

Abstract
Some growth hormone secretagogues act as agonists at the ghrelin receptor and have been described as “ago-allosteric” ligands because of an ability to also modulate the maximum efficacy and potency of ghrelin (Holst et al., 2005). In membranes prepared from cells coexpressing the human ghrelin receptor and the G protein Gαo1, N-[1(R)-1, 2-dihydro-1-ethanesulfonylspiro-3H-indole-3,4′-piperidin)-1′-yl]carbonyl-2-(phenylmethoxy)-ethyl-2-amino-2-methylpropanamide (MK-677), growth hormone-releasing peptide 6 (GHRP-6), and the 2(R)-hydroxypropyl derivative of 3-amino-3-methyl-N-(2,3,4,5-tetrahydro-2-oxo-1-([2′-(1H-tetrazol-5-yl) (1,1′-biphenyl)-4-yl]methyl)-1H-1-benzazepin-3(R)-yl)-butanamide (L-692,585) each functioned as direct agonists, and each displayed higher efficacy than ghrelin. The effect of multiple, fixed concentrations of each of these ligands on the function and concentration-dependence of ghrelin and the effect of multiple, fixed concentrations of ghrelin on the action of MK-677, GHRP-6, and L-692,585 was analyzed globally according to a modified version of an operational model of allosterism that accounts for allosteric modulation of affinity, efficacy, and allosteric agonism. Each of the data sets was best fit by a model of simple competition between a partial and a full agonist. Both positive and negative allosteric modulators are anticipated to alter the kinetics of binding of an orthosteric agonist. However, none of the proposed ago-allosteric regulators tested had any effect on the dissociation kinetics of 125I-[His]-ghrelin, and GHRP-6 and MK-677 were able to fully displace 125I-[His]-ghrelin from the receptor. At least in the system tested, each of the ligands acted in a simple competitive fashion with ghrelin as demonstrated by analysis according to a model whereby ghrelin is a partial agonist with respect to each of the synthetic agonists tested.
The ghrelin receptor (Howard et al., 1996) was identified initially as a regulator of growth hormone release because it acted as the target of synthetic growth hormone secretagogues that induce stimulation of growth hormone release from the anterior pituitary. The endogenous ligand, ghrelin, is a 28-amino acid peptide cleaved from a 117-amino acid precursor (Van der Lely et al., 2004; Kojima and Kangawa, 2005). In addition to key roles produced via ghrelin receptors present on pituitary somatotrophs and on cells in the hypothalamus that trigger release of growth hormone releasing hormone, ghrelin stimulates gastric acid secretion and motility. Furthermore, ghrelin increases food intake, leading to weight gain and reduced fat utilization, and circulating ghrelin levels significantly increase during fasting and decrease as a response to food intake (Van der Lely et al., 2004; Leite-Moreira and Soares, 2007). At the same time, ghrelin levels are low in obese persons and high in lean persons, suggesting that ghrelin is not only important for the acute regulation of food intake but also plays an important role in the regulation of long-term energy homoeostasis; thus, the ghrelin receptor has attracted interest as a potential therapeutic target (Cummings et al., 2005). An intriguing feature of the ghrelin receptor is that it displays a high level of agonist-independent or constitutive activity (Holst et al., 2003), and this seems to be of physiological relevance (Holst and Schwartz, 2006), because mutations that suppress constitutive activity, but not ghrelin-mediated receptor activation, have been associated with both obesity and short stature (Pantel et al., 2006). Hence, inverse agonism (Milligan, 2003) would seem to be required for a ligand to suppress function of the ghrelin receptor.
A series of both growth hormone-releasing peptides and small-molecule growth hormone secretagogues has previously been shown to act as agonists at the ghrelin receptor (Howard et al., 1996; Holst et al., 2005). Moreover, these have recently been described as “ago-allosteric” ligands at the ghrelin receptor (Holst et al., 2005; Schwartz and Holst, 2006) because, in addition to producing direct activation of the receptor, when coadministered with ghrelin, such ligands acted to increase the maximum efficacy of ghrelin (Holst et al., 2005). Furthermore, coadministration of ghrelin with ligands including L-692,429 and GHRP-6 either increased or decreased, respectively, the potency of ghrelin (Holst et al., 2005). Thus, GHRP-6 and L-692,429 seemed to act both as direct agonists of the ghrelin receptor and as allosteric enhancers or allosteric inhibitors of ghrelin function. Allosteric modulators are defined as binding to a site topographically distinct from that of the endogenous ligand (Conn et al., 2009); it is of interest, therefore, that early mutational studies of the ghrelin receptor suggested that the binding sites for GHRP-6, L-692,429 and MK-677 overlap with the binding site for ghrelin (Feighner et al., 1998), and more recent studies have confirmed this (Holst et al., 2009).
Measurement of receptor function can be performed at many levels of signal transduction. However, one of the earliest is receptor-mediated activation of a heterotrimeric G protein. Furthermore, a key feature of allosteric modulators is that they alter the association and/or dissociation kinetics of the binding of orthosteric ligands (Langmead and Christopoulos, 2006). Herein, we use both of these approaches, in combination with data analysis using the operational model of agonist action (Black and Leff, 1983) linked with the allosteric ternary complex model (Ehlert, 1988) to quantify potential allosteric effects on affinity and efficacy as well as allosteric agonism (Leach et al., 2007). All the data produced for combinations of growth hormone secretagogues and ghrelin are best described by a simple, competitive binding model in which ghrelin has lower efficacy to activate the ghrelin receptor than the synthetic ligands.
Materials and Methods
Materials.
Ghrelin and [d-Arg1,d-Phe5,d-Trp7,9,Leu11]-substance P (substance P analog) were purchased from Bachem Bioscience (St. Helens, Merseyside, UK). GHRP-6 was purchased from Sigma-Aldrich (Poole, Dorset, UK). L-692,585 was purchased from Tocris (Avonmouth, Bristol, UK) 125I-[His]-ghrelin was purchased from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK).
Transfections and Tissue Culture.
HEK293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) newborn calf serum and 2 mM l-glutamine. In transient transfection studies, cells were transfected, in 100-cm2 plasticware, with 5 μg of ghrelin receptor cDNA in pcDNA3.1 and/or Gαo1 in pcDNA3.0 using Lipotectamine (Invitrogen, Paisley, UK) according to the manufacturer's instructions. In all other experiments membranes were prepared from HEK293 cells stably expressing Gαo1, which were transfected with a ghrelin receptor BacMam at 5 × 107 plaque-forming units/ml. Sodium butyrate was added to give a final concentration of 2 mM, and the transfection incubated for 24 h at 37°C.
[35S]GTPγS Binding Assays.
Guanosine 5′-O-([35S]thio)triphosphate (GTPγS) binding experiments were performed using two separate methods. In Fig. 1, cell membranes (10 μg) were incubated in 900 μl of buffer [20 mM HEPES, 100 mM NaCl, 6 mM MgCl2, and 0.1% bovine serum albumin (BSA), pH 7.4] containing 10 μM GDP, 0.1 nM [35S]GTPγS, and varying concentrations of ligands. The reaction was incubated at 30°C for 20 min and subsequently terminated by rapid filtration through GF/C filters using a Brandel cell harvester (Brandel, Gaithersberg, MD). Filters were washed three times with 3 ml of ice-cold phosphate-buffered saline, and bound radioactivity was determined by liquid scintillation counting. All other experiments used a [35S]GTPγS scintillation proximity assay in which membranes were resuspended in assay buffer [20 mM HEPES, 10 mM MgCl2, 100 mM NaCl, 0.05% (v/v) BSA, and 0.05% (v/v) Pluronic F-127, pH 7.4 at 25°C] to a concentration of 50 μg/ml and preincubated with 8 μM GDP and 2 mg/ml of wheat germ agglutinin polystyrene LEADseeker imaging beads (GE Healthcare) under gentle agitation for 30 min (25°C). Twenty-five microliters of this mixture and 25 μl of a final concentration of 0.6 nM [35S]GTPγS diluted in assay buffer were added to each well of a 384-well white plate stamped with 0.5 μl of ligand and centrifuged (800g; 2 min). After 80-min incubation, bound [35S]GTPγS was determined by scintillation counting. In experiments designed to examine potential interaction between two compounds, the assay was performed with the compound that was to be kept at a fixed concentration (e.g., ghrelin for Fig. 3, and MK-677, GHRP-6, or L-692,585 for Fig. 4) mixed under gentle agitation for 30 min (25°C) with 50 μg/ml membranes (resuspended in assay buffer as previously detailed), 8 μM GDP, and 2 mg/ml wheat germ agglutinin polystyrene LEADseeker imaging beads. Twenty-five microliters of this mixture and 25 μl of a final concentration of 0.6 nM [35S]GTPγS diluted in assay buffer were added to each well of a 384-well white plate stamped with 0.5 μl of either MK-677, GHRP-6, or L-692,585 (Fig. 3) or ghrelin (Fig. 4) and centrifuged (800g; 2 min). After an 80-min incubation, bound [35S]GTPγS was determined by scintillation counting.
Fig. 1.

The ghrelin receptor is able to cause constitutive activation of Gαo1: substance P analog is an inverse agonist; HEK293 cells were transfected to express Gαo1 (open bars) or Gαo1 and the ghrelin receptor (filled bars). The binding of [35S]GTPγS in membranes of these cells in the absence of ligand or the presence of ghrelin or SPA (both 1 μM) was then assessed. Data are presented as the percentage of the effect of ghrelin in membranes coexpressing Gαo1 and the ghrelin receptor (means ± S.E.M., n = 3). ***, p < 0.001; **, p < 0.01 (one-way ANOVA with Tukey's multiple comparison test).
Fig. 3.

Ghrelin does not alter the EMAX of MK-677, GHRP-6, or L-692,585 to activate Gαo1 via the ghrelin receptor; [35S]GTPγS binding experiments were performed on membranes of HEK293 cells transfected to coexpress Gαo1 and the ghrelin receptor. Data points represent the mean ± S.E.M of three independent experiments performed in triplicate. Data were fitted to eq. 2. Data are shown fitted with αβ = 0 and the slope transducer function constrained to 1. A, A series of concentration-response curves to MK-677 was performed in the absence (control) or presence of varying concentrations of ghrelin (as indicated). B, equivalent studies were performed with GHRP-6 and varying concentrations of ghrelin. C, equivalent studies were performed with L-692,585 and varying concentrations of ghrelin.
Fig. 4.

Growth hormone secretagogues and growth hormone releasing peptides do not alter the EMAX of ghrelin to activate of Gαo1 via the ghrelin receptor; [35S]GTPγS binding experiments were performed on membranes of HEK293 cells transfected to coexpress Gαo1 and the ghrelin receptor. Data points represent the mean ± S.E.M of three independent experiments performed in triplicate. Data were fitted to eq. 2. Data are shown fitted with αβ = 0 and the slope transducer function constrained to 1. A, a series of concentration-response curves to ghrelin was performed in the absence (control) or presence of varying concentrations of MK-677 (as indicated). B, equivalent studies were performed with ghrelin and varying concentrations of GHRP-6. C, equivalent studies were performed with ghrelin and varying concentrations of L-692,585.
125I-[His]-Ghrelin Binding Assays.
Cell membranes (5 μg) were incubated in triplicate with a final concentration of 83 pM 125I-[His]-ghrelin in a final volume of 150 μl of assay buffer (50 mM Tris-base, 2 mM EGTA, 0.1% (w/v) BSA, pH 7.3 at 4°C) (Muccioli et al., 2001). Nonspecific binding was determined by the inclusion of 1 μM ghrelin. Reactions were incubated for 120 min at 4°C and terminated by rapid filtration through GF/B filters presoaked in 0.5% (w/v) polyethylenimine and washed three times with 1 ml of ice-cold assay buffer. Bound 125I-[His]-ghrelin was measured by liquid scintillation counting.
125I-[His]-Ghrelin Competition Binding Assays.
To establish whether the growth hormone secretagogues could compete with 125I-[His]-ghrelin for binding to the ghrelin receptor, various concentrations of GHRP-6, L-692,585, and MK-677 were added to the assay mix, and the experiment was initiated, terminated, and measured as described in the preceding section.
125I-[His]-ghrelin Dissociation Assays.
For dissociation experiments, after binding for 120 min at 4°C, 1 μM ghrelin, with or without varying concentrations of GHRP-6, L-692,585, or MK-677, was added to prevent reassociation of 125I-[His]-ghrelin to the ghrelin receptor after dissociation.
Data Analysis.
Data analysis was performed using Prism software (ver. 4.0 and 5.0; GraphPad Software Inc., San Diego, CA). Unless otherwise stated, concentration-response curve data were analyzed according to a four-parameter logistic fit with data points representing the mean ± S.E.M. of three independent experiments performed in triplicate. Agonist concentration-response curves, in the absence and presence of substance P analog (SPA), were globally fitted to the following logistic equation (eq. 1) (Motulsky and Christopoulos, 2004):
 |
Top represents the maximal asymptote of the curves, Bottom represents the lowest asymptote (basal response) of the curves, LogEC50 represents the logarithm of the agonist EC50 in the absence of antagonist, [A] represents the concentration of the agonist, [B] represents the concentration of the antagonist, nH represents the Hill slope of the agonist curve, s represents the Schild slope for the antagonist, and pA2 represents the negative logarithm of the concentration of antagonist that shifts the agonist EC50 by a factor of 2. If the estimated Schild slope was not significantly different from unity, it was constrained as such, and the estimate of pA2 represented the pKB.
To investigate whether the interaction between the partial agonist, ghrelin, and a higher efficacy agonist (GHRP-6, MK-677, or L-692,585) is allosteric or simply competitive, a more complex model that incorporates the agonist activity of both compounds under test is required. The [35S]GTPγS binding data sets studying the effect of multiple fixed concentrations of ghrelin on concentration-response curves with GHRP-6, MK-677, or L-692,585 were analyzed globally according to a modified version of an operational model of allosterism that accounts for allosteric modulation of affinity, efficacy and allosteric agonism (Leach et al., 2007). The equation represents a simplified model in which it is assumed that the concentration-response curve data are to a full agonist (eq. 2):
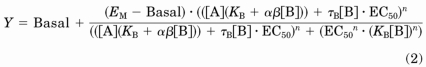 |
Basal is the response in the absence of ligand, EC50 is the midpoint of the full agonist concentration-response curve, KB is the equilibrium dissociation constant of the putative allosteric ligand, τB denotes the capacity of the putative allosteric ligand to exhibit agonism (a function of the intrinsic efficacy and receptor expression) and αβ represents a net affinity/efficacy cooperativity parameter that describes the effect of the putative allosteric ligand on agonist function. The terms EM and n denote the maximal possible system response and the slope factor of the transducer function that links occupancy to response, respectively. In all fits, n was constrained to 1.
If the interaction between ghrelin and GHRP-6, MK-677, or L-692,585 was competitive, then the value of αβ would be zero (because the value of the affinity cooperativity factor, α, would be zero), and eq. 2 would reduce to that for the interaction of a partial agonist and full agonist binding to the same site. Therefore, the datasets were analyzed under two conditions—where the value of αβ was left to float or constrained to zero. Comparisons of the two fits were performed using Akaike's information criterion (AICc) (Motulsky and Christopoulos, 2004) to determine the fit that was most likely to be correct.
To further validate the results of the interaction studies, experiments were performed to study the effects of multiple fixed concentrations of GHRP-6, MK-677, or L-692,585 on concentration-response curves to ghrelin. These data were analyzed using a recast version of eq. 2 such that the concentration of ghrelin was the independent variable on the x-axis (i.e., full agonist versus partial agonist). As before, the datasets were analyzed under two conditions (where the value of αβ was left to float or constrained to zero), and the fits were compared using AICc.
Results
The ghrelin receptor is most widely recognized as a G protein-coupled receptor (GPCR) able to couple effectively to the phosphoinositidase C-linked Gαq/Gα11 family G proteins and hence to the elevation of intracellular Ca2+ levels (Howard et al., 1996; Holst et al., 2003, 2005; van der Lely et al., 2004). However, like many other GPCRs (Gudermann et al., 1996; Wise et al., 1997), it is also able to modulate cellular signaling via pathways initiated via activation of other heterotrimeric G proteins (Holst et al., 2005; Camiña et al., 2007; Dezaki et al., 2007). When membranes of HEK293 cells transfected transiently to express both the ghrelin receptor and the G protein Gαo1 were employed in [35S]GTPγS binding studies, substantial levels of bound [35S]GTPγS were recovered in the absence of addition of ligands (Fig. 1). This was not observed in membranes of equivalent cells transfected to express Gαo1 but not the ghrelin receptor (Fig. 1) and is consistent with the idea that the ghrelin receptor displays significant constitutive capacity to activate Gαo1. Addition of a single, maximally effective concentration of ghrelin (10−6 M) was without effect in the absence of the ghrelin receptor but produced a significant increase (approximately 2-fold) above basal levels of bound [35S]GTPγS in membranes expressing both Gαo1 and the ghrelin receptor (Fig. 1). Further indication of the constitutive capacity of the ghrelin receptor to activate Gαo1 was that a substance P analog [d-Arg1,d-Phe5,d-Trp7,9,Leu11]Substance P (10−6 M), described previously as a ghrelin receptor inverse agonist (Holst et al., 2003, 2005) was able to reduce basal levels of [35S]GTPγS binding substantially in membranes coexpressing Gαo1 and the ghrelin receptor (Fig. 1).
Both growth hormone-releasing peptides (e.g., GHRP-6 and small-molecule growth hormone secretagogues, including MK-677 and L-692,585) have previously been shown to act as agonists at the ghrelin receptor. Each of these ligands, as well as ghrelin, increased binding of [35S]GTPγS in a concentration-dependent manner in membranes of HEK293 cells stably expressing Gαo1 and transfected to express the ghrelin receptor transiently (Fig. 2). Compared with ghrelin, each of these three ligands was a “superagonist,” generating maximal efficacy (EMAX) greater than ghrelin, whereas GHRP-6 and L-692,585 also acted with significantly lower potencies than ghrelin (see Table 1 for potency and efficacy values). To explore these observations and the suggestion that a number of synthetic agonist ligands also act as allosteric regulators of the action of ghrelin and hence as ago-allosteric ligands (Holst et al., 2005), a series of [35S]GTPγS binding studies was performed on membranes of HEK293 cells coexpressing Gαo1 and the ghrelin receptor. Multiple fixed concentrations of ghrelin were added, and concentration-response curves to each of MK-677, GHRP-6, and L-692,585 were then performed. For MK-677, the presence of ghrelin at concentrations ranging from 10−11 M to 3 × 10−10 M, which stimulated [35S]GTPγS binding to between 10 and 50% of the level that could be achieved by a maximally effective concentration of MK-677, did not alter the potency or EMAX of MK-677 (Fig. 3A; Table 2). At ghrelin concentrations of 10−9 and 10−7 M, a significant reduction in potency of MK-677 was observed (Table 2), whereas only in the presence of 10−7 M ghrelin was the EMAX of MK-677 decreased (Table 2). To determine whether the interaction between ghrelin and the secretagogues was likely to be allosteric or merely competitive, analysis of the data were performed using a modified version of an operational model of allosterism (Leach et al., 2007; see Materials and Methods). Comparison of data fits using Akaike's information criterion (Motulsky and Christopoulos, 2004) showed a clear preference for the simpler model with the value of αβ constrained to zero (Fig. 3A, Supplementary Table S1). Therefore, the [35S]GTPγS binding studies with ghrelin and MK-677 do not provide evidence to favor an allosteric mode of interaction between the two ligands but instead favor a competitive model in which a partial agonist (ghrelin) and a full agonist (MK-677) bind to a common site (see Discussion for further details). In equivalent experiments employing GHRP-6 or L-692,585 (Fig. 3, B and C), concentrations of ghrelin up to 3 × 10−10 M again did not alter the potency or EMAX of these ligands (Table 2). Similar to MK-677, only at a concentration of 10−7 M ghrelin was there a reduction in EMAX of L-692,585, whereas varying concentrations of ghrelin did not alter the EMAX of GHRP-6 (Table 2). Comparison of data fits using Akaike's information criterion (Supplementary Table S1) again showed a clear preference for the simpler model with the value of αβ constrained to zero; hence, all the data were consistent with the action of ghrelin at a site that can be considered to be orthosteric with the synthetic compounds tested.
Fig. 2.

A number of growth hormone secretagogues and growth hormone-releasing peptides act as superagonists for ghrelin receptor-mediated activation of Gαo1; The ability of varying concentrations of ghrelin, GHRP-6, MK-677, and L-692,585 (as indicated) to enhance binding of [35S]GTPγS in membranes of HEK293 cells transfected to coexpress Gαo1 and the ghrelin receptor was assessed. Data points represent means ± S.E.M. of four independent experiments performed in triplicate. See Table 1 for quantitative details.
TABLE 1.
Potency and efficacy of ghrelin and the growth hormone secretagogues as measured using a [35S]GTPγS scintillation proximity assay
The potencies and efficacies of GHRP-6, MK-677, and L-692,585 were compared with that of ghrelin. EMAX is the maximum efficacy of each ligand, where 100% equals the maximum efficacy of ghrelin. Data were fitted with concentration-response curves with Hill slopes constrained to 1. Data are presented as mean ± S.E.M.
| Ligand | pEC50 | EMAX |
|---|---|---|
| Ghrelin | 9.11 ± 0.10 | 95.4 ± 3.4 |
| GHRP-6 | 7.85 ± 0.13** | 139.5 ± 5.4* |
| MK-677 | 9.21 ± 0.12 | 139.6 ± 9.9* |
| L-692,585 | 7.60 ± 0.17** | 145.4 ± 14.0* |
P < 0.05, one-way ANOVA with Dunnett's post hoc test.
P < 0.01, one-way ANOVA with Dunnett's post hoc test.
TABLE 2.
Potency and efficacy of MK-677, L-692,585, and GHRP-6 in the presence of increasing concentrations of ghrelin, as measured using a [35S]GTPγS scintillation proximity assay
Data were fitted with concentration-response curves with the Hill slope shared between datasets. The Hill slopes were 0.78 ± 0.10 for MK-677, 0.87 ± 0.10 for L-692,585, and 0.80 ± 0.09 for GHRP-6. EMAX is displayed as percentage of maximum response to each ligand in the absence of ghrelin. Data are presented as mean ± S.E.M.
| Condition | pEC50 | EMAX |
|---|---|---|
| MK-677 | 9.58 ± 0.03 | 99.7 ± 0.8 |
| + 10 pM ghrelin | 9.43 ± 0.03 | 105.1 ± 0.9 |
| + 0.1 nM ghrelin | 9.27 ± 0.06 | 96.8 ± 1.3 |
| + 0.3 nM ghrelin | 9.16 ± 0.13 | 93.7 ± 2.6 |
| + 1 nM ghrelin | 8.32 ± 0.10** | 107.1 ± 1.5 |
| + 100 nM ghrelin | 7.82 ± 0.21** | 94.6 ± 2.3* |
| L-692,585 | 7.77 ± 0.03 | 101.3 ± 1.4 |
| + 10 pM ghrelin | 7.65 ± 0.03 | 111.4 ± 1.7 |
| + 0.1 nM ghrelin | 7.65 ± 0.05 | 102.3 ± 2.0 |
| + 0.3 nM ghrelin | 7.50 ± 0.13 | 102.0 ± 4.4 |
| + 1 nM ghrelin | 6.80 ± 0.12** | 111.5 ± 3.0 |
| + 100 nM ghrelin | 7.84 ± 0.25 | 90.3 ± 1.6* |
| GHRP-6 | 8.55 ± 0.02 | 99.6 ± 1.0 |
| + 10 pM ghrelin | 8.41 ± 0.05 | 110.6 ± 1.9 |
| + 0.1 nM ghrelin | 8.40 ± 0.70 | 106.5 ± 2.4 |
| + 0.3 nM ghrelin | 8.17 ± 0.13 | 105.0 ± 3.8 |
| + 1 nM ghrelin | 7.31 ± 0.22** | 106.5 ± 4.0 |
| + 100 nM ghrelin | 7.55 ± 0.35** | 93.4 ± 2.8 |
P < 0.05, one-way ANOVA with Dunnett's post hoc test.
P < 0.01, one-way ANOVA with Dunnett's post hoc test.
To explore this further, the experimental protocol was reversed and the effect of multiple, fixed concentrations of the synthetic compounds on concentration-response curves to ghrelin was assessed. At 3 × 10−11 M MK-677, the effect of increasing concentrations of ghrelin was still to increase binding of [35S]GTPγS above the level produced by MK-677 (Fig. 4A; Table 3). However, because of the “superagonist” effect of MK-677 compared with ghrelin, at all concentrations of MK-677 greater than or equal to 10−9 M, increasing concentrations of ghrelin caused a decrease in [35S]GTPγS binding (Fig. 4A). Comparison of the data fits was performed using the same model as described above, but recast such that the partial agonist, ghrelin, was the independent variable on the x-axis. As would be expected, the estimates for parameters such as the pEC50 of MK-677 and affinity of ghrelin were similar to the previous estimates, despite the reversed protocol (Supplementary Table S1). As before, the comparison of the data fits using AICc suggested that interaction between ghrelin and MK-677 was likely to be competitive (see Discussion).
TABLE 3.
Potency and efficacy of ghrelin in the presence of increasing concentrations of MK-677, L-692,585, and GHRP-6 as measured using a [35S]GTPγS scintillation proximity assay
Data were fitted with concentration-response curves with the Hill slope shared between datasets.
The Hill slopes were 1.07 ± 0.31 for MK-677, 1.14 ± 0.28 for L-692,585, and 1 for GHRP-6. EMAX is displayed as percentage of maximum response of ghrelin in the absence of growth hormone secretagogues. Data are presented as mean ± S.E.M.
| Condition | pEC50 | EMAX |
|---|---|---|
| Ghrelin only | 8.54 ± 0.02 | 96.0 ± 0.8 |
| + 0.03 nM MK-677 | 7.84 ± 0.06* | 117.2 ± 1.8** |
| + 0.1 nM MK-677 | 8.03 ± 0.31 | 121.5 ± 6.6** |
| + 1 nM MK-677 | 9.07 ± 0.12 | 159.0 ± 1.7** |
| + 3 nM MK-677 | 8.50 ± 0.07 | 190.8 ± 1.3** |
| Ghrelin only | 8.54 ± 0.05 | 97.3 ± 1.8 |
| + 3 nM L-692,585 | 8.03 ± 0.27 | 111.7 ± 7.7 |
| + 10 nM L-692,585 | Not fitted | Not fitted |
| + 30 nM L-692,585 | 9.53 ± 0.78 | 129.8 ± 10.6* |
| + 1 μM L-692,585 | 7.80 ± 0.39 | 177.9 ± 5.6** |
| Ghrelin only | 8.64 ± 0.02 | 100.2 ± 0.7 |
| + 0.1 nM GHRP-6 | 8.32 ± 0.21* | 108.3 ± 7.6 |
| + 1 nM GHRP-6 | 8.01 ± 0.08 | 103.1 ± 1.5 |
| + 10 nM GHRP-6 | 9.01 ± 0.15 | 134.3 ± 2.2** |
| + 100 nM GHRP-6 | 7.67 ± 0.15** | 187.6 ± 2.5** |
P < 0.05, one-way ANOVA with Dunnett's post hoc test.
P < 0.01, one-way ANOVA with Dunnett's post hoc test.
Entirely equivalent data were obtained for ghrelin concentration-response curves performed in the presence of varying concentrations of GHRP-6 (Fig. 4B; Table 3; Supplementary Table 1) and L-692,585 (Fig. 4C; Table 3; Supplementary Table 1). The data for both of these compounds fit better to a competitive, rather than allosteric model, which is consistent with ghrelin's sharing the orthosteric binding site with each of these three ligands.
Both MK-677, GHRP-6 and, less potently, L-692,585 (Fig. 5) were able to compete with 125I-ghrelin and limit its specific binding. Although sufficiently high concentrations of L-692,585 could not be employed in these studies to assess this directly, both MK-677 and GHRP-6 were able to compete fully with 125I-ghrelin and in a monophasic manner (Fig. 5), again consistent with these ligands competing for a common binding site (Table 4). Analysis of the L-692,585 inhibition curve (constraining minimum to zero and using Kd = 250 pM (see below) and 125I-ghrelin = 83 pM) result in an estimated pKi of 5.6 for L-692,585. Such competition binding studies do not, however, provide clear insight into the mechanism of the reduction in specific 125I-ghrelin by these ligands. Allosteric ligands are predicted to alter the kinetics of binding of orthosteric agonists (Langmead and Christopoulos, 2006), an effect that is often monitored by measuring changes in dissociation of a radiolabeled orthosteric ligand. In membranes of HEK293 cells coexpressing Gαo1 and the ghrelin receptor, association of 125I-ghrelin to specific binding sites was fitted adequately by a monophasic hyperbola and reached a plateau within 120 min when incubated at 4°C (Fig. 6A). Dissociation studies were initiated by the addition of 10−6 M ghrelin after an initial 120 min incubation to allow binding of 125I-ghrelin. Under these conditions dissociation of 125I-ghrelin was monophasic (Fig. 6B), and the measured Kobs and Koff values resulted in an estimate for Kd of 2.53 × 10−10 M for 125I-ghrelin. To test potential allosteric effects directly 125I-ghrelin dissociation studies were performed in the presence of L-692,585. This had no effect on the kinetics of 125I-ghrelin dissociation (Fig. 7, A and B). With estimated pKi of 5.6, 1 μM L-692,585 would be predicted to occupy only some 30% of receptors. However, this is the highest concentration of ligand that we could employ for these studies. However, various concentrations of either MK-677 or GHRP-6, consistent with substantially higher receptor occupancy, also failed to alter the rate of dissociation of 125I-ghrelin (Fig. 7B). These data are again consistent with lack of an allosteric effect of these ligands on the binding of 125I-ghrelin.
Fig. 5.

The specific binding of 125I-ghrelin is inhibited by the presence of growth hormone secretagogues and growth hormone-releasing peptides; the specific binding at 4°C of 125I-ghrelin to membranes of HEK293 cells coexpressing Gαo1 and the ghrelin receptor was measured over a 120-min period in the absence and presence of varying concentrations of ghrelin, GHRP-6, MK-677, or L-692,585 (as indicated). Data points represent means ± S.E.M. of three to five independent experiments. Data are fitted to a one-site competition model.
TABLE 4.
pKi and Hill slope values obtained for ghrelin, MK-677, GHRP-6, and L-692,585 competing with 125I-ghrelin binding to the ghrelin receptor
The use of an F test revealed that data were best fit to one-site competition curves. In each instance, the Hill slope obtained was not significantly different from unity. Data are presented as mean ± S.E.M.
| Ligand | pKi | Hill Slope |
|---|---|---|
| Ghrelin | 8.97 ± 0.27 | 0.59 ± 0.41 |
| GHRP-6 | 7.51 ± 0.71 | 0.52 ± 0.63 |
| MK-677 | 8.14 ± 0.08 | 0.67 ± 0.49 |
| L-692,585 | <6.00 | Not fitted |
Fig. 6.

[125I]Ghrelin binds to and dissociates from the ghrelin receptor in a monophasic fashion. A, the specific binding at 4°C of 125I-ghrelin to membranes of HEK293 cells coexpressing Gαo1, and the ghrelin receptor was measured over time. Data were fitted to a monophasic hyperbola consistent with Kobs = 0.029 min−1. Data points represent mean ± S.E.M of three independent experiments performed in triplicate. B, after association of 125I-ghrelin as above for 120 min, dissociation of the ligand was measured over time after addition of 10−6 M ghrelin. Data are presented as a semi-log plot. Koff = 0.02 ± 0.002 min−1. Data points represent the mean of three independent experiments performed in triplicate.
Fig. 7.

Growth hormone secretagogues and growth hormone-releasing peptides do not affect the dissociation of 125I-ghrelin; as in Fig. 6B, the loss of specific binding at 4°C of 125I-ghrelin to membranes of HEK293 cells coexpressing Gαo1 and the ghrelin receptor was assessed over time as a measure of the dissociation rate. A, as well as addition of 10−6 M ghrelin at time 0, L-692,585 (3 × 10−7 M) was also present. Koff = 0.01 ± 0.00 min−1. Data points represent mean ± S.E.M of three independent experiments performed in triplicate. B, varying concentrations of GHRP-6 (top), L-692,585 (middle), or MK-677 (bottom) were added along with 10−6 M ghrelin. The level of specific binding of 125I-ghrelin was then measured at time 0 and at 60 min. Data points represent mean ± S.E.M of three independent experiments performed in triplicate; data are shown as semi-log plots and analyzed using linear regression.
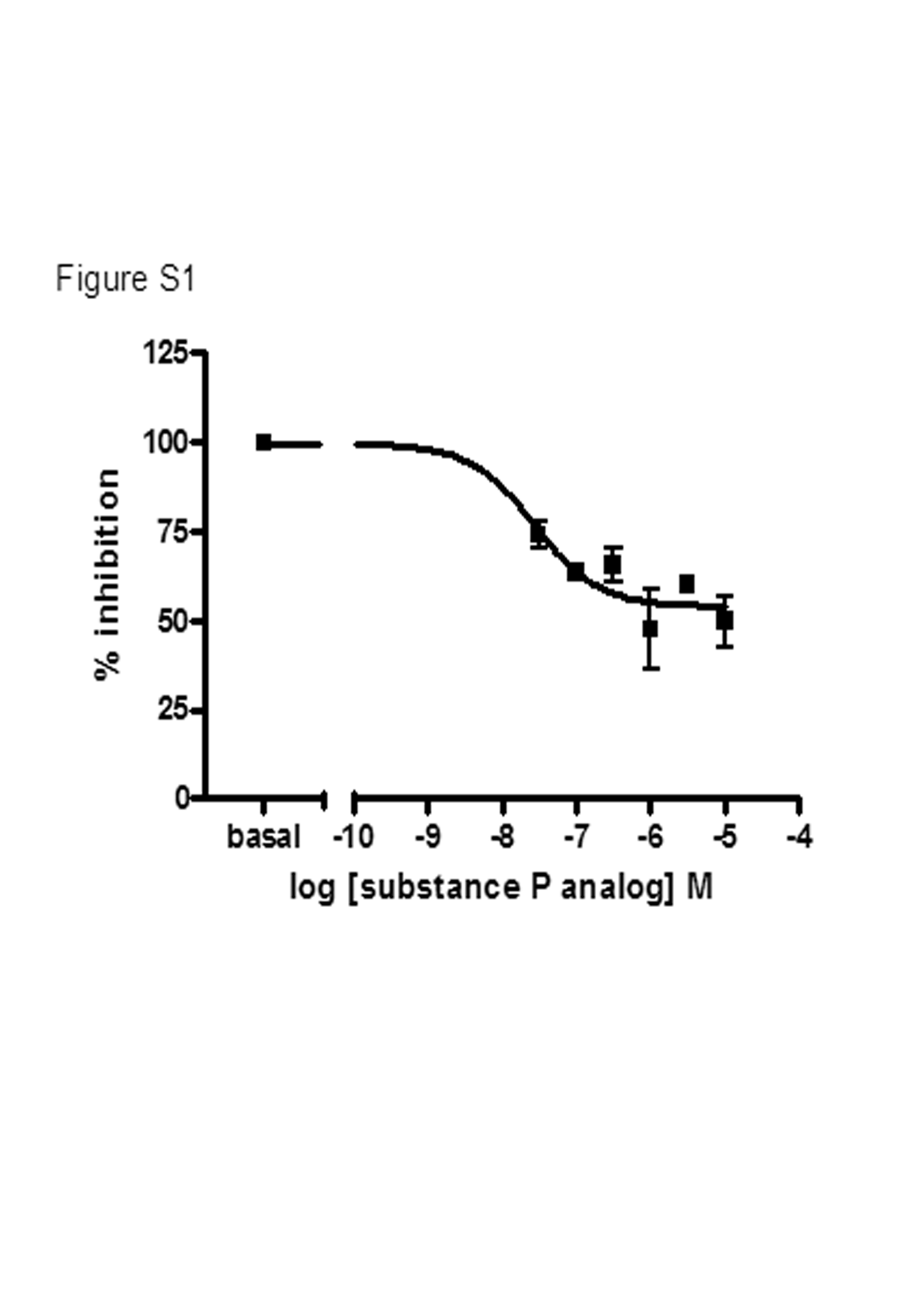
In studies exploring the effect of the substance P analog on ghrelin concentration-response curves for stimulation of [35S]GTPγS binding in membranes of HEK293 cells coexpressing Gαo1 and the ghrelin receptor, increasing concentrations of the substance P analog caused a progressive rightward shift in the EC50 for ghrelin to higher concentrations. However, an associated reduction in apparent ghrelin EMAX was observed, and such an effect could be consistent with a noncompetitive mechanism of inhibition. However, as shown in Fig. 1, the substance P analog acts as an inverse agonist for ghrelin receptor activation of Gαo1; therefore, basal binding of [35S]GTPγS in the absence of ghrelin was reduced by the presence of the substance P analog (Fig. 1) (Holst et al., 2006). When the inverse agonist effect of the substance P analog was accounted for, increasing concentrations of the substance P analog produced parallel and surmountable rightward shifts in the concentration-response to ghrelin (Fig. 8A; Supplementary Table S2) that resulted in Schild plots with slope values not significantly different from 1.0 (0.81 ± 0.18) and an estimated pKB for the substance P analog of 6.58 ± 0.18. Similar data were obtained when varying concentrations of the substance P analog were used to explore the effectiveness of MK-677 (pKB = 7.10 ± 0.10), GHRP-6 (pKB = 7.49 ± 0.09) and L-692,585 (pKB = 7.45 ± 0.10) to stimulate binding of [35S]GTPγS (Fig. 8, B–D). Fitting the basal data to a concentration-response curve revealed that the potency of the substance P analog for reducing the constitutive activity of the receptor was similar to the pKB values obtained from the Schild regression data and, furthermore, revealed that maximally effective concentrations of the substance P analog could reduce the constitutive activity of the ghrelin receptor to a level 54.1 ± 3.2% of that measured in the absence of inverse agonist (Supplementary Fig. 1).
Fig. 8.

Substance P analog is a competitive antagonist of the agonist actions of ghrelin, GHRP-6, L-692,585, and MK-677. Concentration-response curves were generated to ghrelin (A) GHRP-6 (B), L-692,585 (C), or MK-677 (D) in the presence of multiple, fixed concentrations of SPA. Data shown normalized with 0% equal to the basal [35S]GTPγS binding obtained in the presence of SPA. Data were fitted to eq. 2, with the Schild slopes and Hill slopes shared across the data sets (see Supplemental Table 2). Data points represent the mean ± S.E.M of three individual experiments performed in triplicate (black, no substance P analog; red, 30 nM substance P analog; blue, 0.1 μM substance P analog; green, 0.3 μM substance P analog; purple, 1 μM substance P analog; orange, 3 μM substance P analog; pink, 10 μM substance P analog).
Discussion
A series of both nonpeptide growth hormone secretagogues and synthetic growth hormone-releasing peptides are known agonists of the ghrelin receptor. Previous studies exploring the effects of a number of these on both the binding of 125I-ghrelin and the function of ghrelin in COS-7 cells transfected to express the human ghrelin receptor have indicated inconsistencies in their action in different assay end points and shown them to possess characteristics of allosteric regulators of the action of ghrelin (Holst et al., 2005). Such data have resulted in the generation of a complex model that evokes the necessity of the ghrelin receptor's existing as a dimer and in which the various positive and negative allosteric effects on the action of ghrelin may be explained by the growth hormone secretagogues and growth hormone releasing peptides binding in distinct ways to the individual protomers of the ghrelin receptor dimer (Holst et al., 2005; Schwartz and Holst, 2006). This is intriguing because there are a growing number of instances in which ligands with highly selective affinity and/or potency for one GPCR can affect the pharmacology, function, and/or cellular distribution of a second GPCR for which they have no inherent direct affinity if the two GPCRs form a heterodimer (El-Asmar et al., 2005; Ellis et al., 2006; Parenty et al., 2008), and this has been discussed in terms of allosteric effects across the heterodimer interface (Milligan and Smith, 2007). However, such effects are substantially more challenging to explore for potential GPCR homo-dimers unless a mutated receptor, designed to alter its affinity to pharmacological agents, is paired with the corresponding wild-type receptor to generate an asymmetric homodimer or pseudo heterodimer that has distinct pharmacology at each protomer (Damian et al., 2006; Sartania et al., 2007).
It is now becoming obvious that many, and perhaps all, GPCRs are able to regulate a range of intracellular signals, and there is considerable interest in the concept of different agonists being able to selectively modulate one or other pathway (Kenakin, 1995). Such “biased” ligands may offer therapeutic advantage (Michel and Alewijnse, 2007; Urban et al., 2007). Along with the well characterized activation of Gαq/Gα11 family G proteins that results in elevation of intracellular Ca2+ levels (Howard et al., 1996; Holst et al., 2005), activation of the ghrelin receptor has been reported to generate signals mediated via the stimulatory G protein Gαs (Malagón et al., 2003) and pertussis toxin-sensitive G proteins of the Gi-family (Dezaki et al., 2007). There is also great interest in the prospect that “allosteric” ligands, which bind to a site on the receptor distinct from that of the endogenous “orthosteric” ligand, may be able to generate selective effects at individual subtypes of closely related receptors that share a common orthosteric ligand (for example, the muscarinic acetylcholine receptors) (Christopoulos et al., 1998).
In the model used herein, the human ghrelin receptor and the pertussis toxin-sensitive G protein Gαo1 were coexpressed in HEK293 cells. A pair of well studied growth hormone secretagogues and a growth hormone-releasing peptide each acted as a superagonist in activating Gαo1 compared with ghrelin. However, analysis of the data sets according to a modified version of an operational model of allosteric interaction (Leach et al., 2007; see Materials and Methods) provided no evidence to support either positive or negative allosteric effects of the various ligands studied on the action of ghrelin. Instead, such analysis favored a simpler model in which ligands of distinct efficacy compete at the orthosteric site. Allosteric ligands can cause a change in the location of an agonist concentration-response curve. This is usually manifest as a rightward or leftward shift, dependent on the nature of the cooperativity between the agonist and allosteric ligand. Allosteric cooperativity has historically been considered only in terms of effects on ligand affinity, denoted by the parameter α, which is a bidirectional thermodynamic measure of the ratio of affinities of the orthosteric ligand in the presence and absence of the allosteric ligand. Values of α > 1 represent positive cooperativity (and increase in agonist affinity and hence potency), whereas values of α < 1 represent negative cooperativity (and a decrease in affinity and hence potency). A value of α = 1 represents neutral cooperativity; the allosteric ligand does not alter agonist affinity. At very low values (α < 0.01), a negatively cooperative interaction becomes almost indistinguishable from that of simple competition (where α = 0). It is now recognized that in addition to effects on affinity, allosteric ligands can modulate agonist efficacy and even activate receptors in their own right (Langmead and Christopoulos, 2006). From a practical perspective, a number of models have been developed to analyze data sets displaying such a range of behaviors. These models use the operational model of agonist action (Black and Leff, 1983) combined with the allosteric ternary complex model (Ehlert, 1988), to quantify the allosteric effects on affinity and efficacy as well as allosteric agonism (Leach et al., 2007).
One of the hallmarks of an allosteric interaction is that any effects on agonist affinity and/or efficacy, whether positive or negative, are saturable and reflect the degree of cooperativity between the two ligands. This is in contrast to the effects of a competitive antagonist, which is theoretically limitless in its effect on agonist function. Relatively low concentrations of ghrelin had no effect on the location of the agonist curves produced by GHRP-6, MK-677, and L-692,585, but caused increases in [35S]GTPγS binding in its own right. At 10−7 M, ghrelin caused a rightward shift in the concentration-response curve to all three synthetic agonists consistent with a competitive mode of action. However, ghrelin seems to be a high-efficacy partial agonist with respect to all three agonists; as such, the window with which to examine the mechanism of interaction using this assay design is limited. To better profile the mechanism of action of the synthetic agonists, reverse studies were performed to examine the effects of multiple, fixed concentrations of GHRP-6, MK-677, or L-692,585 on a concentration response curve to ghrelin. In the absence of synthetic agonist, ghrelin stimulated [35S]GTPγS binding in a concentration-dependent manner. Increasing concentrations of GHRP-6, MK-677, or L-692,585 also stimulated [35S]GTPγS binding but to a level over and above the maximal ghrelin response. At the highest concentrations of the synthetic agonist, increasing concentrations of ghrelin actually inhibit [35S]GTPγS binding to the same level as the maximal response to ghrelin in the absence of synthetic agonist. Analysis of the data sets according to the operational model described under Materials and Methods showed a clear favor for a competitive fit in preference to an allosteric mechanism of interaction.
These studies do not attempt to replicate the model system used by Holst and colleagues (2005) and thus do not inherently repudiate their conclusions on the ago-allosteric actions at the ghrelin receptor of growth hormone secretagogues and growth hormone releasing peptides. However, these data in combination with the ligand dissociation rate studies provide clear evidence that, at least for direct activation of Gαo1 by the human ghrelin receptor, all three synthetic agonists examined share the orthosteric site with the endogenous ligand, ghrelin. Early studies indicated an overlapping binding site for ghrelin with many of these ligands, based on the similar effect on a Glu3.33 mutation in transmembrane domain III of the receptor (Feighner et al., 1998), and this is certainly also consistent with orthosteric and competitive actions of each ligand. Furthermore, recent mutational studies from Holst et al. (2009) have provided further evidence for the overlap of binding sites of the endogenous agonist ghrelin with growth hormone secretagogues and growth hormone-releasing peptides. The nature of the orthosteric binding site in receptors with large peptide ligands clearly poses a substantial challenge for pharmacological definition of the mode of action of synthetic agonist ligands. Likewise, these studies do not attempt to explore whether the ghrelin receptor acts as a dimer as suggested by the ago-allosteric model (Schwartz and Holst, 2006). Although there are now a number of reports that indicate that purified and reconstituted GPCR monomers can cause activation of G proteins (Whorton et al., 2007, 2008), there is a general consensus that many GPCRs do exist as dimers and/or higher order oligomers (Milligan, 2007, 2008), although the specific relevance of this for pharmacology and function remains a highly active area of research and debate. The current data highlight the contribution pharmacological modeling can provide to understanding and the need to apply Occam's razor to analysis of data sets.
Supplementary Material
Acknowledgments
K.A.B. thanks the Biotechnology and Biosciences Research Council for award of a CASE studentship.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by a Collaborative Awards in Science and Engineering studentship from Biotechnology and Biosciences Research Council (to K.A.B.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- L-692,429
- 3-amino-3-methyl-N-(2,3,4,5-tetrahydro-2-oxo-1-([2′-(1H-tetrazol-5-yl) (1,1′-biphenyl)-4-yl]methyl)-1H-1-benzazepin-3(R)-yl)-butanamide
- AICc
- corrected Akaike's Information Criterion
- GHRP-6
- growth hormone releasing peptide 6
- GPCR
- G protein-coupled receptor
- [35S]GTPγS
- guanosine 5′-O-([35S]thio)triphosphate
- L-692,585
- the 2(R)-hydroxypropyl derivative of 3-amino-3-methyl-N-(2,3,4,5-tetrahydro-2-oxo-1-([2′-(1H-tetrazol-5-yl) (1,1′-biphenyl)-4-yl]methyl)-1H-1-benzazepin-3(R)-yl)-butanamide.
- MK-677
- N-[1(R)-1, 2-dihydro-1-ethanesulfonylspiro-3H-indole-3,4′-piperidin)-1′-yl]carbonyl-2-(phenylmethoxy)-ethyl-2-amino-2-methylpropanamide
- SPA
- substance P analog ([d-Arg1,d-Phe5,d-Trp7,9,Leu11]-substance P)
- ANOVA
- analysis of variance.
References
- Black JW, Leff P. (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162 [DOI] [PubMed] [Google Scholar]
- Camiña JP, Lodeiro M, Ischenko O, Martini AC, Casanueva FF. (2007) Stimulation by ghrelin of p42/p44 mitogen-activated protein kinase through the GHS-R1a receptor: role of G-proteins and beta-arrestins. J Cell Physiol 213:187–200 [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Lanzafame A, Mitchelson F. (1998) Allosteric interactions at muscarinic cholinoceptors. Clin Exp Pharmacol Physiol 25:185–194 [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DE, Foster-Schubert KE, Overduin J. (2005) Ghrelin and energy balance: focus on current controversies. Curr Drug Targets 6:153–169 [DOI] [PubMed] [Google Scholar]
- Damian M, Martin A, Mesnier D, Pin JP, Banères JL. (2006) Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J 25:5693–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezaki K, Kakei M, Yada T. (2007) Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes 56:2319–2327 [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. (1988) Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol 33:187–194 [PubMed] [Google Scholar]
- El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. (2005) Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol 67:460–469 [DOI] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. (2006) Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem 281:38812–38824 [DOI] [PubMed] [Google Scholar]
- Feighner SD, Howard AD, Prendergast K, Palyha OC, Hreniuk DL, Nargund R, Underwood D, Tata JR, Dean DC, Tan CP, et al. (1998) Structural requirements for the activation of the human growth hormone secretagogue receptor by peptide and nonpeptide secretagogues. Mol Endocrinol 12:137–145 [DOI] [PubMed] [Google Scholar]
- Gudermann T, Kalkbrenner F, Schultz G. (1996) Diversity and selectivity of receptor-G protein interaction. Annu Rev Pharmacol Toxicol 36:429–459 [DOI] [PubMed] [Google Scholar]
- Holst B, Schwartz TW. (2006) Ghrelin receptor mutations—too little height and too much hunger. J Clin Invest 116:637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Brandt E, Bach A, Heding A, Schwartz TW. (2005) Nonpeptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol Endocrinol 19:2400–2411 [DOI] [PubMed] [Google Scholar]
- Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. (2003) High constitutive signaling of the ghrelin receptor—identification of a potent inverse agonist. Mol Endocrinol 17:2201–2210 [DOI] [PubMed] [Google Scholar]
- Holst B, Frimurer TM, Mokrosinski J, Halkjaer T, Cullberg KB, Underwood CR, Schwartz TW. (2009) Overlapping binding site for the endogenous agonist, small-molecule agonists, and ago-allosteric modulators on the ghrelin receptor. Mol Pharmacol 75:44–59 [DOI] [PubMed] [Google Scholar]
- Holst B, Lang M, Brandt E, Bach A, Howard A, Frimurer TM, Beck-Sickinger A, Schwartz TW. (2006) Ghrelin receptor inverse agonists: identification of an active peptide core and its interaction epitopes on the receptor. Mol Pharmacol 70:936–946 [DOI] [PubMed] [Google Scholar]
- Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, et al. (1996) A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273:974–977 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (1995) Pharmacological proteus? Trends Pharmacol Sci 16:256–258 [DOI] [PubMed] [Google Scholar]
- Kojima M, Kangawa K. (2005) Ghrelin: structure and function. Physiol Rev 85:495–522 [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. (2006) Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol Sci 27:475–481 [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389 [DOI] [PubMed] [Google Scholar]
- Leite-Moreira AF, Soares JB. (2007) Physiological, pathological and potential therapeutic roles of ghrelin. Drug Discov Today 12:276–288 [DOI] [PubMed] [Google Scholar]
- Malagón MM, Luque RM, Ruiz-Guerrero E, Rodríguez-Pacheco F, García-Navarro S, Casanueva FF, Gracia-Navarro F, Castaño JP. (2003) Intracellular signaling mechanisms mediating ghrelin-stimulated growth hormone release in somatotropes. Endocrinology 144:5372–5380 [DOI] [PubMed] [Google Scholar]
- Michel MC, Alewijnse AE. (2007) Ligand-directed signaling: 50 ways to find a lover. Mol Pharmacol 72:1097–1099 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2003) Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol 64:1271–1276 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2007) G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta 1768:825–835 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2008) A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol 153 (Suppl 1):S216–S229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Smith NJ. (2007) Allosteric modulation of heterodimeric G-protein-coupled receptors. Trends Pharmacol Sci 28:615–620 [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Christopoulos A. (2004) Fitting models to biological data using linear and nonlinear regression. A Practical Guide to Curve Fitting, Oxford University Press, New York: [Google Scholar]
- Muccioli G, Papotti M, Locatelli V, Ghigo E, Deghenghi R. (2001) Binding of 125I-labeled ghrelin to membranes from human hypothalamus and pituitary gland. J Endocrinol Invest 24:RC7–RC9 [DOI] [PubMed] [Google Scholar]
- Pantel J, Legendre M, Cabrol S, Hilal L, Hajaji Y, Morisset S, Nivot S, Vie-Luton MP, Grouselle D, de Kerdanet M, et al. (2006) Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest 116:760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenty G, Appelbe S, Milligan G. (2008) CXCR2 chemokine receptor antagonism enhances DOP opioid receptor function via allosteric regulation of the CXCR2-DOP receptor heterodimer. Biochem J 412:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartania N, Appelbe S, Pediani JD, Milligan G. (2007) Agonist occupancy of a single monomeric element is sufficient to cause internalization of the dimeric beta2-adrenoceptor. Cell Signal 19:1928–1938 [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Holst B. (2006) Ago-allosteric modulation and other types of allostery in dimeric 7TM receptors. J Recept Signal Transduct Res 26:107–128 [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13 [DOI] [PubMed] [Google Scholar]
- van der Lely AJ, Tschöp M, Heiman ML, Ghigo E. (2004) Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev 25:426–457 [DOI] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A 104:7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Jastrzebska B, Park PS, Fotiadis D, Engel A, Palczewski K, Sunahara RK. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem 283:4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Watson-Koken MA, Rees S, Lee M, Milligan G. (1997) Interactions of the alpha2A-adrenoceptor with multiple Gi-family G-proteins: studies with pertussis toxin-resistant G-protein mutants. Biochem J 321:721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}