so-called “skinned” fiber muscles or myocytes, in which the status of the tissue, including frequency of contraction prior to harvesting of the tissue, may play a big role in the outcome of the data28. For instance, if trabeculae would be skinned and their phosphorylation status preserved after contracting at 1 Hz, it may be that there is no difference between SHAM and PAB when assessed, whereas if they were harvested right after contracting at 4 Hz, it may be determined that myofilament calcium sensitivity is increased. If the tissue would be quiescent for a long while, which often is the case, one could possible extrapolate to find a decreased sensitivity. Thus, frequency of stimulation prior to harvesting muscles/myocytes may play an important role in the variability of the results pertaining to myofilament calcium sensitivity in diseased myocardium.
We used the pulmonary banded rabbit as a model of hypertrophy for a few reasons. The linear trabeculae needed for the in vitro study are mainly found in the RV. In the LV such preparations also exist, but are much more sparse, and generally not quite as linear. However, the hypertrophic response of the RV includes increase in myocyte and wall thickness, a similar loss of functional (flattening or reversing of the normally positive FFR), increase in diastolic tension, and the protein expression pattern is also extremely similar to what is observed in LV hypertrophy/failure, including a downregulation of serca29upregulation of NCX, 30, and upregulation of ANP31. Thus, both anatomical, mechanical, and molecular changes after PAB mimic what is found in LV hypertrophy and failure, and thus the outcome of the results likely reflects similarly the response that would have been observed in left ventricular myocardium.
At this point, we can only speculate on potential therapeutic implications of our findings. Alterations in myofilament calcium sensitivity in hypertrophic ventricular tissue could potentially be an underlying mechanism for the diastolic dysfunction observed in some models of hypertrophy and heart failure. Diastolic dysfunction often follows compensatory hypertrophy as one of the first signs of decompensation1 and increased myofilament calcium sensitivity has been proposed as a possible underlying mechanism. In human heart failure, increased myofilament calcium sensitivity was reported in intact trabeculae32 and skinned myocytes, and was accompanied by elevated diastolic tension26. Increased myofilament calcium sensitivity could potentially have detrimental effects on diastolic filling pressures, especially at high heart rates when diastolic filling time is reduced. Thus, in light of the loss of frequency-dependent myofilament desensitization observed in this study, heart rate control is likely beneficial to avoid diastolic complications. Once in future work the molecular mechanisms via which frequency-dependent myofilament desensitization is mediated are unraveled, they may provide a pharmacological approach to re-establish this frequency-dependent process, thereby allowing the heart to use higher pacing rates without an immediate compromise of diastolic function.
In vivo, changes in heart rate are often achieved through activation of the beta-adrenergic system. Thus, the data observed in vivo is in many cases accompanied by PKA-dependent phosphorylation of proteins including the L-type calcium channel, phospholamban, and Troponin-I. Although it may be interesting to observe the frequency-dependent changes in concert with beta-adrenergic stimulation, it is well known that the response to beta-adrenergic stimulation is altered in cardiac disease, and thus would likely overcomplicate the analysis. In addition, the short experimental span of the preparation in light of assessment of calcium transients at body temperature presented technical limitations to assess both processes in our models at this stage.
In conclusion, we have shown that a myofilament contribution to the force-frequency relationship is altered in dysfunctional hypertrophic myocardium. Blunted frequency-dependent myofilament desensitization may be a prominent, previously unrecognized mechanism that results in diastolic dysfunction especially at higher heart rates, and likely involves an impairment of frequency dependent phosphorylation of TnI.
Clinical Perspective.
Not only does the heart need to beat forcefully to provide adequate blood supply to the organism, it is critically important the myocardium relaxes sufficiently fast to allow for filling with blood to be ejected the next beat. The relaxation of the myocardium, especially at increase heart rates, is a critical regulator of cardiac function. Myocardial relaxation has become increasingly clinically relevant, better diagnostic techniques have elucidated that relaxation disorders, be it called diastolic dysfunction, heart failure with preserved ejection fraction, or relaxation-impaired failure, are highly prevalent in patients suffering from cardiac disease. In this study we show that in the rabbit, who’s EC-coupling and contraction-relaxation coupling are very similar to humans, right ventricular hypertrophy results in an impairment of the myofilaments to de-sensitize to the higher diastolic calcium levels that prevail when heart rate increases in large mammals. If myofilament calcium sensitivity fails to be adjusted at high heart rates, the high diastolic calcium concentration will result in residual myofilament activation, and a slower and incomplete relaxation of the myocardium. This incomplete relaxation in turn results in impaired filling of the ventricle, and can lead to worsening of overall cardiac output. The results of the study further indicate that kinase-mediated processes are likely involved in post-translational modification of myofilament proteins when the heart rate increases. Pharmacological modulation of these pathways, once further identified and unraveled, potentially present us with novel therapeutic options for cardiac disorders where the impairment of myocardial relaxation is prominent.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the surgical staff at The Ohio State University Laboratory Animal Resources, especially Valerie Bergdall, Jeanne Greene, Annemarie Hoffman, Erin Yu, and Daise Da Cunha.
FUNDING SOURCES
This investigation was supported by NIH National Heart, Lung, and Blood Institute grants R01 746387 and KO2 83957 (PMLJ), Established Investigator Award 0740040 of the American Heart Association (PMLJ), and American Heart Association Ohio Valley affiliate predoctoral fellowship 0615288B (KDV).
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Pokharel S, Sharma UC, Pinto YM. Left ventricular hypertrophy: virtuous intentions, malign consequences. Int J Biochem Cell Biol. 2003;35:802–806. doi: 10.1016/s1357-2725(02)00344-8. [DOI] [PubMed] [Google Scholar]
- 2.Rossman EI, Petre RE, Chaudhary KW, Piacentino V, 3rd, Janssen PM, Gaughan JP, Houser SR, Margulies KB. Abnormal frequency-dependent responses represent the pathophysiologic signature of contractile failure in human myocardium. J Mol Cell Cardiol. 2004;36:33–42. doi: 10.1016/j.yjmcc.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 4.Quaile MP, Rossman EI, Berretta RM, Bratinov G, Kubo H, Houser SR, Margulies KB. Reduced sarcoplasmic reticulum Ca(2+) load mediates impaired contractile reserve in right ventricular pressure overload. J Mol Cell Cardiol. 2007;43:552–563. doi: 10.1016/j.yjmcc.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc Res. 2003;58:99–108. doi: 10.1016/s0008-6363(02)00854-4. [DOI] [PubMed] [Google Scholar]
- 6.Kinugawa S, Tsutsui H, Satoh S, Takahashi M, Ide T, IgarashiSaito K, Arimura K, Egashira K, Takeshita A. Role of Ca2+ availability to myofilaments and their sensitivity to Ca2+ in myocyte contractile dysfunction in heart failure. Cardiovasc Res. 1999;44:398–406. doi: 10.1016/s0008-6363(99)00205-9. [DOI] [PubMed] [Google Scholar]
- 7.Perez NG, Hashimoto K, McCune S, Altschuld RA, Marban E. Origin of contractile dysfunction in heart failure: calcium cycling versus myofilaments. Circulation. 1999;99:1077–1083. doi: 10.1161/01.cir.99.8.1077. [DOI] [PubMed] [Google Scholar]
- 8.Belin RJ, Sumandea MP, Kobayashi T, Walker LA, Rundell VL, Urboniene D, Yuzhakova M, Ruch SH, Geenen DL, Solaro RJ, de Tombe PP. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am J Physiol Heart Circ Physiol. 2006;291:H2344–H2353. doi: 10.1152/ajpheart.00541.2006. [DOI] [PubMed] [Google Scholar]
- 9.Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 10.Lamberts RR, Hamdani N, Soekhoe TW, Boontje NM, Zaremba R, Walker LA, de Tombe PP, van der Velden J J, Stienen GJ. Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J Physiol. 2007;582:695–709. doi: 10.1113/jphysiol.2007.134486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koumallos N, Antoniades C, Tousoulis D, Shirodaria C, Stefanadis C. Levosimendan: a novel agent in heart failure. Recent Patents Cardiovasc Drug Discov. 2006;1:185–191. doi: 10.2174/157489006777442487. [DOI] [PubMed] [Google Scholar]
- 12.Mulieri LA, Hasenfuss G, Ittleman F, Blanchard EM, Alpert NR. Protection of human left ventricular myocardium from cutting injury with 2,3-butanedione monoxime. Circ Res. 1989;65:1441–1449. doi: 10.1161/01.res.65.5.1441. [DOI] [PubMed] [Google Scholar]
- 13.Varian KD, Janssen PML. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–H2219. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]
- 14.Varian KD, Raman S, Janssen PML. Measurement of myofilament calcium sensitivity at physiological temperature in intact cardiac trabeculae. Am J Physiol Heart Circ Physiol. 2006;290:H2092–H2097. doi: 10.1152/ajpheart.01241.2005. [DOI] [PubMed] [Google Scholar]
- 15.Janssen PML, Stull LB, Marban E. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am J Physiol Heart Circ Physiol. 2002;282:H499–H507. doi: 10.1152/ajpheart.00595.2001. [DOI] [PubMed] [Google Scholar]
- 16.Backx PH, Ter Keurs HE. Fluorescent properties of rat cardiac trabeculae microinjected with fura-2 salt. Am J Physiol Heart Circ Physiol. 1993;264:H1098–H1110. doi: 10.1152/ajpheart.1993.264.4.H1098. [DOI] [PubMed] [Google Scholar]
- 17.Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, Periasamy M. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci U S A. 2007;104:17867–17872. doi: 10.1073/pnas.0707722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiranandani N, Bupha-Intr T, Janssen PML. SERCA overexpression reduces hydroxyl radical injury in murine myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H3130–H3135. doi: 10.1152/ajpheart.01315.2005. [DOI] [PubMed] [Google Scholar]
- 19.Gupta SC, Varian KD, Bal NC, Abraham JL, Periasamy M, Janssen PML. Pulmonary artery banding alters the expression of Ca2+ transport proteins in the right atrium in rabbits. Am J Physiol Heart Circ Physiol. 2009 doi: 10.1152/ajpheart.00026.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leeuwenburgh BP, Helbing WA, Wenink AC, Steendijk P, de Jong R, Dreef EJ, Gittenberger-de Groot AC, Baan J, van der Laarse A. Chronic right ventricular pressure overload results in a hyperplastic rather than a hypertrophic myocardial response. J Anat. 2008;212:286–294. doi: 10.1111/j.1469-7580.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leeuwenburgh BP, Steendijk P, Helbing WA, Baan J. Indexes of diastolic RV function: load dependence and changes after chronic RV pressure overload in lambs. Am J Physiol Heart Circ Physiol. 2002;282:H1350–H1358. doi: 10.1152/ajpheart.00782.2001. [DOI] [PubMed] [Google Scholar]
- 22.Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis. 2006;48:326–341. doi: 10.1016/j.pcad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Bronzwaer JG, Paulus WJ. Matrix, cytoskeleton, or myofilaments: which one to blame for diastolic left ventricular dysfunction? Prog Cardiovasc Dis. 2005;47:276–284. doi: 10.1016/j.pcad.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Givertz MM, Andreou C, Conrad CH, Colucci WS. Direct myocardial effects of levosimendan in humans with left ventricular dysfunction: alteration of force-frequency and relaxation-frequency relationships. Circulation. 2007;115:1218–1224. doi: 10.1161/CIRCULATIONAHA.106.668640. [DOI] [PubMed] [Google Scholar]
- 25.Janssen PM, Periasamy M. Determinants of frequency-dependent contraction and relaxation of mammalian myocardium. J Mol Cell Cardiol. 2007;43:523–531. doi: 10.1016/j.yjmcc.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 27.van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM, Dekkers DH, Stienen GJ, Lamers JM, Duncker DJ. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 28.Marston SB, de Tombe PP. Troponin phosphorylation and myofilament Ca(2+)-sensitivity in heart failure: Increased or decreased? J Mol Cell Cardiol. 2008;45:603–607. doi: 10.1016/j.yjmcc.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, Prestle J, Minami K, Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 30.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 31.Prestle J, Dieterich S, Preuss M, Bieligk U, Hasenfuss G. Heterogeneous transmural gene expression of calcium-handling proteins and natriuretic peptides in the failing human heart. Cardiovasc Res. 1999;43:323–331. doi: 10.1016/s0008-6363(99)00119-4. [DOI] [PubMed] [Google Scholar]
- 32.Brixius K, Savvidou-Zaroti P, Mehlhorn U, Bloch W, Kranias EG, Schwinger RH. Increased Ca2+-sensitivity of myofibrillar tension in heart failure and its functional implication. Basic Res Cardiol. 2002;97 suppl:I111–I117. doi: 10.1007/s003950200039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.