

1. Introduction
The search for natural products of medicinal significance led the Pettit group to isolate the cephalostatins1 (from the hemichordate worm Cephalodiscus gilchristi,2 e.g. cephalostatin 1 (1), and the Fusetani team to the ritterazines3 (from the tunicate Ritterella tokioka, e.g. ritterazine B (2)), respectively. The cephalostatins and ritterazines, are a family of 45 trisdecacyclic bissteroidal pyrazines that display striking cytotoxicity against human tumors (~1 nM in the 2-day NCI 60 cell panel;4 and in some cases ~10 fM 6-day in the Purdue mini panel5), thereby ranking them among the most potent anticancer agents tested by the NCI.
Computer matching at the NCI using the COMPARE program have revealed several additional compounds exhibiting similar profiles to the Cephalostatin/Ritterazine family. These compounds include OSW-16 (3) a monosteroidal saponin glycoside from the garden perennial Ornithogalum saundersiae (GI50 of 0.8 nM in the NCI 60 cancer cell line), and solamargine7 (4) (from Solanum species) as additional possible candidates for cancer therapy. OSW-1 (3) shows low toxicity to normal human pulmonary cells but encouraging activity against malignant solid tumor cells. Solamargine (4), is an active ingredient of crème Curaderm®, claimed to be 100% effective against melanomas in preliminary clinical trials without significant side effects or recurrence of cancer 10 years after treatment (Figure 1).8
Figure 1.

Steroidal anticancer agents.
Following Pettit's seminal report on cephalostatin 1 (1) in 1988,1 several articles9 have reviewed the structure elucidation, biological activities, and syntheses of cephalostatins. This account will focus on the advances in the syntheses of cephalostatins and ritterazines over the past 15 years (up to ~July 2008) emphasizing the different strategies adopted, key transformations, and methods for achieving the late construction of the dissymmetric bissteroidal pyrazine framework.
Classical steroid numbering (carbons 1-27) and ring designations (A-F) are used throughout the text, supplemented by a “prime” designator for the second steroidal hemisphere (e.g. C21′ = 21′Me of the South hemisphere of cephalostatin 1 (1). Steroidal subunit nomenclature follows published practice, e.g. “North 1” indicates the North10 unit of cephalostatin 1 (1), abbreviated to “1N” especially in analog names or tables (Figure 2). Known stereochemistry is always shown. The somewhat controversial use of solid circles and short dashes to indicate β (up, as drawn) and α (down) hydrogens, respectively, will be retained in the absence of a superior alternative.
Figure 2.

Steroid and bissteroid nomenclature and numbering.
2. Isolation and Biological Activity
2.1. Cephalostatin Family
In 1972, Pettit and coworkers first collected a sample of the marine tubeworm Cephalodiscus gilchristi. Two years later, methanol and water extracts proved active invivo in the National Cancer Institute's PS system (murine lymphocytic leukemia) with a significant lifespan increase in mice.1a In 1988, they were “pleased to report that 15 years of relentless research” had culminated in the structure elucidation of the cephalostatins.
Currently, 19 cephalostatins have been reported (Figure 3). All cephalostatins possess two highly oxygenated steroidal spiroketal units linked by a central pyrazine ring. Cephalostatin 1 (1) is among the most powerful anticancer agents ever tested, displaying subnanomolar to picomolar cytotoxicity against much of the National Cancer Institute's (NCI) 60-cell line panel,3 with femtomolar activity against the P388 cell line and in the Purdue Cell Culture Laboratory (PCCL) human tumor panel.4 Four cephalostatins 3, 4, 8, and 9 were as potent vs. P388 (10-4-10-6 nM) but 4-30 fold weaker in the NCI human tumor panel, while three more cephalostatins 10, 11, and 17 displayed 3-10 nM GI50's in both tests. Cephalostatin 16 displayed a mean GI50 (1 nM, NCI) similar to cephalostatin 1 but a 104-106 weaker ED50 (P388). Cephalostatin 7 was assumed to have activity comparable to cephalostatin 1 based on the fact that it was championed along with cephalostatin 1 for clinical trials and was reported to display a comparable ~femtomolar ED50 (P388) as well as “remarkable potency…against a number of cell lines; the mean graphs of cephalostatin 1 (1) and cephalostatin 7 (5) were remarkably similar, if not indistinguishable” in the NCI panel. The cephalostatin's complex, unprecedented structure and promise as an anticancer lead compound inspired attention by several groups.
Figure 3.

Cephalostatin family.
Clinical trials of a cephalostatin (or analog) will require several grams of material. Pettit's fourth and most prodigious collection afforded only ~0.1 g of cephalostatin 1 (1) from half a ton (450 kg) of this tiny (<5 mm) worm, which hides as colonies in small calcium carbonate sheaths. The harvest involved repeated SCUBA operations at ~25 m depth in waters off East Africa patrolled by the great white shark. The bioassay-guided isolation followed a complex, evolving protocol of extraction (whole worm, several months with aq. MeOH), multiple large scale solvent partitionings, and protracted chromatographic separations. Clearly, chemical synthesis is the only solution to the availability problem.
Early speculation on the mode of action of the cephalostatins centered around; i) the likelihood of cell membrane penetration due to the steroidal nature and dimensions (~30 Å x 9 Å x 5 Å) of cephalostatin 1 (1);11 ii) the possibility that the compounds serve as a spatially-defined set of hydrogen-bond donors/acceptors for enzyme binding,12 and iii) the importance of the Δ14 moiety,13 perhaps due to a chemical role of a derived β-epoxide and the C-ring ketone in the South half of cephalostatin 1 or 7 (Scheme 1).14
Scheme 1.

Possible C/D ring alkylating sites generated from a cephalostatin.
The Purdue group initially speculated that reaction of the C/D homoallylic alcohol array of South 7 generated similar potential alkylating centers. However, the 1997 revelation15 that OSW-1 (3), a monosteroidal glycoside lacking a South unit, displayed a profile and potency similar to cephalostatin 1 against human tumor lines, prompting consideration of an equilibrium between the North spiroketal and its E-ring oxacarbenium ion as a potential alkylating agent (Scheme 2).
Scheme 2.
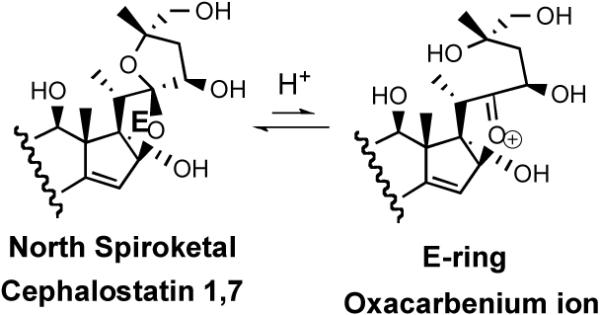
The E-ring oxacarbenium ion.
The antineoplastic mechanism of the cephalostatins is presently largely unknown. The fingerprint of cephalostatin activity in the NCI 60-tumor panel is quite different from known anticancer agents, likely indicating a new mechanism of action. The cephalostatin pattern was most similar to the topoisomerase II inhibitors, but Pettit relates that cephalostatins 1 (1) and 7 (5) are neither topoisomerase inhibitors nor serve as antimicrotuble agents like taxol.16 Studies using synthetic cephalostatin 7 (5) indicate that this compound is not an inhibitor of protein Kinase C nor does it inhibit the tyrosine phosphatase cdc25. A recent biological study17 revealed that cephalostatin 1 affects cells by disrupting the mitochondrial transmembrane potential. Dirsch et al. in collaboration with Pettit documented18 that cephalostatin 1 triggers the release of Smac/DIABLO, a pro-apoptotic mitochondrial signaling factor which induces receptor-independent apoptosis. Müller and coworkers demonstrated16 that cephalostatin 1 inactivate Bcl-2, an anti-apoptotic protein, by activating JNK (c-Jun N-terminal Kinase). In 2006, Vollmar et al. reported19a that cephalostatin 1 utilizes the endoplasmic reticulum stress pathway rather than the intrinsic mitochondrial pathway. Cephalostatin 1 (1) not only induces classical apoptosis parameters (e.g. cell shrinkage, increased cellular granularity, DNA fragmentation, caspase activation) but also shows very unusual apoptosis signaling events (e. g. selective Smac/DIABLO release, no cytochrome c release from mitochondria, and apoptosome-independent activation of caspase-9).18b This unique apoptotic pathway triggered by cephalostatins implies that they could be used to treat drug-resistant cancers.
2.2. Ritterazine Family
During the 1990s, Fusetani's group completed the structure determination of 26 ritterazines2 from extracts of the tunicate Ritterella tokioka collected off the coast of Japan (Figure 4). The ritterazines, found 7000 miles from where the cephalostatins were discovered, are surprisingly similar to the cephalostatins both in structure and bioactivity, again unifying two highly oxygenated steroidal spiroketals by a central pyrazine.
Figure 4.


The ritterazine family.
Isolation of closely related cephalostatins and ritterazines from different phyla raises questions as to the true origin of bissteroidal pyrazines.2 Pettit originally observed that the Cephalodiscus worm is not confined to its coenecium (worm tube) but is independent, able to move in or out of the tube using a sucker-like proboscis, and speculated that exposure to predators during food-harvesting may have necessitated development of the cephalostatins for biological defense.
While the isolation yields of the ritterazines are slightly better than the cephalostatins, they also are too low to supply clinical trials. Ganesan outlined an exciting prospect that has not yet been realized. If the compounds derive from a shared symbiotic microorganism that could be grown in the laboratory, large-scale fermentation might provide much greater quantities of these highly potent agents.8d
This scarcity has been nontrivial to alleviate via synthesis due to the complexity of the steroid substructures, as evidenced by the preparation of cephalostatin 7 (5),20 wherein the 3-ketosteroid South 7 and North 1 precursors required 32 and 33 steps from hecogenin acetate (2 and 3% yields, respectively). Interestingly, several ritterazines, although far less oxygenated, exhibited P388 cytotoxicities approaching the same nanomolar range as some cephalostatins. A COMPARE pattern recognition analysis gave correlation coefficients of ~0.9 between cephalostatins and ritterazines in NCI-10 cell lines, suggesting they share the same mechanism.21 The relative simplicity of ritterazines promises greater synthetic accessibility with probable retention of significant bioactivity.
2.3. OSW-1 and Natural Analogs
The steroidal saponin OSW-1 (3) and its four natural analogs (Figure 5) were isolated by Sashida and his coworkers at Tokyo University from Ornithogalum saundersiae, a perennial cultivated in southern Africa as a cut flower and garden plant.22 These natural products belong to a family of cholestane glycosides. OSW-1 (3) and its analogs i) share the same steroidal unit, namely, 3β,16β,17α-trihydroxycholest-5-en-22-one, ii) have the attachment of a disaccharide to the C-16 position of the steroid aglycone, and iii) have structural variation at the 2′′ position of the disaccharide moiety and the C3 alcohol position of the steroid.
Figure 5.

OSW-1 and its natural congeners.
All five saponins exhibit strong cytotoxicity against leukemia HL-60 cells with IC50 values ranging between 0.1 to 0.3 nM. An in vivo study showed that OSW-1 (3) prolonged the life span of P388 leukemia infected mice by 59% with a single administration at 10 mg/kg. While OSW-1, the major component from the extraction, is exceptionally cytotoxic against various human tumors, it has surprisingly lower toxicity (IC50 1500 nM) to normal human pulmonary cells. The compound was tested in the NCI 60 cancer cell line and showed an average G150 of 0.78 nM. Intriguingly, the cytotoxicity profile of OSW-1, a plant-derived mono steroidal glycoside, is similar to that of cephalostatins, the marine animal-derived bissteroidal pyrazines. COMPARE analysis shows a correlation with cephalostatin 1 (1) of 0.83 for OSW-1,21b suggesting that these two classes might share the same mechanism of action. The Purdue group hypothesized that the C22-oxacarbenium ion, which could be generated from both OSW-1 and cephalostatins, may function as an alkylating agent.23 Loss of the disaccharide, which may be serving as a recognition element or a polarity modifier, from OSW-1 might generate aglycone hemiketal and thence an oxacarbenium ion (Scheme 3).
Scheme 3.
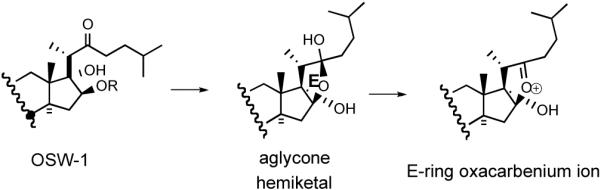
Hypothetical access to an E-ring oxacarbenium ion from OSW-1.
2.4. Solamargine
The solanum alkaloids (Figure 6) have been used for centuries in traditional anticancer folk medicine in China. Cham et al. disclosed24 that solamargine 4 was extraordinarily effective against melanomas in vivo. A crème (called BEC and later Curaderm®) containing solasodine and its dirhamnoglucoside solamargine has demonstrated to be highly efficacious both in mice in the terminal state of murine leukemia and in humans with advanced melanomas,25 with complete remission of the cancers in all human tests (56/56 patients, 181/181 lesions in initial clinical trials). The crème is now being widely tested, especially in Australia.
Figure 6.

Solasodine and the antitumor agent solamargine.
More recently, it has been shown26 that solamargine causes membrane lysis and mitochondria damage and exhibits antiproliferative activity in several cell lines at about 19 µM. Solamargine is now known to trigger apoptosis by up-regulating the expression of external death receptors, such as tumor necrosis factor receptor I and the Fas receptor.27 The rhamnose portion of solamargine has been shown to be a critical recognition element. The susceptible (especially melanoma) cancer cells apparently express a unique endogenous endocytic lectin (EEL) which binds the solamargine molecule prior to membrane penetration. The differential cytotoxicity of solamargine (nontoxic to normal cells both in in vitro tests and when applied to healthy subjects in animal and human trials) may thus be rationalized, since normal mammalian cells do not incorporate rhamnose in glycoconjugates nor express a receptor for such glycals.
In 1996, Kingston et al. reported that steroidal alkaloid solasodine 6, aglycone of solamargine, displayed considerable activity against DNA repair-deficient yeast, and N-acetylation destroyed its DNA alkylating ability.6 Kingston postulated that solasodine acts in a related manner to alkylate DNA via its spiroaminal-derived iminium ion, which is reminiscent of the oxacarbenium ions proposed to account for the cephalostatins/OSW-1 relationship (Figure 6).
2.5. Simple Analogs
Although the biologically hyperactive cephalostatins and ritterazines are asymmetric and structurally complex, some simple symmetrical analogs (Figure 7), exhibited differential cytotoxicity (as well as in vivo anticancer activity in animal trials) for a ras-oncogene transfected cell line. These compounds were tested in mice and found to decrease tumor growth by 50-60%.11 This observation is significant since testing the same compounds in the NCI 60 tumor panel failed to reveal any indication of anticancer activity. Unsymmetrical hydroxyketone 7 showed a low micromolar range of G150 in the NCI 60 cell line panel. Surprisingly, this simple analog displayed the same pattern of bioactivity as cephalostatin 1 (1) suggesting a common mode of action.
Figure 7.

Simple bissteroidal pyrazine analogs.
3. Pyrazine Synthesis
An approach to the cephalostatins must address the central heteroaromatic ring. The dominance of unsymmetrical pyrazines in the cephalostatin family presents a puzzle. Does nature modify symmetrical dimers, couple different subunits, or perform both pre- and post-union modifications? In his seminal contribution describing cephalostatin 1 (1), Pettit hypothesized that the pyrazine core structure was assembled via dimerization and oxidation of steroidal α-amino ketones, a spontaneous reaction in the laboratory.15
The Purdue group outlined two main scenarios distinguished by the timing of the dimerization.28 The first hypothesis posits random coupling at equal rates of previously differentiated North 1 and South 7α-aminoketones to form cephalostatin 7 along with C2 symmetric dimers cephalostatin 12 and ritterazine K. Consonant with the preponderance of the North 1 unit in the cephalostatin branch (present in 18 of 19 cephalostatins) and the 10 fold lower yield of cephalostatin 7 relative to cephalostatin 12, this view requires the presence of North 1 in much greater amounts than South 7. Evidence for trace amounts of ritterazine K (South 7 dimer) was detected among unassigned products from the Cephalodiscus worm, with matching chromatographic properties using synthetic ritterazine K as a guide. The analysis appears to break down with respect to the yield of the cephalostatin 1, which is 100 fold greater than that of cephalostatin 12, the North 1 dimer. Although unnoted in the literature, a similar dilemma attends the South 1 unit (present in 16 of 19 cephalostatins, substantially modified in cephalostatins 5, 6, and 8), but no such South 1 dimer has been reported.
The second biogenetic scenario for cephalostatin 7 (5) projects differentiation of a homodimer, either by C23 monoreduction of cephalostatin 12 or C23 monooxidation of ritterazine K, with subsequent spiroketal isomerization. Ganesan outlined dimerization of a precursor α-amino ketone followed by unselective oxidations to achieve differential functionalization of the steroidal subunits, and cites the similarity between the two halves of cephalostatin 7 and the identification of dimeric cephalostatins 12-13.8c,d It is unclear why the high proportion of South 1/North 1 unions was ignored, although obvious then in 10 of the 17 known members, and despite recognition of the likely derivation of South 5 from South 1. The majority of “South” units in the cephalostatins could be easily derived from South 1 (Figure 8).
Figure 8.

“South 1” similarities among certain cephalostatins.
“Unselective” oxidations in the cephalostatins now appear unlikely. Indeed, modification of a homodimer in this branch would seem to be quite selective. On the other hand, the South 7 type was ubiquitous among ritterazines (13 of 26) but with no majority of any particular union apparent, and four of these (ritterazines J-M) were of high symmetry. Several of the subsequent thirteen ritterazines also possess such symmetry, for a total of nine near or exact homodimers out of twenty-six examples. Additionally, this branch displays consistently lower oxidation levels than the cephalostatins.
Another logical alternative seems worthy of consideration, wherein directed (not random) coupling of at least partially modified units prevails in the worm but not necessarily in the tunicate. Sole responsibility for production of these cytotoxins by putative common and very well-traveled symbiotic microflora25 seems inconsistent with the observed divergence in character of the two pyrazine branches. Perhaps a common organism participates by fusing steroid stocks, which differ between the animals. Whatever the timing, only the “S” type pyrazine has been isolated (Figure 9), consistent with the sole mechanistically possible outcome of reaction between 2-amino-3-ketones. Unfortunately, although several “U” pyrazines have been synthesized, none have been tested for biological activity.30 No analogs featuring alternative fusions (e.g. benzene, pyridine, pyrrole, quinone, dioxane, etc.) have yet been prepared.
Figure 9.

Possible bissteroidal pyrazine geometries.
3.1. Symmetrical Pyrazine Synthesis
Symmetrical steroidal pyrazine synthesis via classical dimerization of an α-aminoketone was first reported in 1968 by Ohta and coworkers (Scheme 4).29 Ohta reduced the 2-oximino-androstan-17β-ol-3-one 8 in alcoholic HCl. Liberation of free amine 9 followed by brief warming in air provided symmetrical pyrazine 10 in fair yield. Smith and Hicks30 partially characterized the intermediate dihydropyrazine and found that catalytic TsOH enhanced air oxidation at ambient temperature.
Scheme 4.

Early approaches for preparation of symmetrical pyrazines.
Disclosure of the cephalostatin structure renewed interest in such pyrazines as evidenced by a report by Pan et al. who developed11 an improved pyrazine synthesis via reductive dimerization of α-azidoketone 11 upon catalytic hydrogenation (Scheme 4). Smith and Heathcock contemporaneously disclosed31 a similar route to α-azidoketone 12 and achieved improved access to pyrazine by a two-step method, Staudinger reaction followed by air oxidation (Scheme 5). Heathcock's second route employed conversion of α-azidoketone 12 to stable α-aminomethoxime 13, which afforded pyrazine 14 in high yield. In 1994 Jeong reported32 synthesis of C2-symmetric cephalostatin analog 17 via tin hydride reduction of α-azidoketone 16 (Scheme 6).
Scheme 5.

Smith/Heathcock routes to symmetrical pyrazines.
Scheme 6.

Jeong synthesis of C2-symmetric cephalostatin North 1 analog.
3.2. Biomimetic Random Coupling of α-Aminoketones
As discussed earlier, the composition of the cephalostatin and ritterazine families implies that nature may utilize random coupling of differentiated steroidal α-aminoketones to produce the bissteroidal pyrazines. Jeong's synthesis prepared unsymmetrical cephalostatin 7 and the dimers of its subunits, cephalostatin 12 and ritterazine K, to explore this `pseudocombinatorial' hypothesis (Scheme 7).19
Scheme 7.

First synthesis of natural bissteroidal pyrazines.
The synthesis featured production of cephalostatins 7 and 12, and ritterazine K in one pot via in situ reduction of α-azidoketones (18 and 19) to α-aminoketones followed by statistical combination of the α-aminoketones. When a 1:1 mixture of the North 1 and South 7 unit was treated with ethanolic NaHTe in the presence of SiO2 and O2, α-aminoketones produced in situ afforded the expected pyrazines. The reaction provided the protected pyrazines cephalostatin 7 (20), cephalostatin 12 (21), and ritterazine K (22) in 35, 14, and 23% isolated yields, respectively. Individual deprotection of pyrazines (20, 21, and 22) with excess TBAF afforded the first synthetic samples of cephalostatin 7 (5), cephalostatin 12, and ritterazine K, respectively.
3.3. Unsymmetrical Pyrazine Synthesis
Earlier cephalostatin studies focused on preparing symmetric pyrazines. However, since most of cephalostatins and ritterazines are unsymmetrical dimers, and symmetrical dimers (e.g. cephalostatin 12, ritterazine K) universally exhibit substantially weaker cytotoxicity, efficient construction of unsymmetrical bissteroidal pyrazines was absolutely essential.
3.3.1 The Heathcock Method
Smith and Heathcock provided the first synthetic route for unsymmetrical bissteroidal pyrazines in 1992 (Scheme 8).30 α-Azidoketone 24 was obtained by α-bromination of 3-cholestanone 23, followed by displacement of the secondary bromide with sodium azide. Treatment of 24 with O-methyl hydroxylamine provided the O-methoxime, which was reduced with triphenylphosphine in aqueous THF to give 2-amino-3-methoxime 25. To prepare a coupling partner, androstanone 26 was converted to enol acetate 27, which was oxidized with dimethyldioxirane to 2β-acetoxy 3-ketone 28. Initial heating at 90 °C of aminomethoxime 25 with 3-ketoacetate 28, followed by heating at 145 °C provided the first unsymmetrical steroidal pyrazine 29, probably via sequential imine formation followed by loss of AcOH and MeOH (Scheme 9).
Scheme 8.

The first unymmetrical pyrazine synthesis by Smith/Heathcock.
Scheme 9.

Proposed mechanism for pyrazine ring formation.
Since the cephalostatins contain spiroketals, the next logical step was to couple a steroid bearing this structural feature. Thus, α-acetoxyketone 30 was prepared by sequential enolate formation, MoOOPh-mediated α-hydroxylation, and acetylation (Scheme 10). Condensation of 30 with α-aminomethoxime 25 under the same conditions furnished spiroketal-containing unsymmetrical pyrazine 31.
Scheme 10.

Coupling of an unsymmetrical spiroketal-bearing steroid.
Although the yield of pyrazine is low, this protocol provided a breakthrough for the construction of unsymmetrical pyrazines, thereby enabling subsequent efforts to target the North and South segments of cephalostatin with expectation of unification late in the synthesis. Indeed, all synthetic trisdecacyclic analogs are prepared by late-stage pyrazine formation.
3.3.2 Winterfeldt Method
With respect to unsymmetrical pyrazine synthesis, the Winterfeldt group pursued both desymmetrization of homodimers and coupling of different steroids. In 1993, they reported33 the first synthetic bissteroidal spiroketal pyrazines containing a Δ14 olefin moiety (Scheme 11). Desymmetrization began by installing the unsaturation into hecogenin acetate 32, in the succinct words of the author, “by a photoprocess.”
Scheme 11.

Winterfeldt desymmetrization of symmetrical pyrazine 34.
Aminoenone 33 was obtained in excellent yield and showed no tendency to thermally dimerize, but Pd-catalyzed hydrogenation afforded pyrazine 34. Exhaustive NaBH4 or L-selectride reductions of 34 furnished symmetrical derivative 35.
Recently, the Winterfeldt group extended the desymmetrization strategy to proximally functionalize C17 via intramolecular alkoxy radical cyclization (Scheme 12).34 After converting diketone 34 to keto-alcohol 36 by NaBH4 reduction, silylation and Wittig olefination, followed by hydroboration/oxidation provided alcohol 37. Exposure of 37 to lead tetraacetate with irradiation furnished a mixture of isomeric tetrahydrofurans 38.
Scheme 12.

Winterfeldt desymmetrization of symmetrical pyrazine 34.
Winterfeldt's 1996 communication 35 disclosed an original protocol for unsymmetrical pyrazine coupling inspired by the thermal stability of α-aminoenones. He reasoned that azirines would be cyclic equivalents of reactive α-aminoketones yet resistant to dimerization, and thus perform well as coupling partners. Stilbene azirine (stilbene/IN3, 0-60°C, 94%) did indeed condense with an α-aminoenone under mild conditions. However, ring-fused azirines proved too strained to isolate, so their generation from steroidal vinyl azides 39 was conducted thermally in the presence of PPTS and aminoenone 33. This strategy successfully furnished pyrazine 40 in 36% yield.
Synthesis of vinyl azide 39 from C3-OH 41 revealed substantial improvements to the Schweng-Zbiral protocols achieved by the German group (Scheme 13). Tosylation of C3-alcohol 41 followed by ALOX B-assisted E2-elimination provided Δ2 olefin 42, which was subjected to sequential treatment with DMDO and Ph3PCl2 to furnish 2β-chloro-3α-alcohol 43. Exposure of 43 to Mitsunobu conditions gave vinyl azide 39 after treatment with KOtBu. The pyrazine synthesis mechanism involves in situ azirine generation via loss of molecular nitrogen, amination of the azirine, imine formation, loss of water, and aromatization (Scheme 14).
Scheme 13.

Pyrazine synthesis via coupling of vinyl azide 39 with αaminoenone 33
Scheme 14.

Proposed mechanism for the pyrazine ring formation
Although Winterfeldt's vinyl azide approach provided unsymmetrical pyrazines, the synthesis suffered from the length of preparing vinyl azide (6 steps from 41) and the low yield in the coupling reaction. The Hanover group later provided a partial remedy to their earlier approach (Scheme 15).36 Hydroxyketone 44 was readily prepared in 3 steps from commercially available hecogenin and coupled with aminoenone 33, pretreated with ammonium acetate to afford asymmetrical bissteroidal pyrazine 46, probably via azapyrylium salt formation and nitrogen incorporation (Scheme 16).
Scheme 15.

Pyrazine synthesis via coupling of hydroxyketone 44 with aminoenone 33.
Scheme 16.

Proposed mechanism for the pyrazine formation
3.3.3. Guo's unsymmetrical pyrazine synthesis
The Purdue group initially employed a biomimetic synthesis of cephalostatin 7, cephalostatin 12, and ritterazine K where the pyrazine ring was constructed by a statistical coupling of North and South α-amino ketosteroids. While the synthesis was informative in probing several biological questions, the strategy adopted was intrinsically incapable of efficiently providing an unsymmetrical coupling product such as cephalostatin 7 (5) since the substantially less active C2-symmetrical pyrazines (cephalostatin 12 and ritterazine K) were also formed. Inspired by Heathcock's concept of using α-amino methoxime as an imine progenitor, which fosters aromatization in the absence of additional oxidation, Guo devised an unsymmetrical pyrazine synthesis via coupling of an α-azidoketone and aminomethoxime in the presence of dibutyltin dichloride (Scheme 17).37 This procedure is milder (80°C, 3-6 h) and better yielding (60-90%, 28 examples) than the Heathcock-Smith protocol, which combined an α-aminomethoxime with an α-acetoxyketone at elevated temperatures (90-140°C) for 2 days with yields of 29 and 43% for the two cases. The seemingly trivial substitution of an α-azidoketone for the α-acetoxyketone led to not only a more efficient preparation of the acceptor (~80%, 2 steps vs. ~40%, 3 steps) but also a probable change of mechanism. The evolution of gas and basic final pH of the reaction medium suggests that the azido moiety may not simply be serving as a leaving group as does the acetate in the Heathcock transformation (Scheme 17).
Scheme 17.

Proposed mechanism for the Guo pyrazine formation
Guo evaluated the scope of the method using donor/acceptor pairs varying in distal functionality to synthesize several simple pyrazines (equimolar partners, ~0.02 M in benzene, 0.1 equiv. Bu2SnCl2, azeotropic removal of water for 7-12 h). Head-to-head comparisons between insoluble acidic and basic additives indicated superior catalysis by Nafion-H, but polyvinylpyridine (PVP) was more routinely utilized since many spiroketals are acid sensitive.
The Guo protocol worked well even for the coupling of highly functionalized steroid spiroketals. For example, South 1 analog 47 and North 1 partner 48 were smoothly united to provide protected dihydrocephalostatin 1 (49) (Scheme 18). The sequence was later used in the synthesis of various natural and unnatural bissteroidal pyrazines, such as cephalostatin 1,38 23′-deoxycephalostatin 1,39 dihydro-ornithostatin O11N, ritterostatins GN1S and GN1N,37 and ritterazine M,40 in good to excellent yields (Figure 10).
Scheme 18.

Guo unsymmetrical pyrazine synthesis.
Figure 10.

Some unsymmetrical bissteroidal pyrazines prepared by Guo coupling.
4. Classical First Generation Syntheses
The two branches of the bissteroidal pyrazine family were isolated from different phyla: from the marine worm Cephalodiscus gilchristi (Hemichordata) in the Indian Ocean and from the tunicate Ritterella tokioka (Chordata) 7000 miles away off the coast of Japan. Surprisingly, they appear closely related, featuring the union of two C27 steroids taken from an array of six major subunits, variously substituted or isomerized. These subunits may themselves be seen as substituted isomers of the abundant plant-derived steroid hecogenin. Cephalostatins 1, 7 and ritterazine G are of particular interest since they feature the four “most active” of the six basic hemispheres common to the entire family (Figure 11).
Figure 11.

Six basic subunits of the cephalostatin family.
Provocatively, the most potent pyrazines of the natural series were seen to utilize only the four basic units North 1, South 1, South 7 and North G. The mild unsymmetrical pyrazine fusions discussed above provided confidence for achieving late-stage coupling of North and South hemispheres derived from 3-ketosteroids. The many unknowns at the time of the first generation cephalostatin syntheses necessitated employing strategies closely based upon steroid degradation of hecogenin acetate to pregnalone for constructing the two different hemispheres required for the late-stage pyrazine formation.
4.1. Synthesis of the C17-Deoxy-C14,15-Dihydro North Cephalostatin 1
Shortly after disclosing the syntheses of several simple, steroid-derived C2 symmetric nonacyclic and trisdecacyclic cephalostatin analogs which possessed modest anticancer activity in animal trials, the Purdue group reported synthesis of the model C17-deoxy-C14,15-dihydro derivative 58 of North unit of cephalostatin 1 (1) and its C2 symmetric dimer by using hecogenin acetate 32 as starting material.31 The 1994 synthesis relied on i) C23 alcohol introduction via TFAA/sulfoxide-mediated allylic oxidation, ii) the establishment of a 5/5 spiroketal through bromoetherification, and iii) stereoselective reduction of the tertiary bromide of 57 with Bu3SnH (Scheme 19).
Scheme 19.

The Jeong strategy for synthesis of C14,15 dihydro, C17 deoxy North 1.
The Jeong synthesis started with the preparation of terminal olefin 51 from hecogenin acetate 32 using the protocol of Micovic and Piatak (Scheme 20).41 Reaction of enol ether 51 with TFAA-activated phenyl methyl sulfoxide afforded the C23 trifluoroacetates 52, which were then hydrolyzed to give a mixture of C23 alcohols 53/54 (23S:23R = 2.2:1). Further supplies of C23R alcohol 54 were secured by Mitsunobu inversion of C23S alcohol 53 using ClCH2CO2H, providing chloroacetate, which was then chemospecifically deacylated42 with thiourea to alcohol 54, which was protected as TBDPS silyl ether 55. Acceptable dihydroxylation stereoselectivity with olefin 55 required doubly-stereoselective stoichiometric osmylation in the presence of (S,S)-Corey ligand to give diastereomeric alcohols 56 in a C25S/C25R ratio of 7.7:1. While formation of a spiroketal from silyl ether diol 56 under acidic conditions was unsuccessful, NBS-mediated cyclization at lower temperature exclusively afforded C20 brominated spiroketal 57, which was then reduced with triphenyltin hydride to give a 4.8:1 mixture of C20α/C20β methyl 58 in essentially quantitative yield. Protection of alcohol 58, hydrolysis of C3 acetate using KHCO3, Brown-Jones oxidation, PTAB-mediated α-bromination, and azide treatment gave α-azidoketone 59. Reduction of 59 with triphenyltin hydride followed by cyclization of the resultant α-aminoketone using PPTS provided trisdecacyclic pyrazine 60, which was then globally deprotected to afford North spiroketal dimer 61. C2 symmetric analog 61 showed far less potency (GI50 = 2.4 μM) than the natural cephalostatins (Scheme 20).43
Scheme 20.

Jeong synthesis of C-17 deoxy C14,15-dihydro North 1 and its dimer.
4.2. Cephalostatin 7, Cephalostatin 12, and Ritterazine K
The Purdue group's biomimetic cephalostatin synthetic strategy27 was based on Pettit's hypothesis1a that the pyrazine core structure was assembled via dimerization and oxidation of steroidal α-aminoketones. The synthesis highlighted a statistical combination of α-aminoenones North 1 and South 7 to concomitantly produce cephalostatins 7 (5) and 12 (62) and ritterazine K (63) in one pot. The key synthetic steps involved; i) transformation of hecogenin acetate 32 to enone 64, ii) pentacyclic dihydrofuranaldehyde 66 formation via rhodium[II] catalyzed intermolecular oxygen alkylation of secondary neopentyl alcohol 65, and iii) subsequent intramolecular Wadsworth-Emmons reaction (Scheme 21). Aldehyde 66 served as a key common intermediate for preparing both hemispheres of the target pyrazines (North 1 (67) and South 7 (68)).
Scheme 21.

Jeong/Guo strategy for the synthesis of cephalostatin 7.
4.2.1. Construction of the North Hemisphere of Cephalostatin 1
The North 1 synthesis44 began with the reduction of hecogenin acetate 32 with DIBAL-H followed by acylation to provide rockogenin diacetate (Scheme 22). Rockogenin diacetate was converted into pseudorockogenin triacetate 69 by pyridinium hydrochloride catalyzed reaction with acetic anhydride. Oxidation of triacetate 69 gave the known ketoester 70, which was then treated with basic alumina to give enone 64 via β-elimination of the pentanoate side chain. Allylic bromination of 64 followed by epoxidation yielded epoxyketone 71. After reacetylation to recover some C3 alcohol that arose in the epoxidation step, bromoepoxide was reductively cleaved with ultrasonicated zinc/copper couple to generate the tertiary allylic alcohol, which was protected as its TMS ether 72. It is interesting to note that no larger silyl ether could be formed, and compound 72 was an easily handled material, presumably due to the crowded nature of its environment. After dihydroxylation of 72, diol 73 was converted to cyclic sulfate 74 via the Sharpless protocol.45 Reaction of sulfate 74 with excess tetrabutylammonium iodide afforded iodo ammonium sulfate 75, which was oxidized with m-CPBA to C14,15 olefin 65 via Reich syn-elimination46 of iodoso intermediate 76. Acidic cleavage of ammonium sulfate 75 to alcohol 65 occurred without any loss of the TMS ether moiety.
Scheme 22.

Preparation of aldehyde 66.
O-H insertion of allylic alcohol 65 with α-diazophosphonate ester using the Moody oxygen alkylation strategy,47 provided phosphonate ester 77 as a 1:1 mixture of diastereomers. Although hotly debated at the planning stage, the key intramolecular Wadsworth-Emmons reaction of 77 took place without difficulty to provide a high yield of complex dihydrofuran-ester 78. Lithium borohydride reduction of 78 afforded a mixture of alcohols that were selectively oxidized to aldehyde 66 with MnO2, although a finishing acetylation was needed to recover some C3 alcohol formed during ester reduction.
Lithium perchlorate mediated reaction of methallyl stannane with aldehyde 66 afforded a 1.3:1 mixture of allylic alcohols 79/80 (Scheme 23). In addition to providing additional supplies of alcohol 80 via Mitsunobu inversion, unnatural diastereomer 79 also served as progenitor of the South hemisphere of cephalostatin 7 via deoxygenation. Dihydroxylation of terminal olefin 80 gave a workable excess of C25S diastereomer, but again required the stoichiometric use of osmium tetroxide in conjunction with the (S,S) Corey ligand.
Scheme 23.

Synthesis of the North hemisphere of cephalostatin 1.
With the inseparable 4:1 mixture of diols 81 in hand, spiroketal ring formation was next surveyed. Once again, direct reaction of the 4:1 diol mixture with a variety of acids was not successful. However, NBS-mediated spirocyclization afforded the C20 brominated 5/5 spiroketal 82 along with diastereomer 83 resulting from cyclization of the minor diol. After protecting the C26 hydroxyl moiety with a TBDMS group, the C3 acetate was cleaved and then subjected to chromic acid oxidation and bis-desilylation with H2SiF6 to provide C17,26-diol 84. The breakthrough to achieve the correct C20 stereochemistry involved conducting the reductive cleavage on the C17 alcohol 84. Inspired by the classic chromium[II]-mediated halohydrin reductions described by Barton,48 bromide 84 was treated with excess CrCl2 in the presence of n-propylmercaptan to selectively deliver reductive cleavage product 86. Completion of the synthesis of the targeted α-azidoketone 67 involved treatment of ketone 86 with phenyltrimethylammonium perbromide (PTAB) to give α-bromoketone, which reacted with tetramethylguanidinium azide (TMGA) to generate North 1 α-azidoketone 67 (Scheme 23). C25-epi North 1 α- azidoketone 85 was also prepared from 83 via a parallel reaction sequence.
4.2.2. The South Unit of Cephalostatin 7
Synthesis of the South hemisphere of cephalostatin 7 exploited the common intermediate 79.49 Deoxygenation was accomplished via the intermediacy of xanthate, via triphenyltin hydride to exclusively provide 87. While osmylation of (R)-configured C23 TBDPS ether 80 resulted in good stereocontrol at C25, C23 unsubstituted substrate 87 suffered poor stereoselectivity (Scheme 24).
Scheme 24.

Synthesis of the South hemisphere of cephalostatin 7.
After a 3-step MTM protection of the C25 tertiary alcohol to avoid 5/5 spiroketal formation, alcohol 88 was subjected to CSA catalyzed cyclization to give three 5/6 spiroketals as an inseparable mixture. Preparation of South 7 α-azidoketone 68 involved pyridine-CrO3 oxidation of C3 alcohol 89, followed by standard treatment of ketone 90 with PTAB and TMGN.47
3
4.3. Cephalostatin 1 and C14′,15′-Dihydrocephalostatin 1
After communicating the synthesis of C14′,15′-dihydro derivative of the South hexacyclic steroid unit of cephalostatin 1 in 1995,13 the Purdue group fully described the synthesis of South 1 (91), and the first total syntheses of cephalostatin 1 and dihydrocephalostatin 1 (92).37 Key transformations included; i) introduction of Δ14 olefin via the Welzel/Prins procedure, ii) methallylation, iii) chemoselective Rh[II] catalyzed intermolecular oxygen alkylation of a primary neopentyl alcohol, iv) intramolecular Wadsworth-Emmons reaction, and v) proximal functionalization of the C-18 methyl group via hypoiodite-mediated alkoxy radical cyclization (Scheme 25).
Scheme 25.

LaCour/Bhandaru strategy for the synthesis of cephalostatin 1.
4.3.1. South Unit of Cephalostatin 1
The South 1 synthesis started with conversion of hecogenin acetate 32 into enone 64 via a modified Dauben protocol (Scheme 26a).50 After ketalization of the C12 carbonyl, enone 64 was stereospecifically reduced to the allylic alcohol, which was then hydrogenated with platinum oxide to give, after ketal deprotection, the saturated alcohol 93. Proximal functionalization of the C18 methyl group of 93 was accomplished via the hypoiodite method of Meystre51 which provided lactone 94 after chromic acid oxidation. Sequential hydrolysis of the C3 acetate group, silylation of the hydroxyl group, and LiAlH4 reduction of the lactone moiety delivered triol 95.
Scheme 26.

Synthesis of the E-ring of dihydrocephalostatin 1.
The key Bhandaru13a transformation employed the unprecedented chemoselective insertion of a diazophosphonate into the primary neopentyl hydroxyl group of triol 95. Slow addition of ethyldiazophosphonate to triol 95 in the presence of catalytic Rh2(OAc)4 regioselectively provided a 1:1 diastereomeric mixture of neopentyl α-alkoxyphosphonoacetates 96 in near-quantitative yield. Brown-Jones oxidation of diol 96 provided diketone 97 as another 1:1 mixture of phosphonate esters. Treatment of the diastereomeric mixture of 97 with sodium hydride effected the intramolecular Wadsworth-Emmons reaction exclusively affording the dihydropyran ester 98. Dihydropyran 98 was reduced by LiAlH4 to a diol mixture which was directly subjected to Swern oxidation generating the key pentacyclic keto-aldehyde 99 (Scheme 26).
Reaction of aldehyde 99 with methallyl stannane in the presence of boron trifluoride etherate quantitatively produced a 1:2.7 mixture of homoallyl alcohols 100/101. Mitsunobu inversion of the undesired isomer 100 afforded additional alcohol 101. After protecting C23 alcohol with a benzyl group, the ketone was reduced with LiAlH4 to provide a diastereomeric mixture (α:β = 1:9 at C12) of diols which were subjected to stoichiometric osmylation. Oxidative cleavage of diols with lead tetracetate gave a mixture (α:β = 1:9 at C12) of keto-alcohols 102. Addition of MeMgBr to C25 ketone 102 resulted in a mixture of diastereomeric diols 103 which were smoothly converted to a mixture of three spiroketals 104/105/106 upon treatment with camphorsulfonic acid. Chromium oxidation followed by acid-catalyzed spiroketal isomerization established the natural C22 stereochemistry. Replacement of the C23 benzyl protecting group with acetate and subsequent removal of TBDPS group with TBAF provided the South hemisphere of dihydrocephalostatin 1 (108) (Scheme 27).
Scheme 27.

Completion of the South 1 hemisphere of dihydrocephalostatin 1.
4.3.2. Cephalostatin 1 and C14',15'-Dihydrocephalostatin 1
In 1996, Guo and Bhandaru36 reported the dihydrocephalostatin 1 (1) synthesis using the Guo unsymmetrical pyrazine coupling protocol (Scheme 27). Alcohol 108 was oxidized to the C3 ketone followed by α-bromination with PTAB and azide substitution to afford α-azido ketone 109. Heating an equimolar mixture of azido ketone 109 and aminomethoxime 110 in the presence of PVP and dibutyltin dichloride with azeotropic distillation provided protected dihydrocephalostatin 1, which was then globally deprotected with TBAF and methanolic K2CO3 to unveil dihydrocephalostatin 1 (111).
The final stage of 1999 cephalostatin 1 synthesis37 involved a crucial three-step Welzel-Prins sequence to introduce the Δ14 olefin moiety present in the South 1 (Scheme 28). Unlike photolysis of hecogenin acetate, the effects of the altered ring strain and steric repulsions on its reactivity during the photolytic opening and acid-catalyzed recyclization steps were nonobvious. Fortunately, photocleavage of ring-strained ketone 108 at 300 nm smoothly provided the desired aldehyde, which was then subjected to Prins reaction cleanly affording the homoallylic alcohol. Subsequent chromic acid oxidation furnished C3,12 diketone 112. Elaboration of ketone 112 to ketone 113 proceeded by the now standard bromination and azide substitution to give azido ketone 113, which was coupled with North 143 (110) to give, after deprotection, the first sample of synthetic cephalostatin 1 (1) (Scheme 28).
Scheme 28.

Synthesis of C14′,15′-dihydro cephalostatin 1 (111) and cephalostatin 1 (1).
5. Second Generation Synthesis
The initial synthetic strategy provided a cumulative total of ~50-300 mg of the key North 1, South 7, and South 1 steroid subunits which permitted exploration of the anticancer structure-activity relationship and completion of a handful of total syntheses. Nevertheless, the first generation approach suffered from the material supply problems associated with any synthesis of ~35 linear steps per subunit (Scheme 29).
Scheme 29.

The first generation “Cut and Paste” synthesis of cephalostatin 1 (1).
The classical synthesis was unattractive at the strategic level, requiring excision of the entire F-ring and subsequent reintroduction of the same atoms (“cut and paste approach”). Clearly, a new synthetic strategy was required to complete the definition of the minimum pharmacophore and provide compounds for clinical trials. The secondgeneration strategy envisaged a highly aggressive plan targeting preparation of both North and South hemispheres of cephalostatin 1 from hecogenin acetate 32 without adding or deleting any carbon atoms. The new approach exploited oxidations, reductions, and spiroketal isomerizations (“Red-Ox” strategy) rather than the degradation/addition sequence used previously (Scheme 30).
Scheme 30.

The second generation “Red-Ox” strategy for synthesis of cephalostatin 1 (1).
5.1. Ritterazine North Hemispheres B, F, G, and H
A 1998 full paper by LaCour et al. detailed the synthesis of the North G hemisphere 114 (Scheme 31).37 Introducing the D-ring olefin at the first stage of this approach was successful but the olefin moiety of 115 was unstable to spiroketal opening. Success was attained by constructing the 5/5 spiroketal ring prior to olefin introduction. Hecogenin acetate 32 was opened to the dichloroacetate, which was subjected to sequential deacylation, tosylation, iodination, and DBU-mediated elimination to provide enolether-olefin 116. Treatment of 116 with hot aqueous acetic acid delivered 5/5 steroidal spiroketal 117 with desired C22S stereochemistry. Spiroketal 117 was photolyzed to secoaldehyde 118, which afforded diol 119 by the Prins reaction. Jones oxidation followed by dehydration of 120 with thionyl chloride generated keto olefin 121. Luche reduction of ketone 121 provided C12 alcohol (12β:12α = 6.5:1), which was further transformed into North G (114) through straightforward functional group manipulations. The North G synthesis was accomplished in 15% yield over 13 operations, substantially better than the syntheses of the highly oxygenated South 7, North 1, and South 1 hemispheres (30-35 operations, ~1%).
Scheme 31.

LaCour North G synthesis.
In 2007 Phillips and Shair reported52 concise synthetic routes to the North hemisphere of ritterazines B, F, G, and H and these syntheses lead to corrections of previously assigned structures of North B and F (Scheme 32). The Shair group's North G synthesis features an early-stage photolysis of the C12-C13 bond and a late-stage spiroketalization by Suárez oxidation. The synthesis began with Winterfeldt's Norrish type I photolysis of hecogenin acetate 32 to form aldehyde 122 which was treated with BF3•OEt2 to stereoselectively give homoallylic alcohol 123. After inversion of the stereochemistry of the C12 alcohol, the 5/6 spiroketal ring was reductively opened and the resulting primary alcohol was converted to a terminal olefin 125 via Grieco's selenylation/oxidation protocol. Oxymercuration-demercuration of the olefin provided tertiary alcohol 126, which was then subjected to Suárez alkoxy radical cyclization to give North G (127). The North G synthesis was, remarkably, accomplished in 31% overall yield over 10 steps from hecogenin acetate 32.
Scheme 32.

Phillips/Shair Ritterazine North B, F, G, and H syntheses.
Shair further manipulated North G (127) to synthesize North B (128), North F (130), and North H (131). North B (128) was prepared in one step from North G (127) by Pt/C-catalyzed hydrogenation (11 steps from hecogenin acetate 32 and 31% overall yield). Interestingly, the hydrogenation took place preferentially from the more hindered β-face of the Δ14 olefin probably via allylic ether-directed hydrogenation. North F (130) was prepared in two steps via Pt/C-catalyzed hydrogenation of North G (127) in acetic acid followed by Suárez oxidation of tertiary alcohol 129 (12 steps from hecogenin acetate 32 and 5% overall yield). North F (130) was further converted into North H (131) via Dess-Martin periodinane oxidation. The Shair group reassigned the spiroketal stereochemistry for North B and North F by comparing 1H NMR chemical shifts for ritterazine B, F, G, and H with those of their synthetic counterparts.
5.2. North M and Ritterazine M
The 2002 Lee ritterazine M synthesis53 depended on Suárez alkoxy radical cyclization to establish the 5/6 spiroketal moiety (Scheme 33). This synthesis enabled correction of the originally assigned stereochemistry at C12, 22, and 25 of the North hemisphere of ritterazine M via comparison of NMR chemical shift differences2c
Scheme 33.

Lee ritterazine M synthesis.
The North M synthesis began with sequential photocleavage of hecogenin acetate 32, Lewis acid-catalyzed ene reaction of aldehyde 122, and was concluded by benzoylation of the homoallylic alcohol. Treatment of 5/6 spiroketal 132 with triethylsilane-BF3•OEt2 stereospecifically provided primary alcohol 133. Conversion of alcohol 133 to the primary iodide, followed by elimination with DBU afforded terminal olefin 134. Catalytic double stereoselective dihydroxylation of the olefin provided a 5.9:1 mixture of inseparable diols 135. Sequential monosilylation of the primary alcohol, benzoylation of the tertiary alcohol with benzoic anhydride and magnesium bromide/triethylamine, followed by removal of TBS protecting group with BF3•OEt2 provided tertiary monoprotected diol 136. Suárez PhI(OAc)2/I2-mediated alkoxy radical cyclization of alcohol 136 provided spiroketals 137 which were hydrolyzed, and then oxidized to the C-3 ketone 138. Lee et al. prepared four other North M spiroketal isomers via similar synthetic routes (not shown) and, based upon NMR difference correlation with the values published by Fusetani, demonstrated that North M possesses C12α-OH, C22α-spiroketal, and C25-axial OH instead of C12β-OH, C22β-spiroketal, and C25-equatorial OH (141 vs. 142). Thus, from hecogenin acetate 32, aminomethoxime North M (139) was prepared in 15% overall yield over 16 steps. The structural assignment was confirmed by providing the first total synthesis of ritterazine M (141) using the standard sequence (Scheme 33).
5.3. North 1 Analogs
Contemporaneously with the Lee ritterazine M synthesis, the Suárez group reported a North 1 analog synthesis featuring their hypoiodite-mediated alkoxy radical cyclization (Scheme 34).54 The synthesis commenced with regioselective C23 oxidation of 3-methoxytigogenin 143 with NaNO2/BF3•OEt2 to give C23-oxotigogenin 144, which was reduced to a mixture of C23 alcohols 145 with L-selectride. Regio- and stereoselective spiroketal ring opening with Ph2SiH2/TiCl4, protection of the resultant primary alcohol with pivaloyl group and secondary alcohol with TBS group, and subsequent removal of pivalate with KOH afforded alcohol 146. Terminal olefin 147 was obtained via nitrophenylselenenylation of primary alcohol 146 followed by H2O2-mediated syn- elimination. Sequential osmylation and acetylation provided tertiary alcohol 148, which was then transformed into a mixture of 5/5 spiroketal 149/150 via the Suarez alkoxy radical cyclization. These analogs are devoid of both the C-12 oxygen functionality and the D-ring olefin present in the natural products.
Scheme 34.

Suárez North 1 analog synthesis.
Shortly after the Suárez report, Lee disclosed55 a more highly functionalized North 1 analog synthesis exploiting the hypoiodite alkoxy radical cyclization to establish the 5/5 spiroketal (Scheme 34). The key transformations featured; i) DMDO-mediated CH oxidation at C16, ii) dehydrative hemiacetal opening with SOCl2/pyr, and iii) C23R alcohol introduction via sequential stereoselective DMDO-mediated epoxidation and regioselective opening of the oxirane.
The analog synthesis began with an improved transformation of hecogenin acetate 32 to β-hydroxyketone 151 in a one-pot 94% yield (cf. 27%).37 Treatment of the 5/6 spiroketal 151 with dimethyldioxirane provided diol 152 in 82% yield. More recently, the inconvenient large-scale DMDO oxidation was avoided by combining the photo/Prins sequence with a ruthenium-catalyzed oxidation that smoothly provided 152 in 88% overall yield on the 100g scale.56 Dehydration of tertiary alcohol 152 with thionyl chloride and pyridine afforded vinyl ether 153, which was then immediately subjected to DMDO oxidation to stereospecifically establish C-23 axial alcohol 155, presumably via the intermediacy of epoxide 154. Treatment of lactol 155 with PhSeH in the presence of boron trifluoride-etherate gave C16-phenylselenide (not shown), which was further reduced with PhSeH with irradiation to give 5/6 spiroketal 156. After unrewarding attempts at C23 alcohol-directed oxygenation at the C-25 position of 155 or 156, Lee returned to the alkoxy radical cyclization strategy used earlier in the ritterazine M synthesis.39
After C-12 reduction and acetylation, the 5/6 spiroketal of 156 was converted into terminal olefin 158 via sequential reductive spiroketal ring opening, iodination, and DBU-mediated elimination. Sharpless asymmetric dihydroxylation of olefin 158 gave C26 acetate 159, presumably via sequential double intramolecular transacylation. Treatment of alcohol 159 with PhI(OAc)2 and I2 induced Suárez alkoxy radical cyclization to preferentially give unnatural 5/5 spiroketal isomer (160/161, unnatural : natural = 12:1). Control experiments revealed that unnatural isomer 160 was the exclusive thermodynamic product (Scheme 35).
Scheme 35.

Lee C17-deoxy North 1 synthesis.
5.4. Interphylal Hybrid Ritterostatins GN1N and GN1S
In 1998 LaCour et al. detailed synthesis of the interphylal hybrids, ritterostatins GN1N and GN1S, where the North G was used as an easily prepared surrogate for the `Southern hemisphere,' to test the hypothesis that mechanism-based biological activity resulted exclusively from the Northern spiroketal and the primary role of the non-polar South spiroketal was for delivery (Scheme 36).37
Scheme 36.

Synthesis of ritterostatins GN1N and GN1S.
After converting ketone 114 (Scheme 31) to azidoketone 162 by the standard procedure, followed by coupling with the North hemisphere of cephalostatin 1 (48) via the Guo protocol, gave the first hybrid ritterostatin GN1N (163) after global deprotection. In a parallel fashion azidoketone 162 was transformed into aminomethoxime 164 and united with the South 1 azidoketone 165 to provide ritterostatin GN1S (166) (Scheme 36).
Testing of the two analogs against natural cephalostatin 1 (1) in the NCI in vitro human cancer cell panel revealed that ritterostatin GN1N displays exceptionally high potency (avg. GI50 12.6 nM). Finding that ritterostatin GN1N retains most of the activity of cephalostatin 1 represents a significant advance, since preparation of 162 requires only a third of the number of steps compared to synthesis of the `real' South 1 hemisphere 113 (Scheme 28), a net 1500% increase in yield. Ritterostatin GN1S, by contrast, was significantly weaker than ritterostatin GN1N (avg. GI50 900 nM), presumably due to lack of a 17-OH group, a feature present in at least one hemisphere of the most active ritterazines and cephalostatins.
In 1999 LaCour et al. reported the preparation of B'/D ring modified analogs starting from 14αH-17-deoxy-North 1 (167) (Scheme 37).42 Desilylation and double Barton deoxygenation gave diacetate 168. Selective hydrolysis of the 3β-acetate followed by Jones oxidation furnished 14-epi-North B as the 3-ketone 169, which was converted to aminomethoxime 170 via standard procedures and then coupled with azidoketone 140 to give 14-epi-7′-deoxyritterazine B (171), after deprotection.
Scheme 37.

LaCour 14-epi-7′-deoxyritterazine B synthesis.
Ritterostatin GN7S, 12′β-hydroxycephalostatin 1,13b and 20- and 25′-epimers57 of cephalostatin 7 were also synthesized via the Guo protocol from the appropriate azidoketones and aminomethoximes followed by standard deprotection (Figure 12).
Figure 12.

Ritterostatin GN7S and B-D ring altered cephalostatin analogs.
6. Third Generation Biomimetic Synthesis
The first generation synthesis of the South 1 subunit employed the traditional Marker spiroketal degradation and a standard Pb-mediated hypoiodite proximal functionalization of the C18 angular methyl group.48 Although this “classical” synthesis provided ~300 mg of South 1, the strategy adopted was far from optimal. Thus, the third generation plan sought to biomimetically synthesize cephalostatins while retaining all 27 carbon atoms present in the hecogenin starting material.
Fusetani proposed58 that biosynthesis of the spiro-C/D junction, which was manifested in thirteen of the twenty-six ritterazines, involved Wagner-Meerwein rearrangement during hydration and oxidation of a hypothetical 22epi-North G (172). Li later proposed that a dyotropic processes, as originally defined by Reetz59 as the “simultaneous” intramolecular migration of two sigma-bound groups, afforded a rationale for biosynthesis of the cephalostatin family (e.g. North I to North D) (Scheme 38).
In 2005, Lee proposed60 biosynthetic pathways for the North 1 and South 7 hemispheres of cephalostatins, which involve; i) electrophilic spiroketal ring opening to form the diene; ii) a [4 + 2] cycloaddition of singlet oxygen; and iii) an acid catalyzed cyclization cascade (Scheme 39).
Scheme 39.

Proposed biosynthetic pathways for South 7 and North 1.
6.1. C23′-deoxy South Unit of Cephalostatin 1
In 2002, Li et al. disclosed38 a biomimetic route to the South 1 hemisphere of cephalostatin 1. The synthesis featured; i) biomimetic proximal functionalization via dyotropic rearrangement, ii) lactone ring opening by SN2′, iii) intramolecular Friedel-Crafts reaction, and iv) transketalizations (Scheme 40).
Scheme 40.

Li biomimetic strategy for the synthesis of C23-deoxy South 1.
The synthesis started with transformation of hecogenin acetate 32 to β-hydroxyketone 151 (Scheme 41).38 Bayer-Villiger oxidation of ketone 151 afforded lactone 174, which was subjected to sequential treatment with catalytic TBSOTf followed by pyridine/SOCl2 to deliver exomethylene spirolactone 177. Interruption of the sequence after rearrangement provided an equilibrium mixture (1:2) of the hydroxy-spirolactones 175 and 176. Elimination of a mixture of these alcohols gave exomethylene spirolactone 177 as a single isomer. Spirolactones 175/176 arose via unprecedented stereospecific dyotropic ring contraction of the seven-membered lactones to their more stable 6-ring counterparts. Smooth SN2′ opening of the spirolactone moiety 177 with formic acid provided an equilibrium mixture (95:5) of allylic formate 178 and starting 177. Polyphosphoric acid trimethylsilyl ester (PPSE) promoted intramolecular Friedel-Crafts acylation of olefin 178 was employed61 to give an intermediate hexacyclic formate, which was deprotected with catalytic bicarbonate to afford alcohol 179. It is noted that the South unit of cephalostatin 8 has the same C18 alcohol, which could undergo transketalization to form E-ring of South 1. The action of warm 75% aqueous AcOH established an equilibrium mixture (1:2.2) of transketalization product 180 and starting material 179.
Scheme 41.

Li synthesis of the C23-deoxy South 1.
Conversion of C26-alcohol 180 to tosylate, iodide substitution, followed by DBU-assisted elimination provided terminal olefin 181, setting the stage for a TMSOTf-mediated rearrangement to transketalized diene 182. Hydrogenation of diene 182 proceeded with reasonable regio- and stereoselectivity to afford 17αH olefin 183 with modest over-reduction. Methanolysis of C3 acetate 183 followed by Jones oxidation gave the C3-ketone. Application of the previously described two-operation method gave azidoketone 184. Guo coupling of azidoketone 184 with the North 1 partner 110 provided masked pyrazine 185, which was globally deprotected to give 23'-deoxy cephalostatin 1 186.
The new South 1 hemisphere synthesis relied on oxidative functionalization of the C18 methyl group via dyotropic rearrangement combined with spiroketal equilibration studies. The synthesis of 23′-deoxy South 1 (183) was accomplished in only 12 operations (23% overall yield) from hecogenin acetate, and also afforded diene 182 in 11 steps (28% overall). The total synthesis of 23′-deoxy cephalostatin 1 (186) was completed in 16 operations from starting material 32 (9% overall; average 86% yield per operation).
Li et al. later reported62 the preparation of C17′-OH-C23′-deoxy cephalostatin 1 starting from diene 182 used in the above synthesis (Scheme 42). Steroidal diene 182 reacted with singlet oxygen to stereospecifically provide [4+2] cycloaddition adduct 187. Reductive cleavage of 187 by treatment with Zn/AcOH gave diol 188 in near quantitative yield. Subjection of alcohol 188 to hydrochloric acid led to syn halohydrin 189, which was exposed to silver oxide to furnish allylic epoxide 190. Oxirane 190 was also obtained directly by regioselective epoxidation of diene 182 with dioxiranes derived from sterically demanding trifluoroacetophenone analogs.63 Regioselective reductive opening of epoxide 190 with DIBAL-H followed by TPAP oxidation afforded diketoalcohol 191.
Scheme 42.

Li synthesis of C17′-OH-C23′-deoxycephalostatin 1.
Compound 191 was converted to azidoketone 192 using standard procedures and then condensed with North 1 coupling partner 110 by the Guo pyrazine protocol to give the protected cephalostatin 1 analog, which was globally deprotected to afford C17′-OHC23′-deoxy cephalostatin 1 (193).
6.2. South Hemisphere of Cephalostatin 7
The 2005 Lee biomimetic South 7 synthesis73 began with preparation of terminal olefin 195 from 5/6 spiroketal 19470, via sequential reductive spiroketal ring opening, iodination, and DBU-mediated elimination (Scheme 43). Treatment of tetrasubstituted tetrahydrofuran 195 with trifluoroacetyltriflate (TFAT)64 in the presence of a hindered base smoothly afforded dienyl trifluoroacteate 196 at -78°C, without affecting the stereochemistry at C20. Removal of the trifluoroacetyl group by mild basic hydrolysis followed by Swern oxidation produced key dienyl ketone 197 in 86% yield over three operations.
Scheme 43.

Lee synthesis of the South 7 hemisphere.
Oxyfunctionalization of D-ring diene 197 again utilized singlet oxygen to give the cycloaddition product in high yield but with no facial selectivity. This selectivity issue was resolved by employing a substrate 198 bearing a C22 propylene glycol ketal. [4 + 2] cycloaddition between D-ring diene 198 and singlet oxygen stereospecifically occurred at -78 °C to furnish only α-face adducts. In stark contrast, the unnatural C-21 β-methyl ketal (not shown) exclusively gave the β-face adduct. This striking reversal of singlet oxygen preference suggests that the stereochemistry of the C-21 methyl moiety determines the facial selectivity of the cycloaddition via conformational control of the sidechain.73,65 Adduct 199 was transformed into differentially protected C-25,26 diol in three operations with a 4.3:1 ratio of C25S:C25R, in favor of the desired stereochemistry. Under the influence of Zn/AcOH, the O-O bond of 199 was reductively cleaved to ketal-diol 200. Treatment of 200 with aqueous DDQ, via slow hydrolytic release of HCN, led to the unexpected formation of hydroxypropyl ether 203, presumably via ketal participation of 201 followed by hydrolysis of intermediate oxonium ion 202. TPAP oxidation of primary alcohol 203 afforded aldehyde 204, which was treated with TBAF to give hemiacetal 205. Acid catalyzed spiroketalization of hemiacetal 205 provided the South 7 hemisphere 206.
The Lee South 7 synthesis paved the way for the multigram synthesis of cephalostatin analogs. The synthesis was completed in 20% overall yield over 16 operations from commercially available hecogenin acetate 32. Compared with the first generation South 7 synthesis (2% overall yield, 25 operations), this synthesis is vastly improved and provides a more practical route for South 7-bearing cephalostatin analogs.
7. Related Syntheses
In 2008, the Taber group reported synthesis of bis-18,18'-desmethyl ritterazine N (227).66 The synthesis involved a key coupling of ABC carbacycle 216 and E-F spiroketal 223, which were prepared from non-steroid starting materials. The ritterazine synthesis began with opening of oxirane 207 with vinylmagnesium chloride to give allylic alcohol 208, which was subjected to sequential allylic oxidation and Diels-Alder cyclization to provide cyclohexene 210. After converting aldehyde 210 to terminal olefin 211 via Wittig olefination, the triene 211 reacted with zirconocene dichloride in the presence of butyl lithium to give a zirconacycle (not shown), which was then treated with carbon monoxide to afford ABC core 212 of ritterazine N. Exposing ketone 212 to mandelic acid and N-bromosuccinimide led to the formation of enantiopure ketone 213 after column chromatography. Saponification of bromomandelate 213 yielded β-epoxide 214, which was inverted by sequential opening of oxirane ring 214 with pmethoxyphenol, mesylation, DDQ oxidation, and base-mediated intramolecular cyclization to give α-epoxide 216.
The construction of spiroketal 222 for ritterazine N started with regioselective opening of oxirane 218 to provide the secondary alcohol 219. After TES ether formation, the nitrile was reduced with DIBAL to furnish aldehyde 220, which was converted to ketone 221 via Grignard addition followed by Dess-Martin oxidation. Treatment of ketone 221 with pPTS removed the both TES protecting groups and the resulting diol (not shown) underwent spiroketal formation to yield the desired E/F-spiroketal 222 as the major product. The TBDPSO group in 222 was converted to triflate 223, which was coupled with ketone 216 to furnish 224 in low yield. Diaxial opening of oxirane 224 with azide delivered C3-alcohol 225, which was transformed into dimer 226 via sequential Dess-Martin oxidation and NaTeH-mediated pyrazine formation. Ozonolysis of alkene 226 followed by base-catalyzed aldol condensation delivered bis-18,18'-desmethyl ritterazine N (227). Although it suffered from low overall reaction yield (0.04%), the Taber synthesis of ritterazine N analog represents the first successful construction of the 6/6/5/5 ring framework present in several ritterazines.
8. Structure Activity Relationships
The forty-five members of the cephalostatin/ritterazine family isolated to date, together with the growing number of analogs (>40) and related monosteroidal antineoplastics (>30), provide the basis for elucidating some structure-activity relationships (SAR) of these potent cytotoxins.
The cephalostatins and ritterazines are bissteroidal pyrazines with pseudo C2-symmetry (see Figure 9). The symmetry arises from the “S” fusion of two C27 steroids, with the 19-Me of each subunit (C19, C19') on the same face of the molecule and each C2-C3 set para to its mate in the pyrazine core. As no variants on this fusion have been tested, the type of attachment required (e.g. rigid, “S”, aromatic or not) is currently unknown. The most active of these pyrazines (≤10 nM) are unsymmetrical, featuring a pair of significantly different steroids taken from the six natural basic subunits (North 1, North A, North G, South 1, South 5, and South 7; see Figure 11).
Consideration of these disparate structures suggests that four features conspire to provide active in vitro materials: (1) a molecular dipole consisting of covalently linked lipophilic “nonpolar” and hydroxylated “polar” domains, with a molecular length of ~30Å; (2) a spiroketal or other latent precursor of an oxacarbenium ion; (3) one or more homoallylic oxygen arrays; and (4) a 17-OH function. The pyrazine ring, though present in most examples, is absent in several subnanomolar active monosteroids. Questions regarding the necessity, location and molecular function for the latter two features remain, but both are present in the most potent natural and analog examples, whereas one or more of these distinctive units are missing in structures with notably inferior in vitro activity including all “simple” cephalostatin analogs and most saponins.
8.1. Appropriate pairing of polar/nonpolar subunits
A covalent union of a polar (hydrophilic) domain with a nonpolar or lipophilic domain appears required, although total polarity may vary widely within certain limits (Figure 13). Tests on free steroids and sugars, alone or together (North 1 & South 7 pentols, the North G diol, solasodine and/or added rhamnose or other sugars, diosgenin, dihydro-OSW-1 aglycone, etc.) show little or no cytotoxicity. Even the best monosteroids (North G aminomethoxime, OSW-1 aglycone-ethylene ketal) are several orders of magnitude less active than cephalostatin 1 (1).
Figure 13.

The polarity groupings of steroidal and glycone subunits.
Solasodine displays a provocative apparent exception to this trend. Although essentially inactive against human cells, it appears quite potent against DNA-repair deficient yeast strains. The activity of this monosteroid is proposed to be related to its spiroaminal function, which can also afford a heterocarbenium ion moiety. These results add weight to the apparent importance of such pro-oxacarbenium sites in steroidal antineoplastics.
All symmetric bissteroidal pyrazines display inferior cytotoxicity (102 to 106 nM). The activity of symmetrical (polar/polar) natural ritterazine K approaches that of unsymmetrical (polar/polar) cephalostatin 7, underscoring the need for pairing of subunits with quite disparate polarities.
Decreased activity associated with a diminished “molecular dipole” is evident with increased polarity in the lipophilic domain (ritterazines D/A and I to ritterazines F/B), or by decreased polarity in the hydrophilic domain, (cf. e.g. ritterazines Y to B or T to A).
Such decreased polarity in the hydrophilic domain may account for the fact that natural South 7 makes a somewhat inferior substitute for North 1 or the 7'OH-South 7 present in the strongest ritterazines (GN1N is more active and affects many more lines (14 nM, 59/60 lines) than does GN7S (>34 nM, 44/60 lines). The latter situation also applies to comparison of cephalostatins 17 vs. 2. Here, removal of the 26-OH from the polar domain in cephalostatin 2 results in a dramatic ≥ 104 loss of potency against P388 but a modest 4-fold drop against the NCI panel for cephalostain 17, which highlights the sometimes disparate SAR indicated for cephalostatins by P388 and the 60-cell NCI panel. Unfortunately, for cephalostatins 10-19, comparison of the SAR indicated by human leukemia lines to that by P388 is not possible, as detailed NCI results have no t been made available.
Excessive disparity also results in inferior in vitro potency. Such may be the case if the hydrophilic domain becomes too polar for its formerly appropriate nonpolar partner. This situation is seen with OSW-1a,b vs. OSW-1 (removal of acyl groups reveals additional free hydroxyl functions). Likewise, when the lipophilic domain becomes too nonpolar relative to its polar partner, decreased potency results (e.g. 12-acetyl-ritterazine B and ritterazine H vs. ritterazine B: loss of the 12-OH function by acetylation or oxidation; ritterostatin GN1N vs. cephalostatin 1, loss of the South ketone and 23'OH functions, retaining only a secondary 12-OH polarizing function). Comparison of the latter pair might be questioned on the grounds that the spiroketal (pro-oxacarbenium ion) moieties of their nonpolar units have different spatial relationships to the common polar unit. However, it will be seen that the comparison is not unreasonable because, like the total polarity of a given union of subunits, the relative locations of the spiroketals have an acceptable range of values (vide infra) and that of ritterostatin GN1N falls within that range.
The high cytotoxicity associated with unsymmetrical pairing of appropriate polar with nonpolar domains occurs for molecules with a range of overall polarity. A “lower” limit is seen for mainly nonpolar unions, whether unsymmetrical or not. The “upper” limit on total polarity is not apparent, which bodes well for possible alterations to give increased water solubility, a desirable feature in drugs administered orally. The most dramatic range of overall polarity is demonstrated by the total hydrophilicity of three highly potent (all ~1 nM) steroidal antineoplastics: ritterazine B, cephalostatin 2, and OSW-1.
8.2. Homoallylic oxygen
No reported bis-trans-saturated C/D bissteroidal pyrazines are highly active (all are poorly differentiated/polarized), but several mono-unsaturated C/D compounds are extremely cytotoxic, most notably ritterazine B (cis) and dihydrocephalostatin 1 (trans). No bis-cis-saturated C/D compound has been prepared. The possibilities of alkylation via oxacarbenium ion, nucleophilically susceptible carbonyl, Wagner-Meerwein or dyotropic rearrangements have been proposed (See Schemes 38 and 39).
Scheme 38.

Proposed mechanisms for biosynthesis of the spiro-C/D junction.
8.3. A 17-OH function is beneficial
A 17-OH function in one hemisphere is beneficial to high in vitro activity. For bissteroidal pyrazines, it is always in the polar domain. Removal of 17-OH results in ~10-100 fold loss of activity (Ritterazine Y 0.0045 nM, Ritterazine B 0.000025 nM - part due to loss of 7'OH; Ritt T >1500 nM, Ritt A 0.007 nM, part due to 7'OH loss). No glaring exceptions to this rule have been noted, but there may be a flaw in the in vitro approach to SAR. Neither saundersioside B nor solamargine are powerful in vitro but solamargine (4680 nM NCI) is extremely (100%) efficacious in vivo and is nontoxic to healthy tissue. Future work is needed to define the role of the 17-OH heteroatom.
8.4. An aromatic moiety is not necessary
An aromatic group appears beneficial to high in vitro activity but is likewise not a requisite for in vivo efficacy. Although one is present in all cephalostatins and some OSW types, solamargine is wholly aliphatic. The aromatic group's main contribution may be hydrophobic attractive interactions, but the ease with which nitrogenous aromatic heterocycles undergo nucleophilic aliphatic substitution should not be ignored. The fully substituted pyrazine has, as its protonated (pyrazinium) salt, pKa ~5, and is known to hydrogen bond over the ring, like benzene, rather than edge-on like pyridines.67 The carbonyl of the pMeOBz group of OSW-1 is calculated to greatly stabilize formation of a 1"-oxacarbenium ion, the lowest energy ion available to OSW 1 (Figure 14).68
Figure 14.
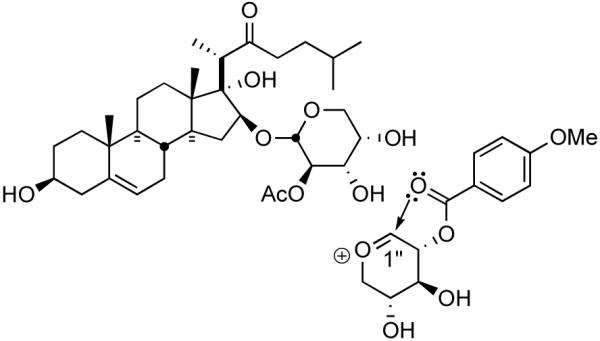
1”-oxacarbenium ion of OSW-1.
8.5. Hydrogen-bonding: Sugars and spiroketals
If, as seems likely, the polar domains in cephalostatin and OSW compounds function as a network of H-bond donors/acceptors and mimic the recognition role demonstrated for solamargine, future computer modeling may reveal critical overlap. A post-entry role for these spatially defined hydroxyl groups may also be important. Attached sugars are often cleaved on admittance within the cell, but the hydroxylated spiroketals of cephalostatins cannot be easily removed. These functions may facilitate transport to the target, binding, or orientation once delivered. The possibility that OSW-1 retains its glycal linkage for such purposes is necessarily considered.
Although NCI COMPARE studies reveal a strong correlation (0.83) with cephalostatins and OSW-1, different biological effects69 of 23'-deoxy-cephalostatin 1 and OSW-1 on mitochondria and cytotoxicity data of C22 deoxy OSW-1 analogs,70 which can not form E-ring oxacarbenium ion (Scheme 3), suggest that the mechanism of action of OSW-1 may be somewhat different from that of cephalostatins. Both OSW-1 and cephalostatin 1 (1) induce apoptosis at similar concentration and exposure,71 but reactive functionality usually associated with anticancer agents is absent in these classes, so particular attention should be paid to the fate of the spiroketals and sugars.
8.6. Two or more pro-(stabilized)carbenium ion moieties
At least two pro-carbenium ion sites with some spatial separation appear requisite for high in vitro activity. It is possible that the interannular homoallylic oxygen (in many cases, the 17OH and the 12OH are both homoallylic) and spiroketal (or equivalent spiroaminal, ketone, imine, hemiacetal, glycosidic acetal, etc.) moieties serve as masked stabilized carbenium ion sites, probably unveiled as alkylation centers via biological acid or metal ion catalyzed processes (Figure 15). Among such bissteroidal pyrazines, one spiroketal is often in a high-energy isomeric form (e.g. 22β) and the other in its thermodynamic form (22α). The requisite spatial relationship of the pro-oxacarbenium ion sites is poorly defined, and in compounds with high activity varies significantly between the glycoconjugate and pyrazine families, and to a lesser degree within the cephalostatin (pyrazine) family itself. In saponin OSW-1, no fixed angle relates the steroid sidechain C22 ketone and the two glycal centers (C1' and C1”), although models indicate that rotation about the pertinent connecting bonds does appear somewhat restricted. Its homoallylic oxygen array, like that of solamargine, bears different spatial relationships compared to the bissteroids. In cephalostatins/ritterazines, the spiroketals are rigidly fixed by the steroid ring system, but small, apparently remote, changes in the hexacyclic system can result in large attenuation of biological activity.13
Figure 15.

Hetero-carbenium ions proposed as potential biological electrophiles.
8.7. Discussion
Limited information regarding effects on activity by stereochemical variation in the spiroketal rings is available from natural epimers such as ritterazine F vs. ritterazine B, and from analogs of cephalostatin 7, 20epi-cephalostatin 7, and 25'epi-cephalostatin 7, and by 5/6 vs. 5/5 isomerization in ritterazine B, ritterazine C (Figure 16). Additional indications of the importance of this functionality are evident by considering cephalostatin 1 (1) vs. its nearly equipotent hemiketal cephalostatin 9 and ritterazine B vs. its dramatically less cytotoxic 22H reduction product 22'-H ritterazine B.
Figure 16.

Bissteroid bioactivity as a function of spiroketal alteration
Surprisingly, inversion at either C20 (as in 20epi-cephalostatin 7) or at C25' (as in 25′epi-cephalostatin 7) similarly diminished the activity. In addition to a substantial increase in many GI50s relative to cephalostatin 7, the number and kinds of tumor lines affected by 20epi-cephalostatin 7 and 25′epi-cephalostatin 7 was considerably reduced, and in strikingly similar fashion. Functionality alteration or polarity match rationales do not apply to 20epi-cephalostatin 7 and 25′epi-cephalostatin 7, and topographical responsibility for their similar loss of activity was deemed unlikely. A simple explanation based on relative hydrophilicity or general hydrogen bonding seemed inadequate. Analysis of altered directional H-bonding capacities likewise did not account for the parallel losses of cytotoxicity. A kinetic protonation argument was considered untenable, especially since the 25'epi-South 7 series was far more acid labile, and stereoelectronics suggest that the axial lone pair of O26' in South 7 units, the one “more hindered” in 25′epi-cephalostatin 7, is kinetically less basic than either the equatorial or O16' (E'-ring) lone pairs.72
Spiroketals (or equivalent functionality) appear to be masked oxacarbenium sites, and biological activity bears an inverse relationship to the calculated energy cost to form such ions for topographically similar series.55 Calculated geometries68 showed only modest conformational alteration by epimerization at C20 in the North 1 subunit of cephalostatin 7 as in 20epi-cephalostatin 7, and virtually no topographical alteration due to epimerization at C25′ as in 25′epi-cephalostatin 7. Rather, it seems 20epi-cephalostatin 7 and 25′epi-cephalostatin 7 suffer unfavorable oxacarbenium ion formation pathways compared to the parent cephalostatin 1 (1), and this rationale may explain their diminished cytotoxicity.
The South units were calculated62 to favor the E'-ring oxonium ion (E'-ox.) via equatorial F'-ring protonation, with E'-ox. formation (relative to pyrazine protonation) 3.1 kcal/mol more endothermic in 10-fold less cytotoxic 25′epi-cephalostatin 7 than in cephalostatin 7 (Figure 17). The computational study showed that the North 1 units also favor F-ring protonation, and E-ox. formation, while more endothermic, lies accessibly within ~1 kcal/mol of the protonated spiroketal. However, the E-ox. of 20epicephalostatin 7 experiences greater steric repulsion than cephalostatin 7 as the ring flattens, with the 21b-Me forced into unfavorable interactions with the concave side of the [3.3.0] D/E moiety. Oxacarbenium ion formation in 20epi-cephalostatin 7 appeared 3.0 kcal/mol less favorable than in cephalostatin 7, about the same increased cost as for 25′epi-cephalostatin 7. The similar potencies of 20epi-cephalostatin 7 and 25′epicephalostatin 7 implies oxacarbenium ion activity in both North and South subunits.
Figure 17.
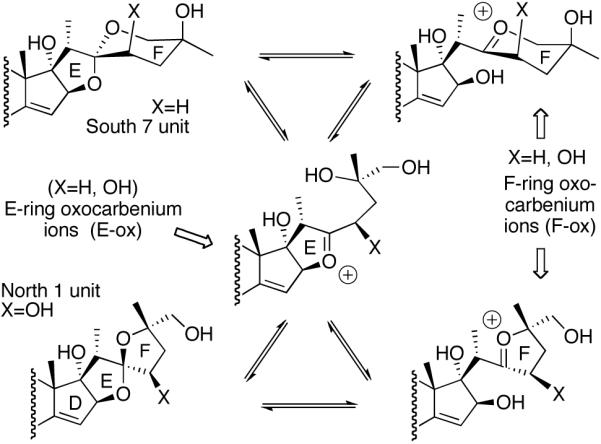
E- and F-ring oxacarbenium ions.
“Pro-oxacarbenium ion moieties” now seem to be critical components in SAR. Can evidence be found that suggests the spiroketals in these small antineoplastics assault the comparably huge tumor target with oxacarbenium ions? If so, does such a moiety constitute a previously unrecognized but widely disseminated medicinally significant function?
Such oxacarbenium ions may serve as direct electron-accepting agents, or may sire rearranged intermediates capable of biological alkylation or oxidation (Scheme 38). At this juncture, the electrophiles will be considered as cations generated upon interaction with a cellular Bronsted or Lewis acid by analogy to known laboratory reactions and biological glycosidations. However, no evidence precludes neutral (zwitterionic) forms stabilized by hydrogen bonding or metal ligation.
Mathematical models relating the energies of chemical interactions to bioactivity are valuable for QSAR and drug design. Active functionality typically associated with antitumor agents includes radical-generating, intercalating, redox-active, and electronic centers, particularly as unveiled in vivo by processes such as bioreductive alkylation.73 By contrast, spiroketals or equivalents (e.g. sugars, spiroaminals, etc.), present in diverse apoptoxins such as cephalostatins, spongistatins,74 and clinically significant solamargine, have not been generally considered of similar consequence. Binding modes with “passive” spiroketal contributing structural rigidity and sometimes ion attraction or hydrogen bonding have been advanced for many classes, e.g. dunaimycins (immunosuppression), 75 and novobiocin (antibiotic). 76 Dependence on spiroketal variations in halichondrins 77 and pectenotoxins 78 was similarly attributed to conformational effects (Figure 18). However, bioactivation of these widely disseminated latent electrophiles by metals, H-bonding, or acids could unmask a cascade of oxacarbenium ions competent to effect toxic modification(s) of susceptible sites in biopolymers.
Figure 18.

Potential heteroatom-stabilized carbenium ion precursors.
Hecht has detailed oxidative alkylation of DNA following metabolic activation (α-oxygenation) of cyclic nitrosamines (Figure 18).79 Iminium ions (cf. solasodine) were implicated in saframycins's reversible covalent binding to double-stranded DNA.80 In the series of events leading to observed cytotoxicity, formation of participating ions might be energetically determinant, therefore predictive of activity and detectable by calculation in silico.
LaCour has proposed a semiempirical calculation57 to rationalize the SAR of the entire 80-compound class of bissteroidal pyrazine antineoplastics. Application of this calculation method indicated an inverse exponential correlation (r2 ~0.970), suggestive in form of the Arrhenius equation, between relative cytotoxicity and endothermicity of oxacarbenium ion formation (Figure 19). The correlation is periodic, and appears regulated by accompanying polar functionality. The correlation originally showed that the biological activities of cephalostatins 8 and 16 and ritterazine M appeared substantially out of place.62 In all three instances, the structures had been assigned incorrectly.81 The calculation method also correctly predicted that compounds 23′-deoxy cephalostatin 138 and 17′-OH-23′-deoxy cephalostatin 137 would exhibit activity within a factor of 10 of cephalostatin 1 (1) (Figure 20).
Figure 19.

Plot of energy (ΔHpr) vs. ln(GI50) with 14 kcal/mol ring-corrections.
Figure 20.

Cephalostatin analogs and their biological activities.
9. Conclusions and Medicinal Prospects
Cephalostatins are among the most powerful anticancer agents tested by the National Cancer Institute. These challenging bissteroidal pyrazine targets have provided a platform for developing new synthetic strategies and methodologies over the last fifteen years. The successful syntheses of these challenging molecular architectures, such as cephalostatin 1, 7, and 12, ritterazine K and M, highlighted the state of the art of contemporary organic synthesis. Significant progress toward developing efficient and scalable synthetic pathways to natural cephalostatins and analogs has been made (e.g. South 7: 25 operations, 2% overall yield (1995); 16 operations, 24% overall yield (2005)).
The growing number of natural cephalostatins and their analogs provides valuable structure-activity relationships which aids the design of future analogs. The ongoing biological study of cephalostatin is gradually unveiling the antineoplastic mechanism of the cephalostatins. The bioactivity pattern of cephalostatins has been found quite different from known anticancer agents, indicating a new mechanism of action, possibly offering the potential for treatment of drug-resistant cancers.
Clinical trials of cephalostatin 1 (1) have been delayed largely due to the supply problem. Progress in the practical and scalable cephalostatin synthesis, should make the bissteroidal pyrazines more accessible, thereby enabling the clinical trials as well as providing tools for probing the biological and biochemical evaluation of the cephalostatins. Identification and structural elucidation of the biological target of cephalostatins coupled with QSAR studies are essential to facilitate the rational design of hyperactive analogs.
Scheme 44.

Preparation of A-B-C carbocyclic core of ritterazine N (216).
Scheme 45.

Taber synthesis of bis-18,18'-desmethyl ritterazine N (227).
Scheme 38.

Oxacarbenium ions as putative alkylating intermediates.
Table 1.
Biological Activity of Cephalostatins.
| pyrazines | P388 | NCI-60a | NCI-10b | PCCLc |
|---|---|---|---|---|
| (nM, IC50,ED50) | (nM, GI50) | (nM, GI50) | (nM, ED50) | |
| Cstat 1 | 10-4-10-6 | 1.2 (4.1) | 0.14-0.77 | 2.4×10-5 |
| Cstat 2 | 10-4-10-6 | 0.78 (6.5) | 0.12 | |
| Cstat 3 | 10-4-10-6 | (4.0) | 0.4 | |
| Cstat 4 | 10-4-10-6 | (36) | 4.0 | |
| Cstat 5 | 4.2 | (130) | 35.5 | |
| Cstat 6 | 22 | (320) | 104 | |
| Cstat 7 | 10-4-10-6 | 76 (6.5) | 16.3-34.4 | 0.052 |
| Cstat 8 | 10-4-10-6 | 29 (9.7) | 3.1 | |
| Cstat 9 | 10-4-10-6 | (6.3) | 0.85 | |
| Cstat 10 | 3.2 | 4.1 | ||
| Cstat 11 | 2.7 | 11 | ||
| Cstat 12 | 76 | 400 | ||
| Cstat 13 | 48 | >1000 | ||
| Cstat 14 | 4.4 | 100 | ||
| Cstat 15 | 27 | 68 | ||
| Cstat 16 | <1 | 1 | ||
| Cstat 17 | 4.6 | 4 | ||
| Cstat 18 | 4.6 | 22 | ||
| Cstat 19 | 7.9 | 17 |
GI50 values of cephalostatins at dosages of 1 μM max. Values in parenthesis were obtained at dosages of 3-10 μM max.
Activity in a 10-line panel of leukemia, brain, renal and breast cancers particularly responsive to this class of cytotoxins.3
Activity in 6-line panel of generally less susceptible breast, renal, lung, prostate, and colon cancers.4
Table 2.
Biological Activity of Ritterazines.
| pyrazines | P388 | NCI-60 | NCI-10a | PCCLb |
|---|---|---|---|---|
| (nM, IC50,ED50) | (nM, GI50) | (nM, GI50) | (nM, ED50) | |
| Ritt A | 3.8 | 24 | 12.7 | 7×10-3 |
| Ritt B | 0.17 | 3.2 | 1.0 | 2.6×10-5 |
| Ritt C | 102 | 115 | 178 | 18 |
| Ritt D | 18 | 102 | 76.8 | 0.012 |
| Ritt E | 3.8 | 37 | 15.9 | 1.9×10-3 |
| Ritt F | 0.81 | 8.3×10-5 | ||
| Ritt G | 0.81 | 5.7×10-5 | ||
| Ritt H | 18 | |||
| Ritt I | 15 | 88 | 47.3 | 0.010 |
| Ritt J | 14 | |||
| Ritt K | 10 | 70 | 4.5 | 5.8×10-3 |
| Ritt L | 11 | 20 | 20.5 | 9.5×10-3 |
| Ritt M | 17 | |||
| Ritt N | 522 | |||
| Ritt O | 2380 | inactive | >625 | 570 |
| Ritt P | 819 | |||
| Ritt Q | 657 | |||
| Ritt R | 2462 | |||
| Ritt S | 539 | |||
| Ritt T | 522 | >590 | >650 | >1500 |
| Ritt U | 2340 | >243 | 272 | 480 |
| Ritt V | 513 | |||
| Ritt W | 3632 | |||
| Ritt X | 3405 | |||
| Ritt Y | 4 | 27 | 13.9 | 4.5×10-3 |
| Ritt Z | 2200 | >722 | inactive | 560 |
10. Acknowledgements
This review covers more than 75 person-years of cephalostatin research in the Fuchs group and is dedicated to the outstanding individuals whose names are given in the accompanying citations. We gratefully thank the National Institutes of Health (CA 60548) for financial support of this project. Additional thanks are due Biogen Idec for recent financial contributions. Dr. Douglas Lantrip has provided extensive technical help on the project and in manuscript preparation. The coverart Earth map is used under terms described in “NASA's Earth Observatory” website. Pettit, Fusetani, and most recently Schiaparelli have provided invaluable assistance during the course of the entire research program.
11. Abbreviations
- AIBN
azobisisobutyronitrile
- CSA
camphorsulfonic acid
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DEAD
diethyl azodicarboxylate
- DIBAL
diisobutylaluminum hydride
- DMAP
4-N,N-(dimethylamino)pyridine
- DMDO
dimethyldioxirane
- HMDS
hexamethyldisilazane
- KHMDS
potassium hexamethyldisilylamide
- LDA
lithium diisopropylamide
- MCPBA
meta-chloroperbenzoic acid
- MOM
methoxymethyl
- MTM
methylthiomethyl
- Ms
methanesulfonyl
- NMO
N-methylmorpholine-N-oxide
- PCC
pyridinium chlorochromate
- PDC
pyridinium dichromate
- PMB
p-methoxybenzyl
- PPTS
pyridinium para-toluenesulfonate
- PTAB
phenyltrimethylammonium tribromide
- PVP
polyvinyl pyridine
- Py
pyridine
- TBAF
tetrabutylammonium fluoride
- TBAI
tetrabutylammonium iodide
- TBDPS
tert-butyldiphenylsilyl
- TBS
tert-butyldimethylsilyl
- TBSOTf
tert-butyldimethylsilyl Trifluoromethanesulfonate
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- TFAT
trifluoroacetyl triflate
- TIPS
triisoproylsilyl
- TMS
trimethylsilyl
- TPAP
tetraisopropylammonium perruthenate
- TPP
5,10,15,20-Tetraphenyl-21H, 23H-Porphine
Biographies

P. L. Fuchs Fuchs' grade school education took place in a four-room, elementary school in Nashotah, Wisconsin (pop. 237). In 1959, eighth Grade graduation saw the stage decorated in a scientific theme complete with a Fuchs-built Boron atom, foreshadowing both his interest in chemistry and his ultimate university. Fuchs' secondary education took place in Hartland, Wisconsin. At the end of his sophomore year Fuchs and another student renovated an old one-room building. At the end of that summer, Willow Brook Laboratory, WBL, complete with epoxy-top lab benches and a homemade fume was in place. During the next two years the neophyte chemists performed reactions from the literature of organic and inorganic synthesis. Fuchs then attended the University of Wisconsin at Madison, and continued summer projects at WBL. WBL sold reagents to Aldrich Chemical Company in Milwaukee and eventually advertised its chemicals on the back page of the Journal of the American Chemical Society. After graduation Fuchs stayed at UW, beginning graduate studies with Edwin Vedejs in 1968. In the summer of 1971, he received his degree as Vedejs' first Ph.D., and moved to Harvard for a two-year postdoctoral fellowship at with E. J. Corey.
Fuchs has been on the Purdue faculty since 1973, and is the current R. B. Wetherill Professor of Chemistry, with 230 papers published and 62 Ph.D.s granted. His awards and honors include an Eli Lilly young faculty fellowship (1975), an Alfred P. Sloan fellowship (1977), a Pioneer in Laboratory Robotics award (1986), a Martin teaching award (1991) and being voted by the students as one of Top 10 Teachers in School of Science at Purdue (1991, 1993, 1995, 1996). He earned Purdue's highest scientific award, the McCoy Research award in 2003. Fuchs has consulted for Pfizer and Eli Lilly, and has served on the editorial board of he Journal of Organic Chemistry. Since 2003 Fuchs has been an executive editor for the John Wiley Encyclopedia of Organic Reagents (EROS).

Seongmin Lee was born in Pohang, South Korea, in 1969. He received his B.S. and M.S. degrees in Chemistry from Seoul National University in 1992 and 1994, respectively. After working for LG Chemical Institute as a medicinal chemist, in 1999, he began his graduate studies at Purdue University under the supervision of Professor Philip Fuchs. During his Ph.D. program, he developed various C-H oxidation protocols and applied them to the synthesis of an anti-cancer steroidal pyrazine, ritterazine M. In 2004, he joined the group of Professor Gregory Verdine at Harvard University as a postdoctoral researcher, where he has been combining tools of synthetic organic chemistry and X-ray crystallography to elucidate molecular mechanisms by which DNA glycosylases, key enzymes in base-excision DNA repair, recognize and repair damaged bases in DNA.

Thomas G. LaCour was born and raised in Dallas, Texas. After wandering Europe, North Africa and North America, he completed degrees in English (B.A., University of Dallas) and Chemistry (B.A., M.A., University of Texas at Austin). He performed patented Process Research at Pfizer (Groton) for several years. Fortune led to the laboratory of Professor Philip Fuchs, who mentored his Ph.D. studies on Steroidal Anticancer Agents. Advances included completion of natural (Cephalostatin 1, several Ritterazines) and potent hybrid (Ritterostatins) bis-steroidal pyrazines. A semi-empirical calculation method precisely relating structure to cytoplastic activity for the entire family was discovered to correct published structures (Cephalostatins 8 & 16, Ritterazine M) and accurately predict new extremely potent analogues. Postdoctoral labors with Professor Larry Overman (UC Irvine), followed by sojourns with Professors J.R. Falck and Patrick Harran at the University of Texas Southwestern Medical Center, rounded out his medicinal chemistry research. Dr. LaCour currently endeavours to enlighten high school (and younger) minds in Dallas and explores history, psychology and predictive statistics of the markets.
Footnotes
This review is dedicated to the community of isolation/identification natural product detectives, with special thanks to the groups of Bob Pettit and Nobuhiro Fusatani.
12. References
- 1.(a) Pettit GR, Inoue M, Herald DL, Krupa TS. J. Am. Chem. Soc. 1988;110:2006. [Google Scholar]; (b) Pettit GR, Inoue M, Kamano Y, Herald DL. J. Chem. Soc. Chem. Comm. 1988:1440. [Google Scholar]; (c) Pettit GR, Xu JP, Schimidt JM. Bioorg. & Med. Chem. Lett. 1995;5:2027. [Google Scholar]
- 2.The unavailability of color photographs of C. gilchristi from the Pettit collection inspired a thorough web search for closely related Cephalodiscus species. A key paper (Schiaparelli S, Cattaneo-Vietti R, Mierzejewski P. Polar. Bio. 2004;27:813.) led us to contact professor Schiaparelli who graciously provided the first three pictures shown in Illustration 1. Comparison of a drawing of C. gilchristi provided by Professor Pettit with C. densus photo 1.3 revealed the minute (3mm) worms to be nearly identical. Schiaparelli collected C. densus from Terra Nova Bay, Ross Sea, Antarctica, begging the question of whether it also hosts an assortment of trisdecacyclic pyrazines.
- 3.(a) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1994;59:6164. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (b) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1995;60:608. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (c) Fukuzawa S, Matsunaga S, Fusetani N. Tetrahedron. 1995;51:6707. [Google Scholar]; (d) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1997;62:4484. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (e) Fukuzawa S, Matsunaga S, Fusetani N. Tetrahedron Lett. 1996;37:1447. [Google Scholar]
- 4.Full NCI-60 results for cephalostatins 1-9 (NSC# 363979-81, 378727-36) are on the Web. http://dtp.nci.nih.gov.
- 5.LaCour TG, Guo C, Ma S, Jeong JU, Boyd MR, Matsunaga S, Fusetani N, Fuchs PL. Bioorg. Med. Chem. Lett. 1999;9:2587. doi: 10.1016/s0960-894x(99)00430-8. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kubo S, Mimaki Y, Terao M, Sashida Y, Nikaida T, Ohmoto T. Phytochemistry. 1992;31:3969. doi: 10.1016/0031-9422(92)83295-a. [DOI] [PubMed] [Google Scholar]; (b) Mimaki Y, Kuroda M, Kameyama A, Sugita K, Beutler JA. Bioorg. Med. & Chem. Lett. 1997;7:633. [Google Scholar]
- 7.Kim YC, Che QM, Gunatilaka AAL, Kingston DGI. J. Nat. Prod. 1996;59:283. doi: 10.1021/np960125a. [DOI] [PubMed] [Google Scholar]
- 8.Daunter B, Cham BE. Cancer Lett. 1990;55:209. doi: 10.1016/0304-3835(90)90121-d. [DOI] [PubMed] [Google Scholar]
- 9.Reviews: Atta-ur-Rahmann, Choudary MI. Nat. Prod. Rep. 1997;14:191. doi: 10.1039/np9971400191.. Atta-ur-Rahmann, Choudary MI. Alkaloids. 1999;52:233.. Ganensan A, Heathcock CH. Angew. Chem. Int. Ed. Engl. 1996;35:611.. Ganensan A, Heathcock CH. Stud. Nat. Prod. Chem. 1996;18:875.. Jacobs MF, Kitching W. Curr. Org. Chem. 1998;2:395.. Urban S, Hickford SJH, Blunt JW, Munro MHG. Curr. Org. Chem. 2000;4:765.. Gryszkiewicz-Wojtkielewicz A, Jastrzebska I, Morzycki JW, Romanowska DB. Curr. Org. Chem. 2003;7:1257.; Flessner T, Jautelat R, Scholz U, Winterfeldt E. Fortschr Chem. Org. Naturst. 2004:1–80. doi: 10.1007/978-3-7091-0581-8_1.. Moser BR. J. Nat. Prod. 2008;71:487. doi: 10.1021/np070536z..
- 10.The “North unit”, the upper steroid nucleus of the cephalostatins, has also been designated “right side” by Pettit and “east unit” by Fusetani. On the other hand, the “South unit” refers to the lower half of the cephalostatins.
- 11.Ganesan A, Heathcock CH. Chemtracts. 1988;1:311. [Google Scholar]; Ganesan A. Stud. Nat. Prod. Chem. 1996;18:875. [Google Scholar]
- 12.Pan Y, Merriman RL, Tanzer LR, Fuchs PL. Bioorg. Med. Chem. Chem. Lett. 1992;2:967. [Google Scholar]
- 13.(a) Kramer A, Ullmann U, Winterfeldt E. JCS Perkin I. 1993:2865. [Google Scholar]; (b) Jautelat R, Müller-Fahrnow A, Winterfeldt E. Chem. Eur. J. 1999;5:1226. [Google Scholar]
- 14.(a) Bhandaru S, Fuchs PL. Tetrahedron Lett. 1995;36:8347. [Google Scholar]; (b) Bhandaru S, Fuchs PL. Tetrahedron Lett. 1995;36:8351. [Google Scholar]
- 15.(a) Tamura K, Honda H, Mimaki Y, Mimaki Y, Sashida Y, Kogo H. Br. J. Pharma. 1997;121:1796. doi: 10.1038/sj.bjp.0701309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuroda M, Mimaki Y, Sashida Y, Hirano T, Oka K, Dobashi A. Tetrahedron. 1997;53:11549. [Google Scholar]
- 16.(a) Pettit GR, Kamano Y, Inoue M, Dufresne C, Boyd MR, Herald CL, Schmidt JM, Doubek DL, Chiriste ND. J. Org. Chem. 1992;57:429. [Google Scholar]; (b) Pettit GR, Tan R, Xu J. -p., Ichihara Y, Williams MD, Boyd MR. J. Nat. Prod. 1998;61:955. doi: 10.1021/np9800405. [DOI] [PubMed] [Google Scholar]
- 17.Müller IM, Dirsh VM, Rudy A, Lopez-Anton N, Pettit GR, Vollmar AM. Mol. Pharmacol. 2005;67:1684. doi: 10.1124/mol.104.004234. [DOI] [PubMed] [Google Scholar]
- 18.Dirsch VM, Müller IM, Eichhorts ST, Pettit GR, Kamano Y, Inoue M, Xu JP, Ichihara Y, Wagner G, Vollmar AM. Cancer Res. 2003;63:8869. [PubMed] [Google Scholar]
- 19.(a) López-Antón N, Rudy A, Barth N, Schmitz LM, Pettit GR, Schulze-Osthoff K, Dirsch VM, Vollmar AM. J. Biol. Chem. 2006;181:33078. doi: 10.1074/jbc.M607904200. [DOI] [PubMed] [Google Scholar]; (b) Rudy A, López-Antón N, Dirsch VM, Vollmar AM. J. Nat. Prod. 2008;71:482. doi: 10.1021/np070534e. [DOI] [PubMed] [Google Scholar]
- 20.Jeong JU, Guo C, Fuchs PL. J. Am. Chem. Soc. 1999;121:2071. [Google Scholar]
- 21.Komiya T, Fusetani N, Matsunaga S, Kubo A, Kaye FJ, Kelley MJ, Tamura K, Yoshida M, Fukuoka M, Nakagawa K. Cancer Chemother Pharmacol. 2003;51:202. doi: 10.1007/s00280-002-0558-8. [DOI] [PubMed] [Google Scholar]
- 22.(a) Kubo S, Mimaki Y, Terao M, Sashida Y, Nikaida T, Ohmoto T. Phytochemistry. 1992;31:3969. doi: 10.1016/0031-9422(92)83295-a. [DOI] [PubMed] [Google Scholar]; (b) Mimaki Y, Kuroda M, Kameyama A, Sashida Y, Hirano T, Oka K, Maekawa R, Wada T, Sugita K, Beutler JA. Biorg. Med. Chem. Lett. 1997;7:633. [Google Scholar]
- 23.Guo C, LaCour TG, Fuchs PL. Bioorg. Med. Chem. Lett. 1999;9:419. doi: 10.1016/s0960-894x(98)00743-4. [DOI] [PubMed] [Google Scholar]
- 24.(a) Cham BE, Daunter B. Cancer Lett. 1990;55:221. doi: 10.1016/0304-3835(90)90122-e. [DOI] [PubMed] [Google Scholar]; (b) Cham BE, Meares HM. Cancer Lett. 1987;36:111. doi: 10.1016/0304-3835(87)90081-4. [DOI] [PubMed] [Google Scholar]; (c) Cham BE, Daunter B, Evans RA. Cancer Lett. 1991;59:183. doi: 10.1016/0304-3835(91)90140-d. [DOI] [PubMed] [Google Scholar]; (d) Beardmore GL, Hart V, Wilson P, Francis D. Med. J. Aust. 1989;150:351. doi: 10.5694/j.1326-5377.1989.tb136329.x. [DOI] [PubMed] [Google Scholar]; (e) Evans RA, Cham B, Daunter B. Med. J. Aust. 1989;150:350. [PubMed] [Google Scholar]
- 25.(a) Evans RA, Cham B, Daunter B. Med. J. Aust. 1990;152:329. [PubMed] [Google Scholar]; (b) Millward M, Powell A, Daly P, Tyson S, Ferguson R, Carter S. J. Clin. Onc. 2006;24:2070. [Google Scholar]
- 26.(a) Hu K, Kobayashi H, Dong A, Jing Y, Iwasaki S, Yao X. Planta Medica. 1999;65:35. doi: 10.1055/s-1999-13958. [DOI] [PubMed] [Google Scholar]; (b) Roddick JG, Weissenberg M, Leonard AL. Phytochemistry. 2001;56:603. doi: 10.1016/s0031-9422(00)00420-9. [DOI] [PubMed] [Google Scholar]; (c) Eschevarria A. J. Braz. Chem. Soc. 2002;13:838. [Google Scholar]
- 27.(a) Hsu S-H, Tsai T-R, Lin C-N, Yen M-H, Kuo K-W. Biochem. Biophys. Res. Comm. 1996;229:1. doi: 10.1006/bbrc.1996.1748. [DOI] [PubMed] [Google Scholar]; (b) Kuo K-W, Hsu S-H, Li Y-P, Lin W-L, Liu L-F, Chang L-C, Lin C-C, Lin C,-N, Sheu H-M. Biochem. Pharmacol. 2000;60:1865. doi: 10.1016/s0006-2952(00)00506-2. [DOI] [PubMed] [Google Scholar]; (c) Liu LF, Liang CH, Shiu LY, Lin WL, Lin CC, Kuo KW. FEBS Letters. 2004;577:67. doi: 10.1016/j.febslet.2004.09.064. [DOI] [PubMed] [Google Scholar]
- 28.Jeong JU, Sutton SC, Kim S, Fuchs PL. J. Am. Chem. Soc. 1995;117:10157. [Google Scholar]
- 29.Ohta G, Koshi K, Obata K. Chem. Pharm. Bull. 1968;16:1487. [Google Scholar]
- 30.Smith HE, Hicks AA. J. Org. Chem. 1971;36:3659. [Google Scholar]
- 31.(a) Smith SC, Heathcock CH. J. Org. Chem. 1992;57:6379. [Google Scholar]; (b) Heathcock CH, Smith SC. J. Org. Chem. 1994;59:6828. [Google Scholar]; (c) Heathcock CH, Smith SC. J. Org. Chem. 1995;60:6641. [Google Scholar]
- 32.Jeong JU, Fuchs PL. J. Am. Chem. Soc. 1994;116:773. [Google Scholar]
- 33.Kramer A, Ullmann U, Winterfeldt E. JCS Perkin I. 1993:2865. [Google Scholar]
- 34.Baesler S, Brunck A, Jautelat R, Winterfeldt E. Helv. Chim. Acta. 2000;83:1854. [Google Scholar]
- 35.Drogemuller M, Jautelat R, Winterfeldt E. Angew. Chem. Int. Ed. Engl. 1996;35:1572. [Google Scholar]
- 36.Haak E, Winterfeldt E. Synlett. 2004:1414. [Google Scholar]
- 37.Guo C, Bhandaru S, Fuchs PL, Boyd MR. J. Am. Chem. Soc. 1996;118:10672. [Google Scholar]
- 38.LaCour TG, Guo C, Bhandaru S, Boyd MR, Fuchs PL. J. Am. Chem. Soc. 1998;120:692. [Google Scholar]
- 39.Li Wei., LaCour TG, Fuchs PL. J. Am. Chem. Soc. 2002;124:4548. doi: 10.1021/ja017323v. [DOI] [PubMed] [Google Scholar]
- 40.Lee SM, Fuchs PL. Org. Lett. 2002;4:317. doi: 10.1021/ol016572l. [DOI] [PubMed] [Google Scholar]
- 41.Micovic IV, Ivanovic MD, Piatak DM. Synthesis. 1990;90:591. [Google Scholar]
- 42.Cook AF, Maichuk DT. J. Org. Chem. 1970;35:1940. doi: 10.1021/jo00831a048. [DOI] [PubMed] [Google Scholar]; Naruto M, Ohno K, Naruse N, Takeuchi H. Tetrahedron Lett. 1979;251 [Google Scholar]
- 43.LaCour TG, Cuo C, Ma S, Jeong JU, Boyd MR, Matsunaga S, Fusetani N, Fuchs PL. Bioorg. Med. Chem. Lett. 1999;9:2587. doi: 10.1016/s0960-894x(99)00430-8. [DOI] [PubMed] [Google Scholar]
- 44.(a) Kim S, Fuchs PL. Tetrahedron Lett. 1994;35:7163. [Google Scholar]; (b) Kim S, Sutton SC, Fuchs PL. Tetrahedron Lett. 1995;36:2427. [Google Scholar]; (c) Kim S, Sutton SC, Guo C, LaCour TG, Fuchs PL. J. Am. Chem. Soc. 1999;121:2056. [Google Scholar]
- 45.Gao Y, Sharpless KB. J. Am. Chem. Soc. 1988;110:7538–7539. [Google Scholar]
- 46.Reich HJ, Peake SL. J. Am. Chem. Soc. 1978;100:4888. [Google Scholar]
- 47.Cox GG, Miller DJ, Moody CJ, Sie ERHB. Tetrahedron. 1994;50:3195. [Google Scholar]
- 48.Barton DHR, McCombie SW. J. Chem. Soc., Perkin I. 1975:1574. [Google Scholar]
- 49.Jeong JU, Fuchs PL. Tetrahedron Lett. 1995;36:2431. [Google Scholar]
- 50.Dauben WG, Fonken G. J. Am. Chem. Soc. 1954;76:4618. [Google Scholar]
- 51.Heusler K, Wieland P, Meystre CH. Organic Synthesis. 1965;45:57. [Google Scholar]
- 52.Phillips ST, Shair MD. J. Am. Chem. Soc. 2007;129:6589. doi: 10.1021/ja0705487. [DOI] [PubMed] [Google Scholar]
- 53.Lee S, Fuchs PL. Org. Lett. 2002;4:317. doi: 10.1021/ol016572l. [DOI] [PubMed] [Google Scholar]
- 54.etancor C, Freire R, Perez-Martin I, Prange T, Suárez E. Org. Lett. 2002;4:1295. doi: 10.1021/ol025580e. [DOI] [PubMed] [Google Scholar]
- 55.Lee JS, Fuchs PL. Org. Lett. 2003;5:2247. doi: 10.1021/ol034551g. [DOI] [PubMed] [Google Scholar]
- 56.Lee JS, Cao H, Fuchs PL. J. Org. Chem. 2007;72:5820. doi: 10.1021/jo070382s. [DOI] [PubMed] [Google Scholar]
- 57.LaCour TG, Guo C, Boyd MR, Fuchs PL. Org. Lett. 2000;2:33. doi: 10.1021/ol991153y. [DOI] [PubMed] [Google Scholar]
- 58.Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1994;59:6164. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]
- 59.Reetz MT. Angew. Chem. Int. Ed. Engl. 1972;11:129. [Google Scholar]
- 60.Lee JS, Fuchs PL. J. Am. Chem. Soc. 2005;127:13122. doi: 10.1021/ja0531935. [DOI] [PubMed] [Google Scholar]
- 61.Li W, Fuchs PL. Org. Lett. 2003;5:4061. doi: 10.1021/ol035425f. [DOI] [PubMed] [Google Scholar]
- 62.Li W, Fuchs PL. Org. Lett. 2003;5:2849. doi: 10.1021/ol034894e. [DOI] [PubMed] [Google Scholar]
- 63.Li W, Fuchs PL. Org. Lett. 2003;5:2853. doi: 10.1021/ol0348957. [DOI] [PubMed] [Google Scholar]
- 64.Lee JS, Fuchs PL. Org. Lett. 2003;5:3619. doi: 10.1021/ol035284h. [DOI] [PubMed] [Google Scholar]
- 65.Fell JD, Heathcock CH. J. Org. Chem. 2001;67:4742. doi: 10.1021/jo011175+. [DOI] [PubMed] [Google Scholar]
- 66.(a) Taber DF, Joerger J-M. J. Org. Chem. 2008;73:4155. doi: 10.1021/jo800454w. [DOI] [PubMed] [Google Scholar]; (b) Taber DF, Joerger J-M. J. Org. Chem. 2007;72:3454. doi: 10.1021/jo0626461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Taber DF, Taluskie KV. J. Org. Chem. 2006;71:2797. doi: 10.1021/jo052656m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K. J. Am. Chem. Soc. 2000;122:11450. doi: 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- 68.LaCour TG. Ph.D. Thesis. Purdue University; 2001. [Google Scholar]
- 69.Zhou Y, Garcia-Prieto C, Carney D, Xu R, Pelicano H, Kang Y, Yu W, Lou C, Kondo S, Liu J, Harris D, Estrov Z, Keating MJ, Jin Z, Huang P. Journal of the National Cancer Institute. 2005;97:1781. doi: 10.1093/jnci/dji404. [DOI] [PubMed] [Google Scholar]
- 70.(a) Deng L, Wu H, Ty B, Jiang M, Wu J. Bioorg. Med. Chem. Lett. 2004;14:2781. doi: 10.1016/j.bmcl.2004.03.081. [DOI] [PubMed] [Google Scholar]; (b) Morzycki A, Wihtiekewicz A, Wolcznski S. Bioorg. Med. Chem. Lett. 2004;14:3323. doi: 10.1016/j.bmcl.2004.03.102. [DOI] [PubMed] [Google Scholar]
- 71.Rudy A, López-Antón N, Dirsch VM. J. Nat. Prod. 2008;71:482. doi: 10.1021/np070534e. [DOI] [PubMed] [Google Scholar]
- 72.Deslongchamps PR. Stereoelectronics in Organic Chemistry. Oxford-Pergamon Press; 1985. p. 53. [Google Scholar]
- 73.Moore HW. Science. 1977:527. doi: 10.1126/science.877572. [DOI] [PubMed] [Google Scholar]
- 74.Pietruszka J. Angew. Chem. Int. Ed. Engl. 1998;37:2629. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2629::AID-ANIE2629>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 75.Burres NS, Premanchandran U, Frigo A, Swanson SJ, Mollison KW, Fey TA, Krause RA, Thomas VA, Lane B, Miller LN, McAlpine JB. J. Antibiotics. 1991;44:1331. doi: 10.7164/antibiotics.44.1331. [DOI] [PubMed] [Google Scholar]
- 76.Bell W, Block MH, Grant A, Timms D. J. Chem. Soc. Perkin 1. 1997:2789. [Google Scholar]
- 77.Hart J, Hart B, Blunt JW, Munro MH. J. Org. Chem. 1996;61:2888. doi: 10.1021/jo951877x. [DOI] [PubMed] [Google Scholar]
- 78.Sasaki K, Wright JLC, Yasumoto T. J. Org. Chem. 1998;63:2475. doi: 10.1021/jo971310b. [DOI] [PubMed] [Google Scholar]
- 79.Wang M, Young-Sciame R, Chung F-L, Hecht SS. Chem. Res. Toxicol. 1995;8:617. doi: 10.1021/tx00046a017. [DOI] [PubMed] [Google Scholar]
- 80.Meyers AG, Plowright AT. J. Am. Chem. Soc. 2001;123:5114. doi: 10.1021/ja0103086. [DOI] [PubMed] [Google Scholar]
- 81.Lee S, LaCour TG, Lantrip D, Fuchs PL. Org. Lett. 2002;4:313. doi: 10.1021/ol0165894. [DOI] [PubMed] [Google Scholar]
Illustration 1.

Hemichordate worms and tunicates.