

Abstract
Among mammalian secreted phospholipases A2 (sPLA2s), the group X enzyme has the most potent hydrolyzing capacity toward phosphatidylcholine, the major phospholipid of cell membrane and lipoproteins. This enzyme has recently been implicated in chronic inflammatory diseases such as atherosclerosis and asthma and may also play a role in colon tumorigenesis. We show here that group X sPLA2 [mouse (m)GX] is one of the most highly expressed PLA2 in the mouse colon and that recombinant mouse and human enzymes stimulate proliferation and mitogen-activated protein kinase activation of various colon cell lines, including Colon-26 cancer cells. Among various recombinant sPLA2s, mGX is the most potent enzyme to stimulate cell proliferation. Based on the use of sPLA2 inhibitors, catalytic site mutants, and small interfering RNA silencing of cytosolic PLA2α and M-type sPLA2 receptor, we demonstrate that mGX promotes cell proliferation independently of the receptor and via its intrinsic catalytic activity and production of free arachidonic acid and lysophospholipids, which are mitogenic by themselves. mGX can also elicit the production of large amounts of prostaglandin E2 and other eicosanoids from Colon-26 cells, but these lipid mediators do not play a role in mGX-induced cell proliferation because inhibitors of cyclooxygenases and lipoxygenases do not prevent sPLA2 mitogenic effects. Together, our results indicate that group X sPLA2 may play an important role in colon tumorigenesis by promoting cancer cell proliferation and releasing various lipid mediators involved in other key events in cancer progression.
Phospholipases A2 (PLA2s) catalyze the hydrolysis of the sn-2 ester bond of glycerophospholipids to generate free fatty acids and lysophospholipids (Schaloske and Dennis, 2006; Lambeau and Gelb, 2008). Over the past few years, it has been realized that PLA2s constitute a superfamily of enzymes comprising several intracellular enzymes and secreted PLA2s (sPLA2s).
The group IVA cytosolic PLA2 (cPLA2α) is the best known intracellular PLA2, and it clearly plays an important, yet not exclusive role in the release of arachidonic acid (AA) and subsequent production of eicosanoids in various biological settings (Kita et al., 2006). In contrast, the biological functions of the different sPLA2s are slowly being unraveled. sPLA2s have been implicated in lipid digestion and obesity; activation of immune cells; asthma; atherosclerosis; acute respiratory distress syndrome; and host defense against bacteria, viruses, and parasites (Touqui and Wu, 2003; Triggiani et al., 2006; Lambeau and Gelb, 2008; Nevalainen et al., 2008). Besides their catalytic activity, sPLA2s are also able to bind to specific soluble and membrane-binding proteins, including the M-type receptor (Rouault et al., 2007).
Several PLA2s have also been implicated in various cancers. Disruption of the cPLA2α gene decreases initiation and growth of intestinal tumors in Apc-mutated mice (Takaku et al., 2000) but increases the number of tumors in the carcinogen azoxymethane-induced mouse model of colon cancer (Ilsley et al., 2005). The gene coding for mouse group IIA (mGIIA)1 sPLA2 was identified as a gene modifier that reduces the number of intestinal polyps in Apc(+/Min) mice, but the mechanism of action is still unclear (Fijneman and Cormier, 2008). Distinct roles have also been proposed for human group IIA sPLA2 in various cancers (Sved et al., 2004; Cummings, 2007). Recent data have shown differential expression of sPLA2s IID, III, and V but not X in human colon cancer (Murakami et al., 2005; Mounier et al., 2008). In ApcΔ716 and azoxymethane mouse models of colon cancer, the expression of group X sPLA2 may be increased in small intestinal tumors but not in colon tumors (Takaku et al., 2000; Ilsley et al., 2005). In vitro, group IB, IIA, and III sPLA2s have been reported to stimulate cell proliferation and activation of MAP kinases in various cancer cells (Kinoshita et al., 1997; Sved et al., 2004; Murakami et al., 2005). Finally, subcutaneous injection into nude mice of colon tumor cells overexpressing mGIIA sPLA2 resulted in a 2.5-fold increase in tumor size (Belinsky et al., 2007).
Over the past few years, the group X sPLA2 has emerged as the most potent sPLA2 capable of hydrolyzing phosphatidylcholine and acting extracellularly on cellular membranes and noncellular phospholipid substrates such as lipoproteins (Lambeau and Gelb, 2008). Consequently, group X sPLA2 has been proposed to play a role in atherosclerosis (Lambeau and Gelb, 2008), asthma (Henderson et al., 2007), and colon cancer (Morioka et al., 2000).
The above-mentioned findings plus the fact that group X sPLA2 is expressed at very high levels in human (Cupillard et al., 1997; Morioka et al., 2000; Mounier et al., 2008) and mouse colon (Eerola et al., 2006; this study) prompted us to analyze the proliferative effect of group X sPLA2 on different colon cancer cells, including the adenocarcinoma-derived mouse Colon-26 cancer cells. Among different mouse sPLA2s, group X sPLA2 was the most potent enzyme to stimulate cell proliferation. Using a combination of tools and methods, we found that its proliferative effect does not depend on binding to the M-type receptor or activation of cPLA2α but rather on its intrinsic catalytic activity and ability to release free fatty acids and lysophospholipids, which most likely act in concert to stimulate cell proliferation.
Materials and Methods
Materials.
The mouse adenocarcinoma cell line Colon-26 was obtained from Cell Lines Service (Heidelberg, Germany). AJ02-nm0 cells (Belinsky et al., 2007), YAMC cells (young adult mouse colon) (Whitehead et al., 1993), and Apc(+/Min) cells (Forest et al., 2003) were generous gifts from Drs. D. W. Rosenberg (University of Connecticut Health Center, Farmington, CT), R. H. Whitehead (Melbourne, Australia), and F. Pierre (Ecole Nationale Vétérinaire de Toulouse, Toulouse, France) and J. Menanteau (Ecole Nationale Vétérinaire de Nantes, Nantes, France), respectively. RPMI 1640 medium and Dulbecco's modified Eagle's medium (DMEM) were from Invitrogen (Cergy Pontoise, France). Fetal calf serum (FCS) was from Dominique Dutscher (Brumath, France). Oleic acid, linoleic acid, linolenic acid, oleoyl-l-α-lysophosphatidic acid sodium salt, 1-oleyl-sn-glycero-3-phosphocholine, PGE2, AA, aspirin, ibuprofen, indomethacin, and FAF BSA were from Sigma-Aldrich (L'isle d'Abeau Chesnes, France). [methyl-3H]Thymidine, [3H]oleic acid, and [3H]AA were from PerkinElmer Life and Analytical Sciences (Courtaboeuf, France). Recombinant murine interferon-γ was from PeproTech EC (London, UK). Insulin, transferrin, selenious acid (ITS) was from BD Biosciences (Le Pont-De-Claix, France). Phospho-specific p42/p44 MAPK antibodies (clone E-4), p42/p44 antibodies, and cPLA2α antibodies were from Santa Cruz Biotechnology, Inc. (Tebu-Bio SA, Le Perray en Yvelines, France). MK886, baicalein, and ATX antibody were from Cayman Chemicals (Interchim, Montluçon, France). Alkaline phosphatase-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories Inc. (Immunotech, Marseille, France), and enhanced chemifluorescence (ECF) substrate was from GE Healthcare (Orsay, France). PGE2 enzyme-linked immunosorbent assay kit was from Assay Designs (Euromedex, Strasbourg, France). Prevalidated siRNAs were from QIAGEN (Courtaboeuf, France). Hoechst was from Invitrogen (Cergy Pontoise, France). LY329722 and Me-indoxam sPLA2 inhibitors were prepared as described previously (Singer et al., 2002; Smart et al., 2006). Lysing Matrix D beads were from Qbiogene (Illkirch, France). Nucleospin RNA extraction kit was from Macherey Nagel (Hoerdt, France). All recombinant mouse and human sPLA2s used in this study and catalytically inactive hGX and mGX H48Q sPLA2 mutants were prepared as described previously (Singer et al., 2002; Rouault et al., 2006, 2007).
sPLA2 Expression in Mouse Intestinal Tract Analyzed by RT-qPCR.
Tissue samples from duodenum, jejunum, ileum, and proximal and distal colon from two C57BL/6J female or three BALB/c female mice were snap-frozen in liquid nitrogen. Total RNA was isolated using the Nucleospin RNA extraction kit with DNase I treatment and Lysing Matrix D beads to homogenize the samples. Reverse-transcription and reverse transcription quantitative polymerase chain reaction (RT-qPCR) were performed as described previously (Eerola et al., 2006). The abundance of each sPLA2 mRNA target was calculated relative to the expression of glyceraldehyde-3-phosphate dehydrogenase mRNA, which was used as a reference gene. The tissue distribution of sPLA2s and cPLA2α was further normalized to the lowest cycle threshold value accurately measured. A relative abundance of 1 (arbitrary unit = 1) was assigned to the expression level of the pancreatic group IB sPLA2 mRNA in the colon, which was detected with cycle threshold values of 33 or higher. Standard deviation calculation was made according to User Bulletin 2 from Applied Biosystems, Foster City, CA.
Expression of mGX sPLA2 by in Situ Hybridization.
Samples for in situ hybridization were obtained from small and large intestines of C57BL/6J mice, fixed in 4% phosphate-buffered formalin, and embedded in paraffin. A 0.45-kilobase mGX sPLA2 cDNA insert was cloned into the pRc/CMV vector (Invitrogen) and used as a template to prepare digoxigenin (DIG)-labeled antisense (test) and sense (control) RNA probes by in vitro transcription using a commercial kit (DIG RNA labeling kit; Roche Diagnostics, Meylan, France) according to the manufacturer's instructions. Nonradioactive in situ hybridization has been described in detail previously (Haapamäki et al., 1999). In brief, 5-μm-thick paraffin sections were placed on silanated slides, dewaxed in xylene, and rehydrated in a graded alcohol series. Pretreatment was performed sequentially with 200 mM HCl for 20 min, 20 μg/ml proteinase K at 37°C for 15 min, 0.2% glycine in phosphate-buffered saline, pH 7.4, two times for 3 min each, freshly prepared 0.4% acetic anhydride in 100 mM triethanolamine two times for 5 min each, and finally with ficin (Digest-All kit; Zymed Laboratories, South San Francisco, CA) at 37°C for 20 min. The sections were then blocked in hybridization solution without probe. The solution was replaced with fresh hybridization solution containing the antisense or sense probe at a final concentration of 60 ng/ml. After overnight hybridization, the sections were washed in 2× standard saline citrate (SSC) for 5 min, in 0.2× SSC, 60% formamide at 37°C three times for 5 min each, and 2× SSC two times for 5 min each. The sections were blocked with 3% bovine serum albumin in Tris-buffered saline, and the hybridized probe was detected with alkaline phosphatase-labeled anti-DIG Fab-fragments (1/2000; Roche Diagnostics) using a substrate solution containing 0.18 mg/ml 5-bromo-4-chloro-3-indolyl-phosphate, 0.34 mg/ml 4-nitro blue tetrazolium phosphate (Roche Diagnostics), and 0.24 mg/ml levamisole (Vector, San Francisco, CA) in 100 mM Tris, pH 9.5, 100 mM NaCl, and 50 mM MgCl2. The color reaction was allowed to develop at 4°C overnight and at 25°C for 6 h. The tissue sections were briefly counterstained with hematoxylin, washed with water, and mounted in aqueous mounting medium.
Cell Culture.
Colon-26 cells were grown at 37°C in DMEM/10% FCS, antibiotics, and 2 mM glutamine. AJ02-nm0 cells were grown in RPMI 1640 medium/10% FCS with antibiotics, glutamine, and supplementation with insulin (6.25 ng/ml), transferrin (6.25 μg/ml), and selenious acid (6.25 ng/ml; ITS). YAMC and Apc(+/Min) cells express the temperature-sensitive mutation of the simian virus 40 large tumor antigen gene (tsA58) and grow well at 33°C but become quiescent at 37°C. YAMC cells were grown at 33°C in RPMI 1640 medium supplemented with 5% FCS, antibiotics, ITS, and 5 U/ml recombinant interferon-γ. Apc(+/Min) cells were grown at 33°C in DMEM supplemented with 10% FCS, antibiotics, 10 U/ml recombinant IFN-γ, and 10 ng/ml recombinant epidermal growth factor. We found that these cells do not release sPLA2 enzymatic activity in cell medium during their normal growth, nor do they express detectable amounts of mGX sPLA2 protein by time-resolved fluoroimmunoassays (Eerola et al., 2006). To determine whether these cell lines express a functional mGIIA sPLA2 gene (MacPhee et al., 1995), we set up conditions for the specific detection of wild-type and mutated mGIIA alleles by polymerase chain reaction on genomic DNA (see Supplemental Data). YAMC cells were found to be heterozygous for the pla2g2a gene, Colon-26 cells were found to be wild type, and AJ02-nm0 and Apc(+/Min) cells were found to contain two mutated pla2g2a alleles (Supplemental Fig. 1S).
[3H]Thymidine Incorporation and Cell Growth Assays.
Colon-26 (5000 cells/well), YAMC (10,000 cells/well), AJ02-nm0 (5000 cells/well), and Apc(+/Min) (4000 cells/well) cells were plated into 48-well culture plates (Falcon; BD Biosciences Discovery Labware, Bedford, MA) that were precoated (Colon-26 and YAMC) or not [AJ02-nm0 and Apc(+/Min)] with rat tail collagen to ensure tight adhesion of cells. Fifty percent (Colon-26) or 80% [YAMC, AJ02-nm0, and Apc(+/Min)] confluent cells were made quiescent by incubating them for 24 h in serum-free medium with 0.02% FAF BSA at 37°C. It is important that we observed that the percentage of cell confluence can dramatically affect the ability of the sPLA2 to trigger cell proliferation. Quiescent cells were treated with medium containing 0.02% FAF BSA and the effectors and were further incubated for 24 h (Colon-26) or 48 h [YAMC, AJ02-nm0, and Apc(+/Min)] at 37°C (Colon-26 and AJ02-nm0) or 33°C [YAMC and Apc(+/Min)]. During the last 4 h, [methyl-3H]thymidine was added to wells to a final concentration of 1 μCi/ml, 1 μM unlabeled thymidine. In some experiments, cell supernatant of Colon-26 cells was collected for PGE2 assays. The cells were then washed twice with ice-cold phosphate-buffered saline, incubated for 30 min in 5% ice-cold trichloroacetic acid, solubilized in 0.2 N NaOH, and analyzed for [3H]thymidine incorporation into DNA. For cell growth assays, cells were seeded as described above, starved at low density, and then treated with sPLA2. Cells were dissociated and counted after trypan blue staining. Assays were done in triplicate.
Immunoblot Analysis.
Colon-26 cells grown and made quiescent as described above were treated with effectors for various times, washed with PBS, lysed in hot Laemmli buffer, and boiled for 5 min at 95°C. Sonicated lysates were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to Immobilon-FL polyvinylidene fluoride membrane (Millipore, Saint-Quentin-en-Yvelines, France). Membranes were blocked for 2 h at room temperature in NETG buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 7.4, 0.05% Triton X-100, and 0.25% gelatin) and probed for 1 h with phospho-specific p42/44 MAPK antibodies or cPLA2α antibodies (1/2000). Blots were washed five times with PBS/0.1% Tween 20, incubated with alkaline phosphatase-conjugated secondary antibodies diluted in PBS/Tween (1/10,000), visualized by incubation for 5 min with ECF substrate, and scanned with a Pro-Xpress imager (excitation, 480 nm; emission, 590 nm). For Western blot of the mouse M-type receptor, cells were lysed in ice-cold 20 mM Tris, pH 7.4, 2 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride and sonicated. Unboiled proteins (60 μg) prepared in nonreducing Laemmli buffer were separated on a 7% SDS-polyacrylamide gel electrophoresis gel and transferred to an Immobilon-FL membrane. The membrane was blocked with 5% blocking agent (GE Healthcare) dissolved in TBS/Tween (25 mM Tris, pH 7.8, 150 mM NaCl, and 0.15% Tween 20) for 1 h and then incubated with the anti-mouse M-type receptor antibody (1/5000 in TBS/Tween; Rouault et al., 2007) overnight at 4°C. The membrane was washed six times for 5 min each and incubated with secondary alkaline phosphatase goat anti-rabbit antibody (1/10,000 in TBS/Tween). The immunocomplex was visualized with ECF substrate as described above.
siRNA Transfection.
siRNAs were designed using the program HP GenomeWide siRNA (HiPerformance Design Algorithm; QIAGEN). Best siRNAs respectively targeting the mouse M-type receptor and mouse cPLA2α were selected from two QIAGEN-designed siRNAs after validation by binding assays and immunocytochemistry for the M-type receptor and Western blot analysis for cPLA2α. For siRNA transfection, duplex siRNAs were first heated for 1 min at 90°C and cooled at 37°C for 30 min to allow for annealing. Cells were seeded on day 0 and transfected on day 1. Duplex siRNAs were preincubated with HiPerFect reagent (QIAGEN) and added dropwise to each well to a final concentration of 5 nM. On day 2, cells were made quiescent for 24 h before stimulation with sPLA2 and subsequent Western blotting, binding assays, proliferation experiments, or immunocytochemistry. Transfection efficiency was evaluated by transfection with an Alexa546-coupled siRNA and fluorescence visualization with a microscope. The effect of the best-selected siRNAs for M-type receptor [forward, r(GGU ACA CUC GAU ACA UUA A)dTdT; reverse, r(UUAAUGUAUCGAGUGUACC)dTdA] and cPLA2α [forward, r(GGA GAU UAA UGA AGA GCU A)dTdT; reverse, r(UAG CUC UUC AUU AAU CUC C)dTdC] were compared with the effect of the nonrelevant GFP-directed siRNA (forward, GCA AGC UGA CCC UGA AGU UCA U; reverse, GAA CUU CAG GGU CAG CUU GCC G).
Binding Assays and Immunocytochemistry.
The expression of the mouse M-type receptor in various cell lines was determined by binding experiments using iodinated OS1 as described previously (Rouault et al., 2007). Cells were seeded in six-well plates, and 90% confluent cells were incubated for 1 h in binding buffer (140 mM NaCl, 1 mM CaCl2, 20 mM Tris, pH 7.4, and 0.1% BSA) at room temperature, with 200,000 cpm iodinated OS1 in the absence (total binding) or presence (nonspecific binding) of 100 nM unlabeled OS1. Cells were washed, lysed with 0.2 M NaOH, and analyzed for bound iodinated OS1. The expression of the mouse M-type receptor after siRNA transfection was evaluated by immunocytochemistry as follows. Colon-26 cells were seeded in 24-well culture plates (4000 cells/well) on coverslips coated with rat tail collagen. Twenty-four hours after transfection with siRNA, the medium was changed, and cells were incubated in FCS-free medium for 24 h to mimic conditions of proliferation assays. Cells were then fixed with 3.7% paraformaldehyde for 25 min at room temperature and washed twice with PBS. Free aldehyde groups were quenched with 50 mM NH4Cl in PBS, and cells were washed once with PBS and permeabilized with 0.5% saponin (two times for 5 min each). Nonspecific sites were blocked with 10% horse serum/0.05% saponin. Anti-mouse M-type receptor rabbit serum (Rouault et al., 2007) was diluted 1/1000 in 5% horse serum/0.05% saponin and incubated for 30 min at room temperature. After three washes with PBS/0.05% saponin, fluorescein isothiocyanate-linked secondary antibodies were diluted 1/600 in 5% horse serum/0.05% saponin and incubated for 1 h at room temperature under dim light. Nuclei were stained with Hoechst 33342 (5 μg/ml). Cells were washed once in PBS/0.05% saponin, twice in PBS, and once in H2O, and then they were mounted with Dako mounting medium (Dako, Trappes, France) and visualized under a microscope.
AA Release.
AA release was performed under the same conditions of culture as those used for proliferation assays. During the starvation time, cells were labeled with [3H]AA (0.2 μCi/well) in FCS-free DMEM/0.02% FAF BSA for 24 h at 37°C. Cells were washed twice with FCS-free medium/0.1% FAF BSA and incubated for the indicated periods in 200 μl of serum-free medium with 200 nM sPLA2. Cell-free supernatants and cell monolayers lysed in 400 μl of 0.2 N NaOH were submitted to scintillation counting. [3H]AA release is expressed as the percentage of cpm in cell supernatant versus total cpm (supernatant and cell lysate).
Eicosanoid Assays.
Colon-26 cells (106 cells) were grown in six-well culture plates for 2 days, starved for 24 h, and stimulated for 24 h with 200 nM mGX sPLA2 in 1 ml of serum-free medium containing 0.02% FAF BSA. Total eicosanoid production in combined supernatant and cell pellet was assayed by high-performance liquid chromatography/mass spectrometry according to the procedure described previously (Kita et al., 2005; Henderson et al., 2007). For specific PGE2 assay, cell supernatants from proliferation assays were centrifuged, appropriately diluted (1/10–1/500), and PGE2 levels were determined using the PGE2 enzyme immunoassay kit (Assay Designs) according to the manufacturer's instructions.
Lysophospholipid Analysis.
Colon-26 cells (2 × 106 cells) were seeded in 10-cm Petri dishes and grown for 2 days. Then, cells were starved for 24 h in FCS-free medium/0.02% FAF BSA and stimulated with effectors in 4 ml of FCS-free medium/0.02% FAF BSA for 6 and 24 h. After incubation, the cell supernatants were collected and lyophilized. The cell monolayer (approximately 107 cells) was scraped in PBS, centrifuged, and stored as a dried pellet at −70°C. The following procedure was adapted from previously published methods of lysophosphatidic acid (LPA) analysis (Baker et al., 2001). Lyophilized supernatants were resuspended in 0.5 ml of Milli-Q water (Millipore), and cell pellets were thawed on ice in 0.5 ml of Milli-Q water. To each sample, 1.5 ml of 30 mM citric acid and 40 mM Na2HPO4, pH ∼4.0, was added and vortexed for 30 s. Samples were then spiked with 500 pmol of 17:0 LPA and 1 nmol of d31-16:0 LPC and extracted using 4 ml of 1-butanol followed by re-extraction with 2 ml of 1-butanol. Organic phases were dried in vacuo overnight. The resulting residues were reconstituted in 1.5 ml of CHCl3:MeOH (2:1), dried again, and finally reconstituted in 100 μl of CHCl3:MeOH (2:1). Liquid chromatography/tandem mass spectrometry analysis was performed using a Micromass Quattro Micro tandem quadrupole mass spectrometer (Waters, Milford, MA) coupled with a 2795 Chromatography System (Waters). Twenty microliters of concentrated lipid extract was injected onto a Luna silica column (5-μm particle size, 4.6 × 250 mm; Phenomenex, Torrance, CA) equilibrated with 65% solvent A (30:40, n-hexane/isopropanol) and 35% solvent B (30:60:15, n-hexane/isopropanol/H2O) at 30°C with a flow rate of 1 ml/min. After injection, 35% solvent B was maintained for 6 min. A gradient of 35 to 100% solvent B was run over 24 min. The gradient was held at 100% solvent B for an additional 20 min. Mass transitions, cone voltages, and collision energies are available on request. Transitions were monitored in the negative mode for the first 30 min, with 40-ms dwell times, and in the positive mode for the final 20 min, with 100-ms dwell times. In both modes, 10-ms interchannel and 100-ms interscan delays were used.
Statistics.
Data are expressed as mean ± S.D. Statistical analyses were performed with Prism (GraphPad Software Inc., San Diego, CA) using Student's t test and one-way analysis of variance (ANOVA), with Bonferroni adjustment for multiple comparisons. P values <0.05 were considered as statistically significant.
Results
Group X sPLA2 Is Expressed at High Levels in Mouse Colon.
We have recently found that mGX sPLA2 is expressed at high levels in the small intestine and colon at both mRNA and protein levels (Eerola et al., 2006). To further analyze the expression of mGX sPLA2 and other PLA2 genes in the different sections of the intestine of C57BL/6J mice, we have measured by RT-qPCR the relative abundance of mRNAs coding for the full set of mouse sPLA2s (except IIC), for cPLA2α (Fig. 1A) and for the M-type receptor (data not shown). Most sPLA2s were expressed in the different gut sections, but marked differences were found. Among catalytically active sPLA2s, mGX sPLA2 is by far the most highly expressed enzyme in the intestine of C57BL/6J mice, with highest expression in the distal colon. For example, its level of expression in the colon is approximately 10-fold higher than that of mGV sPLA2. It is interesting that mGX sPLA2 mRNA levels vary along the intestine with up to 120-fold increase in expression from duodenum to distal colon. Second, the levels of mRNA for the catalytically inactive group XIIB sPLA2-like protein are similar to those of mGX sPLA2, but the expression pattern is the inverse. Third, group IID, IIF, III, and XIIA sPLA2s are expressed in the intestine at significant but low levels, and group IB and IIE sPLA2s are barely detectable. Expression of group XIIA sPLA2 is rather constitutive all along the intestine. Conversely, that of group IIF, III, and V sPLA2s increases, whereas that of group IID sPLA2 decreases when moving from the proximal to the distal part of the gut. Fourth, the expression of cPLA2α is 5-fold lower than that of mGX sPLA2 but follows a similar gradient of expression along the intestine. Finally, the expression of the M-type receptor was low along the gut sections (data not shown), but, interestingly, it follows the same gradient of expression of mGX sPLA2.
Fig. 1.

Expression of the different mouse sPLA2s in the intestine of C57BL/6J and BALB/c mice by RT-qPCR and in situ hybridization of mGX sPLA2. A and B, RT-qPCR in C57BL/6J (A) and BALB/c (B) mouse intestine sections using specific sets of PLA2 primers. To facilitate the comparison of expression between the different sPLA2s, the data were first normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA, which was used as a reference gene, and then expressed relative to the lowest expression level that can be accurately measured in our RT-qPCR assay conditions [i.e., the expression of pancreatic group IB sPLA2 in the colon {relative abundance of 1 (arbitrary unit = 1)}]. Note that two different ordinate axes have been used in B. mGIIE sPLA2 could not be detected in all intestine sections from BALB/c mice (C–F). In situ hybridization of mGX sPLA2 in small intestine (ileum) showing labeling of columnar epithelial cells in mucosal villi (V), Paneth cells (P), and ganglion cells (G) of the myenteric plexus. Hybridized with antisense probe. D, absence of reaction product from ileal tissue when hybridized with sense probe. E, in situ hybridization of mGX sPLA2 in large intestine (cecum) showing labeling in epithelial cells. Hybridized with antisense probe.
It is important that C57BL/6J mice have a disrupted mGIIA sPLA2 gene (MacPhee et al., 1995), which probably explains the low and aberrant amount of mRNA observed in the small intestine (Fig. 1A). When RT-qPCR assays for the different sPLA2s were done as described above on intestinal sections from BALB/c mice expressing a functional gene, the expression of mGIIA sPLA2 in jejunum and ileum was found to be much higher than that of all other sPLA2s, including mGX sPLA2 (Fig. 1B). However, the mRNA levels of mGIIA were fairly low in proximal and distal colon, where mGX sPLA2 remained the most abundantly expressed sPLA2 (Fig. 1B).
To determine more precisely the cellular site of mGX sPLA2 expression in small and large intestine, we performed in situ hybridization. Figure 1, C and D, shows strong labeling of columnar epithelial cells of mucosal villi, Paneth cells in the crypts of Lieberkühn, and ganglion cells of the myenteric plexus between smooth muscle cell fibers. Figure 1E illustrates the absence of labeling from a section of small intestine hybridized with sense probe. Figure 1F shows labeling of epithelial cells in the mucosal glands of the large intestine adjacent to empty-looking goblet cells.
We also analyzed the expression level of group X sPLA2 in the ileum and colon of mouse models of colon cancer (ApcΔ14 and azoxymethane) and found no dramatic change in the expression of group X sPLA2 (Supplemental Fig. 2S).
Group X sPLA2 Stimulates the Proliferation of Mouse Colon Cells.
Because group X sPLA2 is expressed in both normal colon and colon adenocarcinoma (Cupillard et al., 1997; Morioka et al., 2000; Takaku et al., 2000; Osterström et al., 2002; Ilsley et al., 2005; Mounier et al., 2008), we analyzed the effect of exogenous recombinant group X sPLA2 on the proliferation of four different mouse colon cell lines [YAMC, Apc(+/Min), AJ02-nm0, and Colon-26]. Both mGX and hGX sPLA2s were able to stimulate [3H]thymidine incorporation in the four tested cell lines (Fig. 2). At 200 nM, group X sPLA2 increased [3H]thymidine incorporation by up to 3-fold, depending on the cell line and conditions of stimulation. This proliferative effect is important because it corresponds to approximately 50% of the maximal effect observed with fetal calf serum. We decided to use the Colon-26 cell line for the experiments described below because these cells are adenocarcinoma-derived and were easier to grow and transfect than the other cell lines. We also found that these cells do not express mGX sPLA2 at the mRNA level by RT-qPCR analysis and at the protein level by both a highly specific and sensitive time-resolved fluoroimmunoassay (Eerola et al., 2006) and Escherichia coli sPLA2 enzymatic assays (data not shown). Compared with various mouse sPLA2s, we found that group X sPLA2 has the strongest mitogenic capacity on Colon-26 cells (Fig. 3A) and dose-dependently induced cell proliferation (Fig. 3B). mGV and mGIIA sPLA2 also significantly induced cell proliferation, whereas the other sPLA2s were relatively poor inducers. To further demonstrate that group X sPLA2 can trigger cell proliferation, we measured the growth of Colon-26 cells treated with mGX and hGX sPLA2s by direct cell counting. As indicated in Fig. 3C, cells proliferated more rapidly over time in the presence of group X sPLA2. In accordance with their proliferative effects, both mGX and hGX sPLA2s were capable of inducing a time-dependent phosphorylation of p42/44 MAPK, with a sustained induction at 4 h (Fig. 3D).
Fig. 2.

Effect of group X sPLA2 on [3H]thymidine incorporation in mouse colon cell lines. After starvation for 24 h at 37°C in serum-free and additive-free medium, all cell lines were incubated in the presence of mGX and hGX sPLA2s (200 nM) or FCS under the following conditions: Colon-26 were grown at 37°C for 24 h in DMEM and 0.02% FAF BSA (A); AJ02-nm0 cells were grown at 37°C for 48 h in RPMI 1640 medium and 0.02% FAF BSA (B); Apc(+/Min) cells were grown at 33°C in DMEM, 0.02% FAF BSA, and 10 ng/ml recombinant EGF (C); and YAMC cells were incubated at 33°C for 48 h in serum-free RPMI 1640 medium and 0.02% FAF BSA (D). [3H]Thymidine was added during the last 4 h of sPLA2 stimulation, and cells were processed as described under Materials and Methods. The increase in [3H]thymidine incorporation induced by sPLA2s was compared with untreated cells (−) and FCS-treated cells. In all panels, values are representative of at least two experiments performed in triplicates. Significant differences between untreated and sPLA2-treated cells were found (**, P < 0.001; Student's t test).
Fig. 3.

Effect of various mouse sPLA2s and hGX sPLA2 on DNA synthesis, cell growth, and p42/44 MAP kinase phosphorylation in Colon-26 cells. A and B, quiescent cells were incubated for 24 h with the different mouse sPLA2s (A) or various concentrations of hGX sPLA2 (B). [3H]Thymidine incorporation is expressed as -fold increase over untreated cells (incorporation of [3H]thymidine under untreated conditions was 44,962 dpm). Values represent average values of at least three experiments performed in triplicate (A) or are representative of at least three experiments (B). Group IIA, V, and X sPLA2s significantly stimulated [3H]thymidine incorporation compared with untreated cells (**, P < 0.001; one-way ANOVA, with Bonferroni adjustment). No significant difference was found between cells treated with group V and X sPLA2s (P = 0.2176; Student's t test). C, quiescent Colon-26 cells were cultured for up to 3 days in the presence or absence of hGX or mGX sPLA2s (200 nM) and counted every day. The difference in cell number between cells cultured in the absence and presence of sPLA2 at different time points is statistically significant (**, P < 0.05; Student's t test). D, cells were incubated for various times at 37°C with mGX and hGX sPLA2s (200 nM). The cell lysates were subjected to immunoblotting with anti-phospho-specific antibodies that specifically recognize tyrosine-phosphorylated p42/44 MAPK. Equal amounts of proteins were loaded, which was verified by immunoblotting of total p42/44 MAPK proteins.
Group X sPLA2 Stimulates the Proliferation of Colon-26 Cells via Its Catalytic Activity and Independently of Binding to the M-Type Receptor.
We next analyzed the molecular mechanisms involved in the proliferative effect of group X sPLA2. To evaluate the role of sPLA2 enzymatic activity, we first used the specific sPLA2 inhibitors Me-indoxam (Singer et al., 2002) and LY329722 (compound B in Smart et al., 2006), which inhibit the catalytic activity of mGX sPLA2, with IC50 values of 500 and 75 nM, respectively. Both inhibitors clearly suppressed the proliferation and MAP kinase phosphorylation induced by mGX sPLA2 (Fig. 4, A and B). We then tested the mitogenic activity of catalytically inactive mutants of mGX and hGX sPLA2s as well as of OS2, a snake venom sPLA2 with high catalytic activity on phosphatidylcholine (Rouault et al., 2006). The H48Q mutants of mGX and hGX sPLA2s have less than 0.1% of wild-type catalytic activity (data not shown), and the D49K mutant of OS2 is fully inactive (Rouault et al., 2006). Results shown in Fig. 4C further demonstrate that the catalytic activity of group X sPLA2 is required for its mitogenic effect. Finally, we evaluated the role of cPLA2α and found that the mitogenic effect of group X sPLA2 on Colon-26 cells does not depend on cPLA2α activation because siRNA silencing of cPLA2α efficiently suppressed protein expression but has little effect on the -fold increase factor of [3H]thymidine incorporation induced by hGX sPLA2 (Fig. 4D). However, our results also suggest that cPLA2α is involved in the proliferation of Colon-26 in the absence of exogenously added sPLA2 because siRNA silencing slightly decreased the basal level of incorporation of [3H]thymidine (Fig. 4D).
Fig. 4.

The proliferative effect of group X sPLA2 on Colon-26 cells is dependent on sPLA2 catalytic activity. A and B, effect of sPLA2 inhibitors on the proliferative effect of mGX sPLA2 and MAP kinase activation. Me-indoxam (10 μM) and LY329722 (5 μM) were preincubated with mGX sPLA2 (200 nM) in DMEM and 0.02% FAF BSA for 20 min before incubation with Colon-26 cells. Under untreated conditions, incorporation of [3H]thymidine was 54,669 dpm; mGX sPLA2 alone but not mGX sPLA2 preincubated with Me-indoxam or LY329722 significantly stimulated [3H]thymidine incorporation versus untreated cells (**, P < 0.001; one-way ANOVA, with Bonferroni adjustment). Although significative (P < 0.01), the effect of Me-indoxam was probably due to a toxic effect that was not observed with LY329722 (P > 0.05). C, effect of catalytically inactive mGX H48Q, hGX H48Q, and OS2 D49K sPLA2s (200 nM) on the proliferation of Colon-26 cells (**, P < 0.001; one-way ANOVA, with Bonferroni adjustment). Under untreated conditions, the incorporation of [3H]thymidine was 74,679 dpm. D, effect of cPLA2α siRNA silencing on the proliferative effect of hGX sPLA2. Two different commercially available siRNAs (QIAGEN) targeting mouse cPLA2α were tested for silencing by Western blotting (inset), and the effect of the best cPLA2α siRNA (n°1) versus an irrelevant GFP-directed siRNA was tested for incorporation of [3H]thymidine triggered by hGX sPLA2. Colon-26 cells were transfected with siRNA on day 1, starved the next day for 24 h, and then assayed for cPLA2α expression or incorporation of [3H]thymidine triggered by hGX sPLA2 (200 nM). Under untreated conditions, the incorporation of [3H]thymidine was 45,825 dpm. The differences in incorporation of [3H]thymidine in the absence and presence of sPLA2 or between untreated cells transfected with the two siRNA are statistically significant (**, P < 0.01; Student's t test). In all panels, data are representative of two experiments.
Because the M-type sPLA2 receptor was proposed previously to play a role in cell proliferation (Kinoshita et al., 1997) and because both mGX and hGX sPLA2s bind to the mouse M-type receptor (Rouault et al., 2007), we sought to determine whether this receptor is expressed in the above-mentioned different colon cells and plays a role in the proliferative effect of group X sPLA2. We first screened the four colonic cell lines for the expression of the receptor using iodinated OS1, the snake venom sPLA2 that binds to the M-type receptor with very high affinity and specificity (Rouault et al., 2007). Apc(+/Min) cells were found to express high levels of the receptor, whereas Colon-26 and YAMC cells express low but clearly detectable levels (Fig. 5A). Conversely, AJ02-nm0 cells do not express the receptor. Scatchard plot analysis indicated that Colon-26 cells contain a single population of binding sites for iodinated OS1, with a Kd value of 90 pM and a maximal binding capacity (Bmax) of 0.016 pmol/mg total protein (data not shown). These binding data were confirmed by Western blot analysis (Fig. 5B) and RT-qPCR analysis (data not shown). The fact that group X sPLA2 stimulates the proliferation of AJ02-nm0 cells (Fig. 2B), which do not express the M-type receptor (Fig. 5A), represents a first indication that the M-type receptor is not required for the proliferative effect of group X sPLA2. This view was further supported by siRNA experiments targeting the M-type receptor expressed in Colon-26 cells. As shown in Fig. 5C, we observed by both binding assays and immunocytochemistry that the expression of the M-type receptor was dramatically reduced at 48 and 72 h after siRNA transfection, at the time window where the Colon-26 cells were stimulated with exogenous hGX and mGX sPLA2s for cell proliferation (Fig. 5D). The specificity of siRNA silencing of the receptor was validated using a nonrelevant GFP-siRNA that did not suppress M-type receptor expression (Fig. 5C). The capacity of group X sPLA2 to increase Colon-26 proliferation was unaffected by knocking-down the M-type receptor, indicating that receptor binding is not required for the proliferative effect of group X sPLA2 (Fig. 5D).
Fig. 5.

The proliferative effect of group X sPLA2 on Colon-26 cells is not dependent on binding to the M-type receptor. A and B, expression of the M-type receptor in Colon-26, YAMC, AJ02-nm0, and Apc(+/Min) cells. Mouse M-type receptor expression was measured by binding experiments (A) using the specific ligand 125I-OS1 and by Western blotting (B) of cell lysates as described under Materials and Methods. C, validation of M-type receptor silencing by siRNA. Two different commercially available siRNAs (QIAGEN) targeting the mouse M-type receptor were tested for silencing by binding assays and immunocytochemistry to measure the percentage of receptor silencing. The two siRNAs were effective, and representative results of at least two experiments obtained with siRNA1 versus an irrelevant GFP-directed siRNA are shown. Colon-26 cells were transfected with siRNAs on day 1, starved on day 2 for 24 h, and then assayed on day 3 (48 h) and day 4 (72 h) after transfection for M-type receptor expression by binding experiments using 125I-OS1 ligand. The decrease in binding at 48 and 72 h compared with GFP-siRNA is statistically significant (**, P < 0.01; Student's t test). Immunocytochemistry using mouse M-type receptor antiserum was performed 48 h after transfection. D, effect of M-type receptor siRNA1 versus GFP-directed siRNA on incorporation of [3H]thymidine triggered by hGX sPLA2. Colon-26 cells were transfected with siRNAs on day 1, starved on day 2 for 24 h, and then stimulated on day 3 for 24 h in the absence (−) or presence of mGX and hGX sPLA2s (200 nM). [3H]Thymidine was added during the last 4 h of sPLA2 stimulation, and cells were processed as described under Materials and Methods. Under untreated conditions, the incorporation of [3H]thymidine was 42,395 dpm. Incorporation of [3H]thymidine in sPLA2-treated cells over untreated is statistically significant (**, P < 0.05; Student's t test), but no significant difference was found between the two siRNAs. Data are representative of two experiments.
Group X sPLA2 Produces Various Lipid Mediators from Colon-26 Cells.
Based on the above-mentioned findings, we sought to determine whether group X sPLA2 can release various lipid mediators, including free AA, eicosanoids, and lysophospholipids from Colon-26 cells. As found previously in other cells (Singer et al., 2002), exogenously added mGX, but not mGIB, mGIIA, mGV, and mGXIIA sPLA2s, can release significant amounts of free AA from Colon-26 cells radiolabeled with [3H]AA (Fig. 6A). Similar results were obtained when cells were radiolabeled with [3H]oleate (data not shown). We also monitored the ability of group X sPLA2 to produce various eicosanoids derived from free AA by combined liquid chromatography/mass spectrometry after stimulation of Colon-26 cells by the sPLA2 for 6 h (data not shown) and 24 h (Fig. 6B). Group X sPLA2 increases the production of several eicosanoids, with PGE2 being by far the most prominent eicosanoid product, with up to 74 ng/106 cells. PGD2, PGF2α, and 5-, 12-, and 15-hydroxyeicosatetraenoic acids, and to a lesser extent leukotriene C4 and leukotriene E4, are also produced by group X sPLA2 at 6 h (data not shown) and 24 h.
Fig. 6.
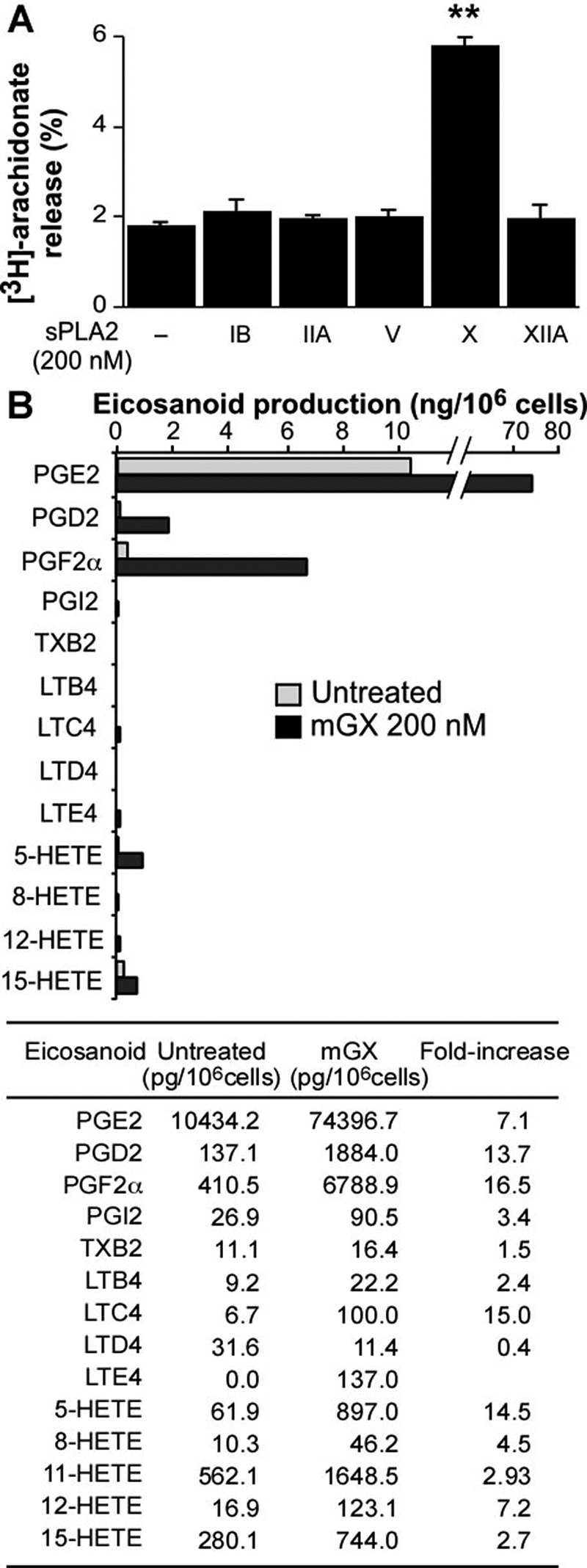
Release of AA by various mouse sPLA2s and production of eicosanoids triggered by mGX sPLA2 from Colon-26 cells. A, release of AA by various mouse sPLA2s was measured on quiescent Colon-26 cells prelabeled with [3H]AA for 24 h as described under Materials and Methods. Mouse recombinant sPLA2s (200 nM) were incubated with cells for 6 h in DMEM with 0.02% FAF BSA. [3H]AA release is expressed as the percentage of radioactivity present in cell medium relative to the total radioactivity incorporated into cells. sPLA2-X-treated cells release significantly more [3H]AA (**, P < 0.001; one-way ANOVA, with Bonferroni adjustment). A representative experiment of at least two experiments is shown. B, eicosanoid production triggered by mGX sPLA2. Colon-26 cells (106 cells) were starved for 24 h and treated with mGX sPLA2 (200 nM) for 24 h in DMEM and 0.02% BSA. The cell medium and the cell monolayer were collected, extracted with organic solvent, combined, and analyzed for eicosanoid production by liquid chromatography/mass spectrometry as described under Materials and Methods. A representative experiment of two experiments is shown.
In parallel experiments, we monitored the ability of group X sPLA2 to release various lysophospholipids, namely, LPA, LPC, LPE, lysophosphatidylglycerol, lysophosphatidylinositol, and lysophosphatidylserine. We found that group X sPLA2 can release a variety of lysophospholipids with different fatty acids at the sn-1 position from Colon-26 cells at 6 h (Fig. 7) and 24 h (data not shown). The lysophospholipids produced in highest quantities were LPC and LPE, a result that is in accordance with the relative abundance of these lipids in cell membranes and the ability of group X sPLA2 to efficiently hydrolyze zwitterionic phospholipids (Singer et al., 2002; Mitsuishi et al., 2007). For example, treatment with group X sPLA2 for 6 h led to 25- and 36-fold increases in the release of total LPC and LPE compared with untreated cells (Fig. 7A). It is important that we also found that group X sPLA2 increases by 3.7-fold the overall production of LPA. The detailed data for production of the different acyl chain species for LPC and LPA are shown in Fig. 7B. They indicate, for example, 41- and 26-fold increases for the two main products oleoyl-LPC and palmitoyl-LPC.
Fig. 7.

Release of lysophospholipids by group X sPLA2 from Colon-26 cells. A, total amount of LPX acyl chain species released by hGX sPLA2. B, release of the different LPC and LPA acyl chain species by hGX sPLA2. Colon-26 cells were starved for 24 h and treated with hGX sPLA2 (200 nM) for 6 h in DMEM and 0.02% BSA. The cell medium and the cell monolayer were collected, extracted with organic solvent, combined, and analyzed for lysophospholipid production by liquid chromatography/mass spectrometry as described under Materials and Methods. A representative experiment of two experiments is shown.
Effects of Various Lipid Mediators Released by Group X sPLA2 on the Proliferation of Colon-26 Cells.
The above-mentioned results led us to analyze the proliferative effect of each type of lipid mediators that are generated by the action of group X sPLA2. Because LPC can be further converted into LPA by autotaxin (Ferry et al., 2008), we also tested the effect of LPA on cell proliferation. As shown in Fig. 8, free AA, LPC, and LPA can all stimulate the incorporation of [3H]thymidine in Colon-26 in a dose-dependent manner. Various other fatty acids were also able to stimulate cell proliferation, although with a lower efficacy than free AA (data not shown). For example, oleic (50 μM), linoleic (50 μM), and α-linolenic (20 μM) acids could stimulate the incorporation of [3H]thymidine by factors of 1.4, 1.6, and 2.1, respectively. Furthermore, both AA and LPA were able to phosphorylate p42/44 MAPK (Fig. 8). To test the hypothesis that PGE2 production could explain at least in part the mitogenic effects of group X sPLA2, we treated Colon-26 cells with four nonselective or COX-2-selective inhibitors (aspirin, indomethacin, ibuprofen, and rofecoxib). We found that all four inhibitors were able to dramatically reduce PGE2 production (Fig. 9A) but were unable to suppress the cell proliferation induced by mGX sPLA2 (Fig. 9B). It is interesting that the COX inhibitors were also ineffective at suppressing the proliferative effect of free AA (Fig. 9C). Furthermore, we found that exogenously added PGE2 (1–1000 nM) did not induce cell proliferation (data not shown). Finally, incubation of Colon-26 cells with MK886 or baicalein, that inhibit 5- and 12-LOXs, respectively, did not alter the effect of mGX sPLA2 on cell proliferation (Fig. 9D). Together, these results indicate that although group X sPLA2 can release large amounts of PGE2 (and small amounts of other eicosanoids) from Colon-26 cells (Fig. 6), this PGE2 is at best a minor effector of the sPLA2 proliferative effect. Rather, the sPLA2 proliferative effect seems to be due to the combined production and direct action of free AA and lysophospholipids, including LPC and LPA.
Fig. 8.

Effect of AA, LPA, and LPC on Colon-26 proliferation. Effect of free AA (A), LPA (B), and LPC (C) on [3H]thymidine incorporation and p42/44 MAPK phosphorylation. Quiescent Colon-26 cells were stimulated with various concentrations of the different lipid mediators, and [3H]thymidine incorporation was evaluated as described under Materials and Methods. Under untreated conditions, the incorporation of [3H]thymidine was 42,493 dpm. The effect of AA and LPA on p42/44 phosphorylation was measured by incubating Colon-26 cells for the indicated periods at 37°C with 20 μM AA and 5 μM LPA as described in Fig. 3 legend. A representative experiment of at least two separate experiments is shown (*, P < 0.05; **, P < 0.001 over untreated cells; one-way ANOVA, with Bonferroni adjustment).
Fig. 9.

Effect of COX and LOX inhibitors on PGE2 release and proliferation in Colon-26. A, effect of COX inhibitors on PGE2 release. Quiescent Colon-26 cells were preincubated with 20 μM aspirin, ibuprofen, indomethacin, or rofecoxib for 20 min before adding mGX sPLA2 (200 nM) for 24 h. Supernatants were collected and analyzed for PGE2 by enzyme-linked immunosorbent assay as described under Materials and Methods. B and C, effect of COX inhibitors on mGX- and AA-induced cell proliferation. Quiescent Colon-26 cells were preincubated with COX inhibitors for 20 min before adding mGX sPLA2 (B) or AA (C). The -fold increase in [3H]thymidine incorporation over untreated cells is shown. Under untreated conditions, the incorporation of [3H]thymidine was 41,719 dpm. D, effects of the 5-LOX inhibitor MK886 (10 μM) and the 12-LOX inhibitor baicalein (10 μM) on Colon-26 cell proliferation. Under untreated conditions, the incorporation of [3H]thymidine was 39,550 dpm. A representative experiment of at least two experiments is shown in the different panels, and results statistically significant over their respective controls are indicated (**, P < 0.001; one-way ANOVA, with Bonferroni adjustment).
Discussion
This work shows for the first time that group X sPLA2 can stimulate the proliferation of colon cancer cells and activate the phosphorylation of p42/44 MAPK via its potent catalytic activity and ability to produce various lipid mediators. We found that the proliferative effect of group X sPLA2 was much higher than that of other mouse sPLA2s. This effect was as strong as or higher than that of sPLA2-IB, -IIA, or -III acting on different cell types (Kinoshita et al., 1997; Sved et al., 2004; Murakami et al., 2005; Belinsky et al., 2007). mGV and mGIIA sPLA2s, but not other enzymes, were also able to promote cell proliferation. Except for mGIIA, the ability of the different sPLA2s to stimulate proliferation seems to be linked to their direct capacity to hydrolyze phosphatidylcholine and release AA and LPC from cells (Singer et al., 2002; Masuda et al., 2005). As found previously in LNCaP prostatic cancer cells (Sved et al., 2004), it is possible that the proliferative effects mediated by mGIIA sPLA2 is due to activation of cPLA2α, but we did not further investigated this point.
The proliferative effect of various sPLA2s has been associated with binding to the M-type receptor (Kinoshita et al., 1997) or enzymatic activity of sPLA2 and subsequent activation of cPLA2α (Sved et al., 2004; Murakami et al., 2005). We found here that the proliferative effect of group X sPLA2 fully depends on its intrinsic catalytic activity and not on binding to the M-type receptor or cPLA2α activation. First, two specific sPLA2 inhibitors suppressed the proliferative effect of group X sPLA2. Second, the H48Q mutants of mGX and hGX sPLA2s and the D49K mutant of OS2 (Rouault et al., 2006), which have less than 0.1% of wild-type enzymatic activity but still bind with high affinity to the M-type receptor (data not shown; Rouault et al., 2006), had no or very modest effects on cell proliferation. Third, mGX and hGX sPLA2s can trigger proliferation on cells expressing or not the M-type receptor. Fourth, siRNA silencing of the M-type receptor in Colon-26 cells did not affect group X sPLA2-induced cell proliferation. Finally, because of the possible role of cPLA2α (Sved et al., 2004), we analyzed its role by siRNA silencing and found that group X sPLA2 stimulates cell proliferation independently of cPLA2α.
In line with the role of group X sPLA2 enzymatic activity in proliferation of Colon-26 cells, we found that group X sPLA2 can release relatively large quantities of free AA, LPC, and LPA, which are all able to stimulate cell proliferation and activation of MAP kinase phosphorylation. AA is the precursor of numerous eicosanoids, including PGE2 and leukotriene D4, which contribute to colon cancer cell proliferation (Wang and Dubois, 2006). Group X sPLA2 increases the production of various prostaglandins, leukotrienes, and hydroxyeicosatetraenoic acids, but these mediators do not explain the sPLA2 mitogenic effect (Fig. 9). It has been shown that Colon-26 cells express the four PGE2 receptors (EP1–4) and that PGE2 is mitogenic on these cells (Pozzi et al., 2004). That exogenous PGE2 or sPLA2-produced PGE2 did not trigger the proliferation of our Colon-26 cells may be linked to the high basal concentrations of PGE2 (approximately 30 nM) that already saturate EP receptors. Treatment of Colon-26 cells with 5- and 12-LOX inhibitors also does not prevent the mitogenic effect of sPLA2 or free AA, indicating that the proliferative effect of the sPLA2 is in part mediated by free AA without conversion into prostaglandins or leukotrienes.
Based on lysophospholipid analyses of cells treated with hGX sPLA2 (Fig. 7), we could estimate that the sPLA2 can release LPA and LPC at nanomolar and low micromolar concentrations, respectively. Although these concentrations are lower than those of exogenously added LPA and LPC (Fig. 8), lysophospholipids produced endogenously may be more effective because they are released locally at the cell surface. The proliferative effect of LPA is probably explained by its binding to receptors LPA1, LPA2, and LPA4, which are expressed in Colon-26 cells (Supplemental Fig. 3S). Whether the proliferative effects of exogenous LPC is due to conversion into LPA by ATX and subsequent binding to LPA receptors or to a direct action via other G protein-coupled receptors and/or transactivation of tyrosine kinase receptors (Ikeno et al., 2005; Fujita et al., 2006) remains an open question. We analyzed Colon-26 cells for expression of ATX at the mRNA and protein levels (Supplemental Fig. 3S). Although detectable amounts of ATX mRNA were observed by RT-qPCR analysis, no ATX protein could be detected by Western blot analysis using two distinct antibodies and a radioactive lysophospholipase D enzymatic assay (Ferry et al., 2008). We also analyzed the effect of the recently described ATX inhibitor S32826 (Ferry et al., 2008) on the production of LPA triggered by hGX sPLA2 in conditions identical to those used in Fig. 7. The ATX inhibitor S32826 had no effect on LPA production at 1 and 10 μM (Supplemental Fig. 3S), further indicating that Colon-26 cells do not express ATX. It thus seems unlikely that the LPA produced by group X sPLA2 comes from released LPC being converted into LPA by ATX. Although we cannot rule out the presence of another lysophospholipase D-like activity in Colon-26 cells, our finding raises the possibility that group X sPLA2 directly hydrolyzes cellular PA to generate LPA by acting either at the plasma membrane or after shuttling into intracellular compartments enriched into PA. It is noteworthy that the total amount of LPA released by group X sPLA2 was less than 3% of the LPC produced (Fig. 7A), suggesting that the sPLA2 may have access to little amounts of PA substrate. We have shown that group X sPLA2 can efficiently hydrolyze PA in mixed phospholipid vesicles (Singer et al., 2002), but there has been so far no report on the capacity of this enzyme to release LPA from cells. Only a few studies have suggested that group IIA sPLA2 may be involved in LPA release (Fourcade et al., 1995; Snitko et al., 1997). Together, the above-mentioned data indicate that the proliferative effects of group X sPLA2 are likely to be dependent on the combined production of both free fatty acids, including AA, and lysophospholipids, including LPC and LPA.
We also found by RT-qPCR that group X sPLA2 is expressed at very high levels in the small intestine and more particularly in the colon of C57BL/6J and BALB/c mice. The cellular sites of mGX sPLA2 expression include columnar epithelial cells, Paneth cells, and ganglion cells. It should be noted that the very low level of expression measured for mGIIA sPLA2 mRNA in C57BL/6J mice is due to the natural disruption of the pla2g2a gene. In the small intestine of BALB/c mice harboring a functional gene, the expression level of mGIIA was much higher than those of group X sPLA2 and other sPLA2s (Fig. 1B). However, the expression of mGIIA dramatically decreased in the colon, whereas that of mGX sPLA2 increased, making mGX sPLA2 among the most highly expressed sPLA2 gene in the proximal and more particularly in the distal colon of both C57BL/6J and BALB/c mice (Fig. 1, A and B). In good accordance, group IIA and X sPLA2s are also the most highly expressed sPLA2s in human colon (Mounier et al., 2008). These observations raise the question as to whether the two enzymes play redundant or divergent functions within the small intestine and colon. Based on their unique molecular and functional features, it is likely that the two sPLA2s have distinct roles. First, group IIA sPLA2 is a very basic protein, whereas group X is the most acidic sPLA2 (Lambeau and Gelb, 2008). Second, group IIA sPLA2 binds tightly to anionic phospholipid interfaces but not zwitterionic interfaces, whereas group X sPLA2 shows similar binding (Singer et al., 2002). Third, their expression in normal intestine is different in both mouse and human species (see above). Fourth, there is a strong up-regulation of group IIA sPLA2 at either mRNA or protein levels in inflammatory bowel diseases and probably in mouse and human colon tumors (Ilsley et al., 2005; Cummings, 2007). Alternatively, there is no conclusive evidence for up-regulation of group X sPLA2 in the small intestine and colon of ApcΔ716 (Takaku et al., 2000) and ApcΔ14 mice (Supplemental Fig. 2S), two models of human familial adenomatous polyposis. No up-regulation was observed in the colon tumors of mice treated with the carcinogen azoxymethane (Ilsley et al., 2005) and in human colorectal adenocarcinomas (Osterström et al., 2002; Mounier et al., 2008). This is probably reminiscent of the fact that the catalytic activity of group X sPLA2, but not group IIA, may be regulated at the post-translational level by maturation of its N-terminal propeptide by a still poorly defined proteolytic mechanism (Cupillard et al., 1997; Masuda et al., 2005). Finally, group IIA sPLA2 probably plays an important antibacterial role in the intestine and is accordingly highly expressed in Paneth cells of the small intestine and epithelial cells of colonic mucosa (Nevalainen et al., 2008). Its antitumoral role in colon cancer has been proposed to be linked to this antibacterial activity, but the mechanism is still enigmatic (Fijneman and Cormier, 2008). Conversely, group X sPLA2 has been proposed to play a central role in AA release and PGE2 production in the colon but not in the small intestine where cPLA2α would play the major role (Morioka et al., 2000; Takaku et al., 2000). This hypothesis fits well with our RT-qPCR data showing a 5-fold higher expression of group X sPLA2 over cPLA2α in the colon but not in the small intestine (Fig. 1). Whether autotaxin and/or LPA receptors, which are overexpressed in cancer (Parrill and Baker, 2008), contribute to the effects of group X sPLA2 is unknown. That group X sPLA2 can produce various lipid mediators suggests that this enzyme may also regulate other key events in tumorigenesis (Wang and Dubois, 2006). Finally, the presence of mGX sPLA2 mRNA in ganglion cells may suggest additional functions in the enteric nervous system, including neuritogenesis (Masuda et al., 2005), peristaltic reflex, or nociception.
In conclusion, we have shown that group X sPLA2 can stimulate the in vitro proliferation of colon cancer cells via its enzymatic activity and production of free AA and lysophospholipids. The high expression of group X sPLA2 in normal colon and tumors in both mouse and human species and its ability to produce various lipid mediators suggests that this enzyme plays a similar role in vivo.
Supplementary Material
Acknowledgments
We are particularly grateful to Catherine Le Calvez and Dr. Sabine Scarzello for the production and characterization of several recombinant sPLA2 proteins and to Dr. Morgane Rouault for the OS2 proteins. We thank Drs. F. Pierre, R. H. Whitehead, D. W. Rosenberg, and C. Perret for providing Apc(+/Min), YAMC, AJ02-nm0 cells and mouse colon tumor samples, and ApcΔ14 mice, respectively. We greatly acknowledge Drs. Gilles Ferry and Jean A. Boutin (Institut de Recherches SERVIER, Croissy-sur-Seine, France) for providing recombinant autotaxin, a specific antibody, and its inhibitor S32826. We also thank Nina Niemi for in situ hybridization experiments on mouse colon and Drs. D. W. Rosenberg, J. Pannequin, J. S. Saulnier-Blache, and D. Wendum for helpful discussions.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported in part by the Centre National de la Recherche Scientifique and the Association pour la Recherche sur le Cancer [Grant 3977] (to G.L.) and the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL36235, HL50040] (to M.H.G.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
1 A comprehensive abbreviation system for the various mouse and human sPLA2s is used. Each sPLA2 is abbreviated with the lowercase letter indicating the sPLA2 species (m and h for mouse and human, respectively) followed by uppercase letters identifying the sPLA2 group (GIB, GIIA, GIIC, GIID, GIIE, GIIF, GIII, GV, GX, GXIIA, GXIIB).
- PLA2
- phospholipase A2
- sPLA2
- secreted phospholipase A2
- cPLA2
- cytosolic phospholipase A2
- Apc
- adenomatous polyposis coli
- AA
- arachidonic acid
- m
- mouse
- MAP
- mitogen-activated protein
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- PGE2
- prostaglandin E2
- FAF BSA
- fatty acid-free bovine serum albumin
- ITS
- insulin, transferrin, selenious acid
- MAPK
- mitogen-activated protein kinase
- MK886
- 3-[1-(p-chlorophenyl)-5-isopropyl-3-tert-butylthio-1H-indol-2-yl]-2,2-dimethylpropanoic acid
- ATX
- autotaxin
- ECF
- enhanced chemifluorescence
- siRNA
- small interfering RNA
- h
- human
- RT-qPCR
- reverse transcription quantitative polymerase chain reaction
- DIG
- digoxigenin
- SSC
- standard saline citrate
- PBS
- phosphate-buffered saline
- TBS
- Tris-buffered saline
- GFP
- green fluorescent protein
- OS1
- Oxyuranus scutellatus scutellatus sPLA2-1
- OS2
- Oxyuranus scutellatus scutellatus sPLA2-2
- LPA
- lysophosphatidic acid
- LPC
- lysophosphatidylcholine
- ANOVA
- analysis of variance
- LPE
- lysophosphatidylethanolamine
- COX
- cyclooxygenase
- LOX
- lipoxygenase
- S32826
- [4-(tetradecanoylamino)benzyl]phosphonic acid
- PA
- phosphatidic acid
- LY329722
- 3-(3-aminooxalyl-1-benzyl-2-ethyl-6-methyl-1H-indol-4-yl)propionic acid.
References
- Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ. (2001) Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal Biochem 292:287–295 [DOI] [PubMed] [Google Scholar]
- Belinsky GS, Rajan TV, Saria EA, Giardina C, Rosenberg DW. (2007) Expression of secretory phospholipase A2 in colon tumor cells potentiates tumor growth. Mol Carcinog 46:106–116 [DOI] [PubMed] [Google Scholar]
- Cummings BS. (2007) Phospholipase A2 as targets for anti-cancer drugs. Biochem Pharmacol 74:949–959 [DOI] [PubMed] [Google Scholar]
- Cupillard L, Koumanov K, Mattéi MG, Lazdunski M, Lambeau G. (1997) Cloning, chromosomal mapping, and expression of a novel human secretory phospholipase A2. J Biol Chem 272:15745–15752 [DOI] [PubMed] [Google Scholar]
- Eerola LI, Surrel F, Nevalainen TJ, Gelb MH, Lambeau G, Laine VJ. (2006) Analysis of expression of secreted phospholipases A2 in mouse tissues at protein and mRNA levels. Biochim Biophys Acta 1761:745–756 [DOI] [PubMed] [Google Scholar]
- Ferry G, Moulharat N, Pradère JP, Desos P, Try A, Genton A, Giganti A, Beucher-Gaudin M, Lonchampt M, Bertrand M, et al. (2008) S32826, a nanomolar inhibitor of autotaxin: discovery, synthesis and applications as a pharmacological tool. J Pharmacol Exp Ther 327:809–819 [DOI] [PubMed] [Google Scholar]
- Fijneman RJ, Cormier RT. (2008) The roles of sPLA2-IIA (Pla2g2a) in cancer of the small and large intestine. Front Biosci 13:4144–4174 [DOI] [PubMed] [Google Scholar]
- Forest V, Pierre F, Bassonga E, Meflah K, Olivier C, Menanteau J. (2003) Apc+/Min colonic epithelial cells express TNF receptors and ICAM-1 when they are co-cultured with large intestine intra-epithelial lymphocytes. Cell Immunol 223:70–76 [DOI] [PubMed] [Google Scholar]
- Fourcade O, Simon MF, Viodé C, Rugani N, Leballe F, Ragab A, Fournié B, Sarda L, Chap H. (1995) Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 80:919–927 [DOI] [PubMed] [Google Scholar]
- Fujita Y, Yoshizumi M, Izawa Y, Ali N, Ohnishi H, Kanematsu Y, Ishizawa K, Tsuchiya K, Tamaki T. (2006) Transactivation of fetal liver kinase-1/kinase-insert domain-containing receptor by lysophosphatidylcholine induces vascular endothelial cell proliferation. Endocrinology 147:1377–1385 [DOI] [PubMed] [Google Scholar]
- Haapamäki MM, Grönroos JM, Nurmi H, Alanen K, Nevalainen TJ. (1999) Gene expression of group II phospholipase A2 in intestine in Crohn's disease. Am J Gastroenterol 94:713–720 [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, Chi EY, Bollinger JG, Tien YT, Ye X, Castelli L, Rubtsov YP, Singer AG, Chiang GK, Nevalainen T, et al. (2007) Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med 204:865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno Y, Konno N, Cheon SH, Bolchi A, Ottonello S, Kitamoto K, Arioka M. (2005) Secretory phospholipases A2 induce neurite outgrowth in PC12 cells through lysophosphatidylcholine generation and activation of G2A receptor. J Biol Chem 280:28044–28052 [DOI] [PubMed] [Google Scholar]
- Ilsley JN, Nakanishi M, Flynn C, Belinsky GS, De Guise S, Adib JN, Dobrowsky RT, Bonventre JV, Rosenberg DW. (2005) Cytoplasmic phospholipase A2 deletion enhances colon tumorigenesis. Cancer Res 65:2636–2643 [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Handa N, Hanada K, Kajiyama G, Sugiyama M. (1997) Activation of MAP kinase cascade induced by human pancreatic phospholipase A2 in a human pancreatic cancer cell line. FEBS Lett 407:343–346 [DOI] [PubMed] [Google Scholar]
- Kita Y, Ohto T, Uozumi N, Shimizu T. (2006) Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim Biophys Acta 1761:1317–1322 [DOI] [PubMed] [Google Scholar]
- Kita Y, Takahashi T, Uozumi N, Shimizu T. (2005) A multiplex quantitation method for eicosanoids and platelet-activating factor using column-switching reversed-phase liquid chromatography-tandem mass spectrometry. Anal Biochem 342:134–143 [DOI] [PubMed] [Google Scholar]
- Lambeau G, Gelb MH. (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem 77:495–520 [DOI] [PubMed] [Google Scholar]
- MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. (1995) The secretory phospholipase A2 gene is a candidate for the mom1 locus, a major modifier of Apcmin-induced intestinal neoplasia. Cell 81:957–966 [DOI] [PubMed] [Google Scholar]
- Masuda S, Murakami M, Takanezawa Y, Aoki J, Arai H, Ishikawa Y, Ishii T, Arioka M, Kudo I. (2005) Neuronal expression and neuritogenic action of group X secreted phospholipase A2. J Biol Chem 280:23203–23214 [DOI] [PubMed] [Google Scholar]
- Mitsuishi M, Masuda S, Kudo I, Murakami M. (2007) Human group III phospholipase A2 suppresses adenovirus infection into host cells. Evidence that group III, V and X phospholipase A2s act on distinct cellular phospholipid molecular species. Biochim Biophys Acta 1771:1389–1396 [DOI] [PubMed] [Google Scholar]
- Morioka Y, Ikeda M, Saiga A, Fujii N, Ishimoto Y, Arita H, Hanasaki K. (2000) Potential role of group X secretory phospholipase A2 in cyclooxygenase-2-dependent PGE2 formation during colon tumorigenesis. FEBS Lett 487:262–266 [DOI] [PubMed] [Google Scholar]
- Mounier CM, Wendum D, Greenspan E, Fléjou JF, Rosenberg DW, Lambeau G. (2008) Distinct expression pattern of the full set of secreted phospholipases A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. Br J Cancer 98:587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Masuda S, Shimbara S, Ishikawa Y, Ishii T, Kudo I. (2005) Cellular distribution, post-translational modification, and tumorigenic potential of human group III secreted phospholipase A2. J Biol Chem 280:24987–24998 [DOI] [PubMed] [Google Scholar]
- Nevalainen TJ, Graham GG, Scott KF. (2008) Antibacterial actions of secreted phospholipases A2. Review. Biochim Biophys Acta 1781:1–9 [DOI] [PubMed] [Google Scholar]
- Osterström A, Dimberg J, Fransén K, Söderkvist P. (2002) Expression of cytosolic and group X secretory phospholipase A(2) genes in human colorectal adenocarcinomas. Cancer Lett 182:175–182 [DOI] [PubMed] [Google Scholar]
- Parrill AL, Baker DL. (2008) Autotaxin inhibition: challenges and progress toward novel anti-cancer agents. Anticancer Agents Med Chem 8:917–923 [DOI] [PubMed] [Google Scholar]
- Pozzi A, Yan X, Macias-Perez I, Wei S, Hata AN, Breyer RM, Morrow JD, Capdevila JH. (2004) Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem 279:29797–29804 [DOI] [PubMed] [Google Scholar]
- Rouault M, Le Calvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Bollinger J, Gelb MH, et al. (2007) Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry 46:1647–1662 [DOI] [PubMed] [Google Scholar]
- Rouault M, Rash LD, Escoubas P, Boilard E, Bollinger J, Lomonte B, Maurin T, Guillaume C, Canaan S, Deregnaucourt C, et al. (2006) Neurotoxicity and other pharmacological activities of the snake venom phospholipase A2 OS2: the N-terminal region is more important than enzymatic activity. Biochemistry 45:5800–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. (2006) The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761:1246–1259 [DOI] [PubMed] [Google Scholar]
- Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, et al. (2002). Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem 277:48535–48549 [DOI] [PubMed] [Google Scholar]
- Smart BP, Oslund RC, Walsh LA, Gelb MH. (2006) The first potent inhibitor of Mammalian group X secreted phospholipase A2: elucidation of sites for enhanced binding. J Med Chem 49:2858–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitko Y, Yoon ET, Cho W. (1997) High specificity of human secretory class II phospholipase A2 for phosphatidic acid. Biochem J 321:737–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved P, Scott KF, McLeod D, King NJ, Singh J, Tsatralis T, Nikolov B, Boulas J, Nallan L, Gelb MH, et al. (2004) Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res 64:6934–6940 [DOI] [PubMed] [Google Scholar]
- Takaku K, Sonoshita M, Sasaki N, Uozumi N, Doi Y, Shimizu T, Taketo MM.2000. Suppression of intestinal polyposis in ApcΔ716 knockout mice by an additional mutation in the cytosolic phospholipase A2 gene. J Biol Chem 275:34013–34016 [DOI] [PubMed] [Google Scholar]
- Touqui L, Wu YZ. (2003) Interaction of secreted phospholipase A2 and pulmonary surfactant and its pathophysiological relevance in acute respiratory distress syndrome. Acta Pharmacol Sin 24:1292–1296 [PubMed] [Google Scholar]
- Triggiani M, Granata F, Frattini A, Marone G. (2006) Activation of human inflammatory cells by secreted phospholipases A2. Biochim Biophys Acta 1761:1289–1300, 2006 [DOI] [PubMed] [Google Scholar]
- Wang D, Dubois RN. (2006) Prostaglandins and cancer. Gut 55:115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. (1993) Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A 90:587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.