

Abstract
We evaluated the ability of different trypsin-revealed tethered ligand (TL) sequences of rat proteinase-activated receptor 2 (rPAR2) and the corresponding soluble TL-derived agonist peptides to trigger agonist-biased signaling. To do so, we mutated the proteolytically revealed TL sequence of rPAR2 and examined the impact on stimulating intracellular calcium transients and mitogen-activated protein (MAP) kinase. The TL receptor mutants, rPAR2-Leu37Ser38, rPAR2-Ala37–38, and rPAR2-Ala39–42 were compared with the trypsin-revealed wild-type rPAR2 TL sequence, S37LIGRL42—. Upon trypsin activation, all constructs stimulated MAP kinase signaling, but only the wt-rPAR2 and rPAR2-Ala39–42 triggered calcium signaling. Furthermore, the TL-derived synthetic peptide SLAAAA-NH2 failed to cause PAR2-mediated calcium signaling but did activate MAP kinase, whereas SLIGRL-NH2 triggered both calcium and MAP kinase signaling by all receptors. The peptides AAIGRL-NH2 and LSIGRL-NH2 triggered neither calcium nor MAP kinase signals. Neither rPAR2-Ala37–38 nor rPAR2-Leu37Ser38 constructs recruited β-arrestins-1 or -2 in response to trypsin stimulation, whereas both β-arrestins were recruited to these mutants by SLIGRL-NH2. The lack of trypsin-triggered β-arrestin interactions correlated with impaired trypsin-activated TL-mutant receptor internalization. Trypsin-stimulated MAP kinase activation by the TL-mutated receptors was not blocked by inhibitors of Gαi (pertussis toxin), Gαq [N-cyclohexyl-1-(2,4-dichlorophenyl)-1,4-dihydro-6-methylindeno[1,2-c]pyrazole-3-carboxamide (GP2A)], Src kinase [4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine (PP1)], or the epidermal growth factor (EGF) receptor [4-(3′-chloroanilino)-6,7-dimethoxy-quinazoline (AG1478)], but was inhibited by the Rho-kinase inhibitor (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide, 2HCl (Y27362). The data indicate that the proteolytically revealed TL sequence(s) and the mode of its presentation to the receptor (tethered versus soluble) can confer biased signaling by PAR2, its arrestin recruitment, and its internalization. Thus, PAR2 can signal to multiple pathways that are differentially triggered by distinct proteinase-revealed TLs or by synthetic signal-selective activating peptides.
Proteinase-activated receptors (PARs) are unique members of the G-protein-coupled superfamily of receptors (GPCRs), modeled as seven transmembrane domain cell-surface receptors that mediate diverse signaling events in response to proteolytic exposure of an N-terminal tethered ligand (TL) sequence. PAR2, the second member of this family to be cloned (Nystedt et al., 1994; Bohm et al., 1996) is a serine proteinase-activated class A GPCR with an unusual TL receptor activation mechanism (Coughlin, 2000; Ramachandran and Hollenberg, 2008). Proteolytic cleavage by serine proteinases, prototypically trypsin for PAR2, reveals a sequence within the receptor N terminus (Ser37LIGRLDTP45— in rodent PAR2), that then acts as a TL, presumably by interacting with other cell surface receptor domains. It is noteworthy that PAR2 is also triggered by soluble synthetic receptor-activating peptides with sequences corresponding to that of the proteolytically revealed TL (e.g., SLIGRL-NH2).
Our previous work, aimed at identifying PAR2 TL amino acids responsible for signaling, has delineated residues within the trypsin-revealed receptor TL sequence that are critical for triggering elevations in intracellular calcium (Al-Ani et al., 2004). The first two amino acids of the trypsin-revealed TL (Ser37Leu38) were found to be important, such that a revealed rat PAR2 mutated TL sequence, SLAAAA—, was able to stimulate increases in intracellular calcium, whereas the revealed mutated TL sequences LSIGRL— and AAIGRL— were not. Furthermore, we showed that, whereas the sequence SLAAAA— could activate calcium signaling as a tethered ligand, the corresponding soluble synthetic peptide, SLAAAA-NH2, was unable to do so. These data pointed to differences in signal trafficking by PAR2 depending on whether it is activated by its own proteolytically revealed TL or by an analogous synthetic peptide. For many other GPCRs, such as those for angiotensin II, dopamine, serotonin, and adrenergic ligands, it is now accepted that there can be differential signaling, depending on the activating ligand. This agonist-dependent differential signaling has been termed “agonist-biased signaling” or “functional selectivity” (Wei et al., 2003; Galandrin et al., 2007; Kenakin, 2007; Urban et al., 2007). We therefore hypothesized that, in a unique way, the trypsin-revealed PAR2 TL might, depending on its sequence, exhibit functional selectivity, so as to target signaling to distinct responses (e.g., calcium versus MAP kinase). To test this hypothesis, we examined rat PAR2 receptor mutants with different revealed TL sequences (described above) for their ability to signal (or not) by 1) either elevating intracellular calcium or activating ERK/p42/44 MAP kinase (or both), 2) recruiting β-arrestins-1 and -2, and 3) promoting endocytosis. These three responses were monitored upon activating the wild-type and TL-mutated receptors both by a proteinase (trypsin) and by a series of soluble synthetic PAR-AP analogs with amino acid sequences corresponding to those of the N-terminal proteinase-revealed wild-type or mutated tethered ligands.
Materials and Methods
Chemicals and Other Reagents.
Enzyme inhibitors (PP1 for Src; Y27362 and H1152 for Rho kinase; doxycycline for metalloproteinases; PD153035/AG1478 for the EGF receptor kinase) were obtained from Calbiochem (La Jolla, CA). The Gαq inhibitor GP2A (used at a concentration of 10 μM) was from Tocris Bioscience (Ellisville, MO). Culture medium (Dulbecco's modified Eagle's medium) and fetal bovine serum were from Invitrogen Life Sciences (Burlington, ON, Canada). Pertussis toxin and calcium ionophore A23187 were from Sigma (St. Louis, MO). Porcine trypsin (approximately 14,900 units/mg) was also obtained from Sigma (St. Louis, MO). A maximum specific activity of 20,000 units/mg was used to calculate the approximate molar concentration of trypsin in the incubation medium (1 unit/ml ≅ 2 nM). All PAR-activating peptides and analogs were synthesized as C-terminally amidated products by the Faculty of Medicine Peptide Synthesis facility at the University of Calgary with purity verified by amino acid analysis and mass spectral analysis.
Expression Vectors.
The plasmid encoding the rat PAR2 receptor (wt-rPAR2) and the tethered ligand mutated rat PAR2 receptor variants LSIGRL (rPAR2-Leu37Ser38) or AAIGRL (rPAR2-Ala37–38) (Table 1) were constructed as described previously (Al-Ani et al., 2004). The YFP-tagged variants for all the constructs were generated by ligating the PAR genes into the pEYFP-N1 vector (Clontech, Mountain View, CA). Plasmids encoding the Gα12 and Gα13 genes in the pCDNA3.1+ vector were obtained from the University of Missouri S&T cDNA Resource Center (Rolla, MO). The sequences for all constructs were confirmed by sequencing.
TABLE 1.
wt-rPAR2 and tethered ligand mutated rPAR2 variants showing the cleavage-activation sequences resulting from trypsin exposure and the corresponding synthetic agonist peptides
| Clone | Cleavage Activation Site | TL-Derived Peptide |
|---|---|---|
| wt-rPAR2 | R/SLIGRL … | SLIGRL-NH2 |
| rPAR2-L37S38 | R/LSIGRL …. | LSIGRL-NH2 |
| rPAR2-A37–38 | R/AAIGRL …. | AAIGRL-NH2 |
Cell Lines and Transfection.
KNRK or HEK-293 cells were routinely cultured as outlined previously and passaged without the use of trypsin using an isotonic EDTA-containing dissociation buffer (Kawabata et al., 1999; Al-Ani et al., 2004) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% sodium pyruvate, and 1% antibiotic antimycotic solution. The KNRK cells do not express functional PAR2, as do the HEK-293 cells, and were thus used to generate the permanent PAR2-expressing cell lines (Table 1) for an evaluation of calcium signaling and MAP kinase activation, in keeping with previous work (Al-Ani et al., 2004). The studies of transiently transfected cells [bioluminescence resonance energy transfer (BRET) and internalization] were done with the HEK-293 cells because of their greater transfection efficiency compared with the KNRK cells. Mouse embryonic fibroblasts, wild type (MEF WT) and β-arrestin double knockout (MEF DKO), were a gift generously provided by Dr. Robert Lefkowitz (Duke University, Durham, NC) and were routinely cultured as above, except that cells were passaged by trypsin dissociation followed by repeated feeding with trypsin-free growth medium. Transfections were performed on cells that were approximately 80% confluent with Lipofectamine (Invitrogen) or FuGENE6 (Roche, Laval, QC, Canada). MEF cells were transfected by the calcium phosphate method as described previously (Chen and Okayama, 1987). KNRK cells stably expressing equivalent levels of wild-type or the TL-mutant rPAR2 were selected by quantifying cell surface receptor expression levels with flow cytometry using the B5 PAR2 selective antibody as described previously (Al-Ani et al., 2004). To test Gα12 and Gα13 involvement in rPAR2 signaling, KNRK cells stably expressing wt-rPAR2, rPAR2-Leu37Ser38, or rPAR2-Ala37–38 were transiently transfected with plasmids encoding the Gα12 and the Gα13 genes for 48 h before use in experiments studying cell signaling.
Calcium Signaling.
Calcium signaling responses were monitored as described previously (Kawabata et al., 1999). In brief, cells were grown to 80% confluence, lifted in enzyme-free cell dissociation buffer (Invitrogen), and incubated with 5 μM Fluo3-acetoxymethyl ester (Invitrogen) for 30 min at room temperature in the presence of sulfinpyrazone (0.25 mM). Fluo3-loaded cells, washed free of extracellular calcium indicator, were placed in stirred plastic cuvettes (VWR, West Chester, PA) and agonist-stimulated calcium mobilization and concomitant increases in fluorescence were monitored with an Aminco Bowan series II fluorimeter using the AB2 software (Thermo Fisher Scientific, Nepean, ON, Canada). Calcium signals monitored using an excitation wavelength of 480 nm and an emission wavelength recorded at 530 nm were expressed as a percentage of the emission fluorescence caused by 2 μM calcium ionophore A23187 (Kawabata et al., 1999).
Western Blotting.
KNRK cells transfected with wt-rPAR2, rPAR2-Leu37Ser38, or rPAR2-Ala37–38 were plated in 35-mm dishes and grown for 24 h in full 10% serum-containing medium before a switch to serum-free medium for a further 16 h. Cells were then incubated with the PAR2 proteinase agonist trypsin (10 nM) or with the synthetic TL sequence-derived peptides such as SLIGRL-NH2 (10 μM) at 37°C for 10 min. Growth medium was then removed by aspiration, and cells were lysed immediately with the addition of cold detergent-containing lysis buffer (complete Lysis-M; Roche, Laval, QC, Canada). Lysates were cleared by centrifugation at 4°C for 10 min before quantifying protein concentration with a bicinchoninic acid protein assay kit (Pierce, Rockford, IL) using bovine albumin as a standard. Lysates were resolved by electrophoresis in SDS-containing polyacrylamide gels (Biorad, Mississauga, ON, Canada) and proteins were transferred to PVDF membranes (GE Healthcare, Baie d'Urfé, QC, Canada) and blocked in detergent-containing Tris buffer [TBST: 50 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% (v/v) Tween 20] supplemented with 5% nonfat milk for 1 h at room temperature. ERK/MAP kinase phosphorylation was detected by incubating membranes with phospho-ERK1/2-specific antibodies (New England Biolabs, Pickering, ON, Canada) (1/2000 in TBST/5% milk) overnight at 4°C. phospho-ERK immunoreactivity was detected using the secondary horseradish peroxidase (HRP)-conjugated anti mouse antibody (1/15,000 in TBST/5% milk for 1 h) and the peroxidase activity was detected by chemiluminescence reagent ECL-plus (GE healthcare, Baie d'Urfé, QC, Canada) and film (Kodak). Blots were then stripped by incubation with stripping buffer (Pierce) for 15 min at RT and blocked in TBST/5% milk for 1 h before incubation with total ERK1/2 antibody (New England Biolabs) (1/2000 in TBST/5% milk) overnight at 4°C. Total-ERK immunoreactivity was detected using the secondary horseradish peroxidase conjugated anti-rabbit antibody (1/15,000 in TBST/5% milk for 1 h), and the peroxidase activity was detected using a chemiluminescence reagent (ECL-plus; GE Healthcare) with X-ray film detection (Kodak) or using a chemiluminescent gel doc system (Bio-Rad Laboratories, Mississauga, ON, Canada). Images were digitized and band intensities were quantified using the ImageJ quantification software (http://rsbweb.nih.gov/ij/). Phospho-ERK1/2/phospho-p42/44MAP kinase levels were normalized for differences in protein loading by expressing the data as a percentage of the corresponding total ERK1/2/p42/44 MAP kinase signal (pERK/tERK; Pp43/44 MAP kinase/Tp43/44 MAP kinase) detected with a pan-MAP kinase/ERK antibody.
In-Cell Western Analysis.
For all in-cell western analysis, calcium phosphate-transfected WT or DKO MEF cells were seeded into clear-bottomed black 96-well tissue culture-treated plates (Nalge Nunc International, Rochester, NY), grown to confluence and serum-starved overnight, treated with 10 nM trypsin for 0 to 90 min at 37°C, and then fixed in normal buffered formalin. Cells were gently permeabilized with phosphate-buffered isotonic saline, pH 7.4 (PBS) supplemented with 0.1% Triton X-100, then blocked for 1 h with PBS + 1% fish gelatin (Sigma). Plates were incubated overnight at 4°C with primary antibody solutions (pERK, 1:500; Cell Signaling Technology, Danvers, MA) and (tERK, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA) in PBS containing 1% (w/v) fish gelatin. Plates were washed three times with PBS, supplemented with 0.5% Tween 20, and then incubated with IR680 and IR800-conjugated secondary antibodies (1:2000; LI-COR Biosciences, Lincoln, NE) diluted in PBS/1% (w/v) fish gelatin for 1 h at room temperature. Plates were washed three times in PBS-Tween 20 and once in PBS only and then analyzed using the Odyssey infrared imaging system (LI-COR). Odyssey software was used to calculate integrated intensities of each well for quantification. A -fold increase in ERK1/2 phosphorylation (normalized to total ERK levels) over baseline was calculated.
BRET-Based Detection of β-Arrestin Interaction with wt-rPAR2 and TL-Mutated Receptors.
The wild-type and mutated receptors (wt-rPAR2, rPAR2-Leu37Ser38, and rPAR2-Ala37–38 constructs) were conjugated with YFP at the C terminus (rPAR2-YFP, rPAR2 Leu37Ser38-YFP, and rPAR2 Ala37–38-YFP) and BRET to Renilla reniformis luciferase-tagged β-arrestin-1 and -2 was determined essentially as described previously (Hamdan et al., 2007). HEK-293 cells were used instead of the KNRK cell line, because the HEK cells provide for a much greater transfection efficiency, which is required for the BRET studies. In brief, HEK-293 cells were transiently transfected with 1 μg of either rPAR2-YFP, rPAR2 Leu37Ser38-YFP, or rPAR2 Ala37–38-YFP along with 0.1 μg of the R. reniformis luciferase-tagged β-arrestin-1 and -2. Cells were plated in white 96-well culture plates (PerkinElmer Life and Analytical Sciences, Waltham, MA) and interactions between the receptors and β-arrestin were detected by BRET following the addition of 5 μM coelenterazine (Promega, Madison, WI) on a Mithras fluorescence plate reader (Berthold Technologies, Bad Wildbad, Germany) in luminescence mode using the appropriate filters. The kinetics of β-arrestin recruitment to the receptors was monitored over 20 min.
Microscopy.
HEK cells were plated on glass-bottomed Petri dishes (MatTek Corp., Ashland, MA) and transiently transfected with 1 μg of rPAR2-YFP, rPAR2-Leu37Ser38-YFP, or rPAR2-Ala37–38-YFP constructs. After 48 h, the media in the Petri dish was replaced with serum-free medium, and the cells were treated with Trypsin (10 nM) or SLIGRL-NH2 (10 μM) for 30 min at 37°C. After fixation of cells with 4% formaldehyde, the cellular localization of the receptor tag YFP signal was detected using an Olympus FV1000 confocal system on an Olympus IX70 microscope with the Fluoview system software (Olympus, Tokyo, Japan). Internalization of cell surface receptor was quantified morphometrically by counting the increased number of intracellular fluorescent speckles per cell in the images, indicative of receptor internalization to endocytic vesicles. To this end, speckles in all cells were counted in a randomly selected representative 40× image, with area dimensions approximately 100 × 100 μm (Fig. 9). The average number of speckles per cell was calculated, and the observation was repeated for comparable fields for cells observed in three independently conducted experiments. Data represent the averages of speckles per cell ± S.E.M. for three or more independent experiments.
Fig. 9.

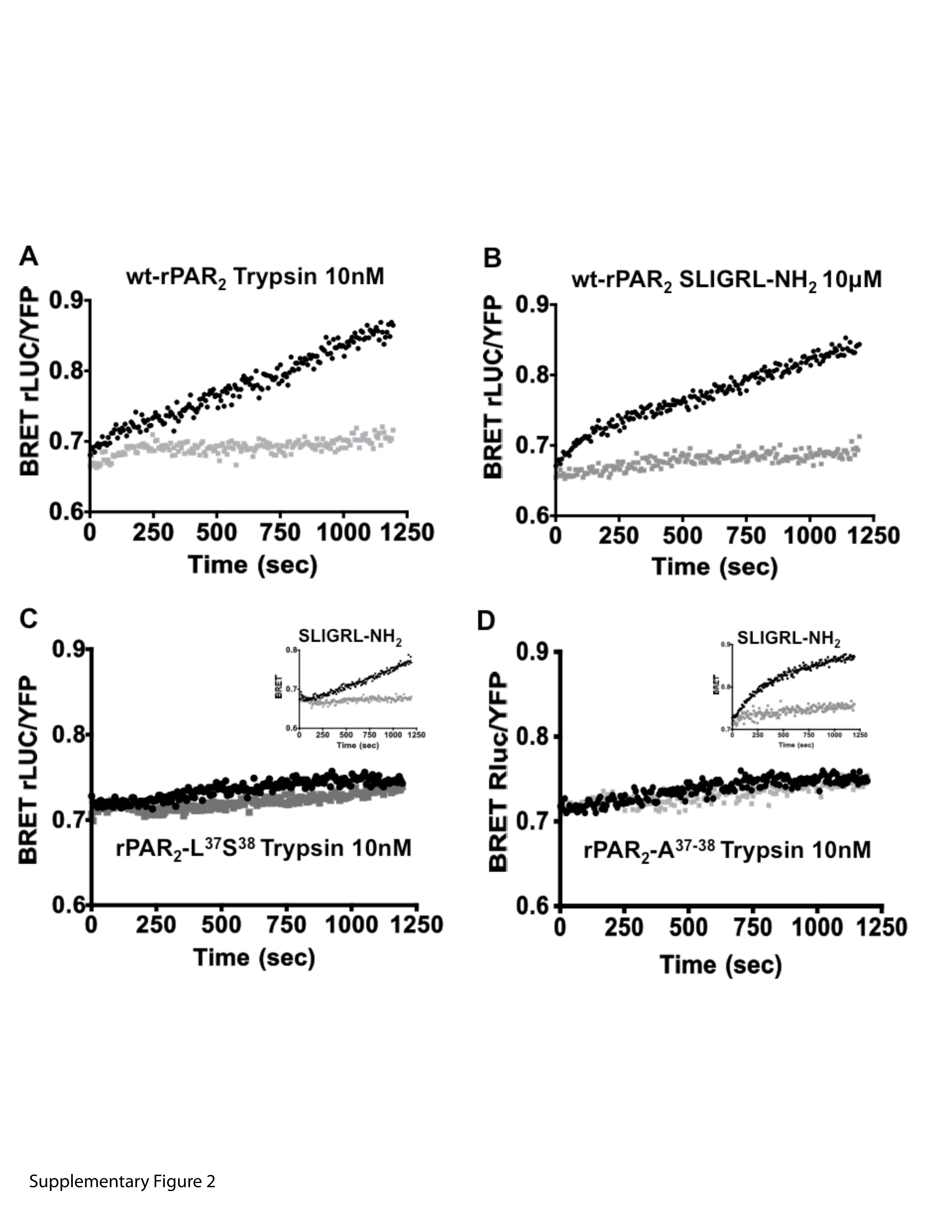
BRET measurements show a lack of trypsin-stimulated recruitment of β-arrestin-2 to rPAR2-Leu37Ser38 (C) and rPAR2-Ala37–38 (D), compared with trypsin-stimulated wt-rPAR2 (A) and SLIGRL-NH2-activated receptors (A, C, inset, and D, inset). BRET measurements (BRET rLUC/YFP) were performed as described under Materials and Methods to monitor recruitment of β-arrestin-2 to wt-rPAR2 (A and B), rPAR2-Leu37Ser38 (C), and rPAR2-Ala37–38 (D). As outlined under Materials and Methods, HEK cells transiently expressing the receptors were activated either by trypsin (A, C, and D) or by the PAR2-activating peptide (B, C, inset, and D, inset) and the BRET signals (light and dark tracings) reflecting a receptor–β-arrestin-2 interaction were monitored continuously (seconds) over a 20-min time period. An agonist-mediated increase in the BRET signal (agonist-stimulated dark tracings, A, B, C, inset, and D, inset) relative to baseline (light tracings) indicates a recruitment of β-arrestin-2 to the activated receptor. No increased BRET was observed for trypsin-treated TL-mutant receptors (coincidence of dark and light tracings, C and D). Data are representative of three independently conducted experiments.
Statistical Analysis.
Statistical analysis of data and curve fitting were done with Instat and Prism 5 software (GraphPad Software, San Diego, CA). Statistical significance was assessed using the Student's t test. In the histograms representing the Western blot data, averages are shown ± S.E.M. for three or more independent measurements of band density.
Results
Calcium Signaling.
In keeping with our previous work (Kawabata et al., 1999), we observed that activation of wt-rPAR2 by either trypsin or the receptor-selective PAR2-activating peptide SLIGRL-NH2 stimulates an increase in intracellular calcium in a concentration-dependent manner, with an EC50 for trypsin of approximately 1 nM (Fig. 1A) and an EC50 for SLIGRL-NH2 of approximately 5 μM (not shown; Al-Ani et al., 2004). Reversal of the first two amino acids of the rPAR2 tethered ligand sequence to generate the construct rPAR2-Leu37Ser38 resulted in a receptor that was unable to recruit calcium signaling after activation by trypsin but still remained responsive in terms of calcium signaling upon stimulation by SLIGRL-NH2 (Fig. 1B). Alanine substitution of the first two amino acids in the TL sequence of rPAR2 generated the construct rPAR2 Ala37–38, which, like the rPAR2 Leu37Ser38 construct, failed to trigger an elevation of intracellular calcium when cleaved by trypsin (Fig. 1A) but did yield an intracellular calcium signal in response to SLIGRL-NH2 activation (Fig. 1B). The EC50 for SLIGRL-NH2-stimulated calcium signaling by the TL-mutant receptors (EC50 ∼ 5 μM: data not shown) was equivalent to that for the wild-type receptor, illustrating that the mutations did not affect the ability of the receptor to respond to activating stimuli but rather that the modified trypsin-revealed TL could not promote calcium signaling. Our previous work established that trypsin treatment was able to unmask the mutant TL sequences (Table 1) after the cleavage at the target Arg36/Ser37 residues (Al-Ani et al., 2004). Alanine substitutions at other positions within the tethered ligand sequence, as previously reported (Al-Ani et al., 2004), resulted in constructs (e.g., rPAR2-Ala39–42, rPAR2-A37–42) that show a varying ability to trigger calcium signaling in response to trypsin (data not shown; Al-Ani et al., 2004) and were not investigated further. In keeping with our previous work, all of the synthetic peptides with sequences corresponding to those of the mutated TL receptor sequences (SLAAAA-NH2, AAIGRL-NH2, and LSIGRL-NH2) (Table 1) at concentrations up to 300 μM failed to trigger a calcium signal (Supplementary Information; Al-Ani e al., 2004). In view of our results, the focus turned to an evaluation of the two constructs, rPAR2-Leu37Ser38 and rPAR2-Ala37–38, which were defective in terms of calcium signaling in response to trypsin, in contrast with the wt-rPAR2 constructs rPAR2-A37–42 and rPAR2-Ala39–42. Thus, as a TL, the sequences revealed in the clones rPAR2-Leu37Ser38 and rPAR2-Ala37–38 were unable to activate calcium signaling, whereas rPAR2-A37–42 and rPAR2-Ala39–42 could. In contrast, none of the corresponding soluble PAR-activating peptides were able to mobilize calcium signaling.
Fig. 1.

Trypsin does not activate calcium signaling in the TL-mutant PAR2 receptors as it does in wt-rPAR2: comparison with activation by SLIGRL-NH2. A, concentration-effect curves for trypsin-triggered calcium signaling. Receptor-expressing KNRK cell lines (■, wt-PAR2; ●, rPAR2-Ala37–38; ♦, rPAR2-Leu37Ser38; □, pcDNA3) were exposed to trypsin at increasing concentrations and the elevation of intracellular calcium relative to the signal caused by 1 μM A23187 (% A23187) was monitored as described under Materials and Methods. Trypsin concentration-effect curve for calcium signaling (A) and calcium signaling (% A23187) (B) triggered by SLIGRL-NH2 (300 μM) in wt-rPAR2 (■), rPAR2-Leu37Ser38 (♦), and rPAR2-Ala37–38 (●) transfected KNRK cells. Values at each trypsin concentration (A) and in the histograms (B) represent the averages (± S.E.M., bars) for three or more independently conducted experiments.
ERK p42/44 MAP Kinase Signaling.
MAP kinase signaling responses were evaluated in cells expressing the wt-rPAR2 receptor as well as the calcium signaling-defective constructs rPAR2-Leu37Ser38 and rPAR2-Ala37–38. Using increasing concentrations of trypsin, we established that 10 nM trypsin consistently stimulated activation of the ERK/p42/44 MAP kinase pathway in the wt-rPAR2 transfected KNRK cells but not in the empty vector-transfected KNRK cells (not shown). All other experiments therefore used this concentration of trypsin to activate the cells. To test the hypothesis that the rPAR2 tethered ligand mutants rPAR2-Leu37Ser38 and rPAR2-Ala37–38, although unable to trigger calcium signaling in response to trypsin, might activate MAP kinase, we incubated KNRK cells expressing these constructs with 10 nM trypsin. We observed that trypsin stimulated a rapid increase in phospho-p42/44 levels (maximal at 5 to 10 min after trypsin exposure; see Figs. 2 and 3) that was not observed in the empty vector-transfected KNRK cells (Fig. 2). The maximal MAP kinase response seen in cells expressing the TL-mutated PAR2 constructs was approximately 70% of the response of the wt-rPAR2 transfected cells. Because the persistence of the MAP kinase signal is thought to be important for a number of physiological responses, we investigated the time course of MAP kinase activation in cells transfected with wild-type and TL-mutated PAR2 constructs (Fig. 3). We observed that in all cell lines, 10 nM trypsin stimulated a robust increase in MAP kinase that was maximal after approximately 5 min, persisted for 40 min, and began to decline thereafter, but remained above baseline for at least 1 h.
Fig. 2.

Trypsin-mediated activation of MAP kinase in KNRK cell lines expressing wild-type and TL-mutant PAR2. A, top, representative Western blots showing activation of p42/44 MAP kinase in wt-rPAR2, rPAR2-Leu37Ser38, rPAR2-Ala37–38, and pcDNA3-transfected KNRK cell lines in response to stimulation by 10 nM trypsin for 10 min. B, bottom, quantitative densitometric image analysis of p42/44 MAP kinase activation (Phospho p42/44 Western blot signal), relative to total p42/44 MAP kinase (total-p42/44 signal, top) in wt-rPAR2, rPAR2-Leu37Ser38, rPAR2-Ala37–38, and pcDNA3-transfected KNRK cells in response to stimulation by trypsin (10 nM) for 10 min. *, significant (P < 0.01) increase in phospho-p42/44 compared with baseline values. Data are representative of three independent experiments.
Fig. 3.

Time course of trypsin-triggered MAP kinase activation by wild-type and TL-mutant PAR2. A, representative Western blots showing the time course (minutes) for p42/44 MAP kinase activation (P-p42/44) by trypsin in wt-rPAR2 and TL-modified rPAR2 transfected KNRK cell lines, compared with total MAP kinase (T-p42/44). B, quantitative densitometric image analysis for the percentage increase of MAP kinase activation of P-p42/44 over baseline, normalized to T-p42/44 levels after activation by trypsin in wt rPAR2 and TL-modified rPAR2 transfected KNRK cell lines. Data represent the average values (± S.E.M., bars) for three or more independently conducted experiments.
Inhibition of Gq and Gi Modestly Attenuates MAP Kinase Activation by Trypsin in wt PAR2 but Not in the TL-Mutant Cell Lines.
Because PAR2-dependent calcium signaling is reported to be Gq-coupled, we evaluated whether the MAP kinase signaling observed in response to activation of the wt-rPAR2 as well as the tethered ligand mutants rPAR2-Leu37Ser38 and rPAR2-Ala37–38 by trypsin was dependent on Gq coupling. To suppress Gq signaling, cell lines were treated with the Gαq inhibitor GP2A (10 μM) (Zoudilova et al., 2007) before activation with the same concentration of trypsin that stimulated MAP kinase activation. As a control, the ability of GP2A to block trypsin-triggered calcium signaling in the wt-rPAR2 KNRK cell line was tested. Because neither of the mutated TL PAR2 cell lines signal via calcium when activated by trypsin, they were not tested in this way. Efficient (>90%) inhibition of Gq-mediated calcium signaling was observed in terms of the complete suppression of trypsin-triggered effects by 10 μM GP2A in the wt-rPAR2 cell line (not shown). However, in the wt-PAR2 cell line, the Gq inhibitor caused only a modest inhibition (approximately 30%) of trypsin-triggered MAP kinase activation (Fig. 4b). In contrast, under the same conditions in the presence of the Gq inhibitor GP2A, we observed no difference in the ability of trypsin to activate the p42/44 MAP kinase pathway in the two cell lines with mutated TL sequence (rPAR2 Leu37Ser38 and rPAR2 Ala37–38; Figs. 5 and 6). This lack of inhibition of MAP kinase activation was in contrast with the ability of GP2A to block SLIGRL-NH2-mediated calcium signaling in the rPAR2 Leu37Ser38 and rPAR2 Ala37–38-expressing cells (not shown). Thus, although upon PAR-AP activation the mutant receptors can engage Gq to promote calcium mobilization, the activation of Gq was not involved in the ability of these receptors to activate ERK1/2 when stimulated by trypsin. Furthermore, treatment of cells with pertussis toxin (PTX; 100 ng/ml; 16 h) to compromise Gi coupling had no effect on the ability of trypsin or SLIGRL-NH2 to cause calcium signaling in all three cell lines (not shown). In the TL-mutant cell lines, PTX treatment was similarly ineffective in blocking trypsin-activated MAP kinase signaling, although in the wt-PAR2 cell line, PTX caused a small (approximately 30%) inhibition of trypsin-triggered MAP kinase activation (Figs. 4–6). Thus, we concluded that in the TL-mutant cell lines, trypsin-mediated MAP kinase activation was independent of both Gi and Gq coupling, whereas in the wt-PAR2 cell line, MAP kinase activation by trypsin was partially Gq- and Gi-dependent.
Fig. 4.

Effects of inhibitors on trypsin-stimulated MAP kinase signaling in wt-rPAR2 KNRK cells. KNRK cells expressing wt-rPAR2 were activated by trypsin (10 nM, 10 min.) in the absence or presence of the indicated inhibitors, followed by Western blot analysis of MAP kinase activation (P-p42/44), relative to total enzyme (T-p42/44). A, representative Western blots visualizing activated (top gel, P-p42/4) and total enzyme (bottom gel, T-p42/44). B, quantitative densitometric image analysis for the percentage increase over baseline of P-p42/44 normalized to T-p42/44, integrating the signal for both bands visualized for P-p42/44 relative to T-p42/44. Asterisks denote a significant increase (*, P < 0.05; **, P < 0.01; ***, P < 0.001) compared with unstimulated cells (NT). Pound signs denote a significant decrease (#, P < 0.15; ##, P < 0.01) between the stimulation by trypsin in the presence of the Rho-kinase inhibitor Y27362 compared with cells stimulated by trypsin in the absence of the inhibitor. Data are representative of three independent experiments.
Fig. 5.

Selective impact of a Rho kinase inhibitor compared with other inhibitors on trypsin stimulated MAP kinase signaling in rPAR2-Leu37Ser38 KNRK cells. Experiments identical to the ones outlined in Fig. 4 were done using rPAR2-Leu37Ser38 KNRK cells. A, representative Western blots visualizing activated MAP kinase (top gel, P-p42/4) and total enzyme (bottom gel, T-p42/44). B, quantitative densitometric image analysis of MAP kinase activation expressed as the percentage increase over baseline (no treatment: NT) of P-p42/44 normalized to T-p42/44. Asterisks denote a significant increase (*, P < 0.05; **, P < 0.01; ***, P < 0.001) compared with unstimulated cells (NT). Pound signs denotes a significant decrease (##, P < 0.01) for the stimulation by trypsin (10 nM) in the presence of the Rho-Kinase inhibitor Y27362 (10 μM) compared with cells stimulated by trypsin in the absence of the inhibitor. Data are representative of three independent experiments.
Fig. 6.

Selective impact of a Rho kinase inhibitor compared with other inhibitors on trypsin-stimulated MAP kinase signaling in rPAR2-Ala37–38 KNRK cells. Experiments identical to the ones outlined in Fig. 4 were done using rPAR2-Leu37Ser38 KNRK cells. A, representative Western blots visualizing activated (top gel, P-p42/44) and total enzyme (lower gel, T-p42/44). B, quantitative densitometric image analysis of MAP kinase activation expressed as the percentage increase over baseline (NT, no treatment) of P-p42/44 normalized to T-p42/44. Asterisks denote a significant increase (*, P < 0.05; **, P < 0.01; ***, P < 0.001) compared with unstimulated cells (NT). Pound signs denote a significant decrease (#, P < 0.01) for the stimulation by trypsin (10 nM) in the presence of the Rho-Kinase inhibitor Y27362 (10 μM) compared with cells stimulated by trypsin in the absence of the inhibitor. Data are representative of three independent experiments.
PAR2 Mediated Activation of MAP Kinase Does Not Involve Transactivation of the EGFR.
In view of the ability of PAR2, like other G-protein-coupled receptors, to cause signaling by transactivation of the EGF receptor (EGFR) (Daub et al., 1996; McCole et al., 2002; Darmoul et al., 2004; van der Merwe et al., 2008), we tested the hypothesis that EGFR transactivation triggered by the metalloproteinase-catalyzed release of an EGFR ligand (heparin-binding EGF or transforming growth factor-α) might be involved in trypsin-mediated activation of either calcium signaling or MAP kinase in the wild-type or mutant TL-PAR2 cell lines. In KNRK cells transfected with wt-rPAR2, neither the EGFR kinase inhibitor AG1478 (1 μM) nor the broad-spectrum matrix metalloproteinase (MMP) inhibitor doxycycline (0.1 mM) inhibited calcium signaling activated by trypsin (not shown). Furthermore, neither AG1478 nor doxycycline had any effect on MAP kinase activation in any of the cell lines tested (Figs. 4–6). These results argued strongly against a role for transactivation of the EGFR in the activation of MAP kinase by trypsin in the PAR2-expressing KNRK cell lines.
Involvement of Src and Rho-Kinase in Trypsin-Mediated Activation of MAP Kinase.
To assess the potential role of Src and Rho kinase in mediating PAR2 signaling, we tested the ability of the Src-selective inhibitor PP1 (Hanke et al., 1996) and the Rho kinase inhibitor Y27362 (10 μM) (Uehata et al., 1997) to attenuate trypsin-mediated MAP kinase activation. As shown in Figs. 4 to 6, the Src-kinase inhibitor inhibited MAP kinase activation by approximately 30% in the wt-rPAR2 cell line (Fig. 4) but had no effect on trypsin-mediated MAP kinase activation in the TL mutant cell lines (Figs. 5 and 6). In contrast, the Rho kinase inhibitor blocked trypsin-triggered activation by more than 80% in all cell lines. Similar results were observed with the more potent Rho-kinase inhibitor H1152 (1 μM, not shown). To determine whether the Rho kinase-dependent signal observed was downstream of Gα12/13, we overexpressed constructs encoding Gα12 and Gα13 into KNRK cells stably expressing rPAR2-Leu37Ser38. We note an increased sensitivity to trypsin activation of MAPK in these cells with a signal observed with exposure to 5 nM trypsin, which is absent in control cells not overexpressing Gα12 and Gα13 (Fig. 7). Similar results were observed with the rPAR2-Ala37–38 receptor (not shown).
Fig. 7.

Overexpression of Gα12 and Gα13 increases sensitivity to trypsin for MAPK activation in rPAR2-Leu37Ser38 transfected KNRK cells. A, representative Western blots visualizing activated (top gel, P-p42/44) and total enzyme (bottom gel, T-p42/44) in control cells or cells transiently overexpressing Gα12 and Gα13 and activated with 5 or 10 nM trypsin. B, quantitative densitometric image analysis of MAP kinase activation expressed as the percentage increase over basal levels of P-p42/44 normalized to T-p42/44 in response to activation with 5 or 10 nM trypsin. Asterisks indicates a significant increase over basal levels (*, P < 0.05). Data are representative of two independently conducted experiments.
β-Arrestin Interactions and PAR2-Mediated MAP Kinase Signaling.
Previous work has pointed to a role for β-arrestin in facilitating a prolonged activation of MAP kinase by PAR2 (Ge et al., 2003), and it has been proposed that PAR2 may trigger responses distinct from a Gq-coupled elevation of intracellular calcium by a β-arrestin-mediated pathway (Zoudilova et al., 2007). We therefore tested the potential role(s) of β-arrestins in trypsin-mediated MAP kinase activation in the different PAR2 cell lines. To evaluate the role(s) of β-arrestins, we assessed MAP kinase signaling triggered by trypsin in DKO MEFs transfected with the wt-rPAR2-YFP and the TL mutated constructs rPAR2-Leu37Ser38-YFP and rPAR2-Ala37–38-YFP. When expressed in the DKO MEF background, all receptor constructs showed an increase in MAP kinase activation in response to trypsin as was observed in the KNRK cells. MAP kinase activation was greater for wt-rPAR2 (approximately 6-fold over baseline) than for both of the TL-mutated constructs (2–4-fold over baseline) (Fig. 8, A–C, top). However, in the DKO MEF cells expressing wt-rPAR2, a reduction in the magnitude of the trypsin-activated MAP kinase signal was observed (∼2-fold over baseline), whereas MAP kinase activation by the TL-mutated receptors was similar to that observed in wt-MEFs (∼2-fold for rPAR2-Ala37–38-YFP and ∼3-fold for rPAR2-Leu37Ser38-YFP) (Fig. 8, B and C). Thus, the MAP kinase activation by wt-rPAR2 seemed to be partially β-arrestin-dependent, whereas the magnitude of MAP kinase activation caused by trypsin activation of the PAR2-Ala37–38 and PAR2-Leu37Ser38 receptors seemed to be principally independent of the expression of the two β-arrestins.
Fig. 8.

Trypsin stimulates MAP kinase signaling in β-arrestin-null DKO MEF cells transfected with wt-rPAR2 (A), rPAR2-Leu37Ser38 (B), or rPAR2-Ala37–38 (C) MAP kinase activation, expressed as a -fold increase (P-p42/44) relative to total enzyme (T-p42/44), as monitored at timed intervals for receptors expressed in either wild-type fibroblasts (○, MEF wt) or β-arrestin-null fibroblasts (■, MEF DKO). Data points showing the average values for triplicate measurements (± S.E.M., bars) are representative of three independent experiments.
Trypsin-Activated wt-rPAR2-YFP Interacts with β-Arrestins, Whereas Trypsin-Activated rPAR2-Leu37Ser38-YFP and rPAR2-Ala37–38-YFP Do Not.
The above result suggested that in contrast to the wt-rPAR2, the trypsin-activated mutant forms of the receptor might not recruit β-arrestin. To test this possibility, we used a BRET approach to monitor the interactions of β-arrestin-1 and -2 with wt-rPAR2, rPAR2-Leu37Ser38-YFP, or rPAR2-Ala37–38-YFP upon activation of the receptors either by trypsin or by the PAR2-activating peptide, SLIGRL-NH2. We observed that when stimulated with trypsin (Fig. 9 and Supplementary Information), only the wild-type rPAR2 showed a time-dependent increase in BRET, indicating interactions with both β-arrestin 1 (Supplementary Information) and β-arrestin 2 (Fig. 9A). The BRET signals for the interaction of wild-type rPAR2 with both β-arrestins-1 and -2 were comparable, whether activated by trypsin or the PAR-activating peptide SLIGRL-NH2 (Fig. 9B versus 9A and data not shown for β-arrestin-1; see Supplementary Information). In contrast, activation of rPAR2-Leu37Ser38-YFP or rPAR2-Ala37–38-YFP with trypsin failed to generate a BRET signal with either of β-arrestin-1 or -2 (Fig. 9, C and D, and data not shown for β-arrestin-1; see Supplementary Information). Notwithstanding, activation of the TL-mutant receptors with the PAR2-activating peptide SLIGRL-NH2 was able to generate a BRET signal for the recruitment of either β-arrestin-1 (not shown and see Supplementary Information) or β-arrestin-2 (Fig. 9, C and D, insets), respectively. Thus, although the two TL mutant receptors were in principle able to interact with both β-arrestin-1 and -2 (activation by SLIGRL-NH2), the trypsin-exposed mutant TL sequences failed to promote a receptor–β-arrestin interaction, as reflected by the BRET measurements.
Impaired Internalization of TL-Mutated PAR2 Receptors after Trypsin Activation.
β-Arrestin interaction is known to be critical for internalization of PAR2 (Ge et al., 2003). Because we observed impaired β-arrestin interaction with the TL-modified PAR2 constructs, we wished to investigate the internalization of wt-rPAR2, rPAR2-Leu37Ser38, and rPAR2-Ala37–38 after activation with trypsin and SLIGRL-NH2. To evaluate agonist-triggered receptor internalization, we elected to use an HEK cell background because of the much higher efficiency of receptor transfection in these cells compared with the KNRK cells. wt-rPAR2-YFP, rPAR2-Leu37Ser38-YFP, or rPAR2-Ala37–38-YFP transiently transfected in HEK cells were treated with agonists for 30 min, and receptor internalization was quantified by morphometric analysis as described under Materials and Methods. We observed that the wt-rPAR2 receptor showed significant internalization after both trypsin (10 nM) and SLIGRL-NH2 (10 μM) activation, with the magnitude of trypsin-triggered internalization approaching that of internalization triggered by the synthetic activating peptide (Fig. 10, A, ii and iii, and B). In contrast, trypsin-mediated internalization of the TL-mutant receptors was markedly diminished, relative to that of the wild-type receptor (Fig. 10, A, v, vi, viii, and ix, and B), whereas SLIGRL-NH2-activated internalization of the rPAR2-Leu37Ser38-YFP and rPAR2-Ala37–38-YFP receptors was equivalent to peptide-triggered internalization of the wild-type receptor.
Fig. 10.

Lack of internalization of trypsin-activated rPAR2-Leu37Ser38 (v) and rPAR2-Ala37–38 (viii) compared with wt-rPAR2 (ii) and with SLIGRL-NH2-activated receptors (iii, vi, ix). Transiently transfected receptor-expressing HEK cells, either untreated (NT; i, iv, and vii) or treated for 30 min at 37°C with either trypsin (ii, v, and vii) or SLIGRL-NH2 (iii, vi, inset, and ix, inset) were then fixed (4% formalin, 5 min) to enable visualization of the YFP-tagged receptors, as outlined under Materials and Methods. Top, A, representative images for wt-rPAR2 (i–iii), rPAR2-Leu37Ser38 (iv–vi), and rPAR2-Ala37–38 (vii–ix). Bottom (histograms), B, quantitative morphometric analysis of receptor internalization done as outlined under Materials and Methods for either untreated cells (NT, open histograms) or after treatment with either trypsin (gray histograms) or SLIGRL-NH2 (filled histograms). Internalization, measured in three independently conducted experiments, is expressed as the average number of internalized fluorescent speckles per cell (± S.E.M., bars). Asterisks denote a significantly reduced (*, P < 0.01) trypsin-activated internalization in the TL-mutant receptor-expressing cells compared with cells expressing the wild-type receptor.
SLAAAA-NH2 as a Biased Agonist for PAR2.
We observed that the trypsin-revealed N-terminal sequence of the PAR2 mutant PAR2-Ala39–42 (exposed TL sequence: SLAAAA—) could signal to both calcium (data not shown; Al-Ani et al., 2004) and MAP kinase (Supplementary Information) but that the corresponding TL-derived synthetic soluble peptide SLAAAA-NH2 could not cause PAR2-mediated calcium signaling (data not shown; Al-Ani et al., 2004). We thus wondered whether this peptide might be able to cause biased signaling by PAR2 so as to activate MAP kinase. Although it could not cause an elevation of intracellular calcium, this peptide was able to stimulate MAP kinase activation in both the wild-type and mutated receptor constructs (Fig. 11). In contrast, the peptides AAIGRL-NH2 and LSIGRL-NH2, corresponding to the revealed TL sequences in PAR2-Ala37–38 and PAR2-Leu37Ser38, were not able to activate either calcium (not shown) or MAP kinase signaling (Fig. 10, top).
Fig. 11.

Selective activation of p42/44 MAP kinase in PAR2-transfected KNRK cells by SLAAAA-NH2 compared with the TL mutant-derived synthetic peptides AAIGRL-NH2 and LSIGRL-NH2. KNRK cell lines expressing wt-rPAR2, rPAR2-Leu37Ser38, and rPAR2-Ala37–38 were treated for 10 min at 37°C with the synthetic PAR2 tethered ligand-related peptides, SLAAAA-NH2, LSIGRL-NH2, and AAIGRL-NH2 or with trypsin, and the activation of MAP kinase was monitored (P-p42/44 relative to T-p42/44) by Western blot analysis as outlined by the legend to Fig. 2. A, representative Western blot detection of the activation of MAP kinase (P-42/44, relative to T-p42/44) in wt-rPAR2 by SLAAAA-NH2 (third lane from left) but not by LSIGRL-NH2 and AAIGRL-NH2 (second and fourth lanes from left). B, representative western blots for the activation of MAP kinase (P-p42/44, relative to T-p42/44) in wt-rPAR2 (top), rPAR2-Leu37Ser38 (middle), and rPAR2-Ala37–38 (bottom) by trypsin (10 nM), SLIGRL-NH2 (10 μM), or SLAAAA-NH2 (10 μM). C, quantitative densitometric image analysis for the percentage increase over baseline (no treatment, NT) of P-p42/44, normalized to T-p42/44, as shown in B, for activation of wt-rPAR2 (solid histograms), rPAR2-Leu37Ser38 (open histograms), and rPAR2-Ala37–38 (gray histograms). The quantitative densitometry values (histograms) are expressed as averages (± S.E.M., bars) for three or more independently conducted experiments.
Discussion
The main finding of our study was that distinct trypsin-revealed PAR2 tethered ligand sequences can act as biased agonists to activate different signaling pathways selectively. Furthermore, we found that selective activation of MAP kinase versus calcium signaling can occur either via a proteolytically revealed tethered ligand with a mutated receptor-activating sequence or via a synthetic soluble PAR2 peptide agonist. Given the unusual proteolytic mechanism of activation of PAR2, these results represent a unique tethered ligand mechanism of biased agonism that has been documented for a variety of other GPCRs that are activated by soluble ligands (Wei et al., 2003; Galandrin et al., 2007; Kenakin, 2007; Urban et al., 2007).
Structure-Activity Profile for the Trypsin-Revealed Tethered Ligand Sequence and MAP Kinase Activation.
In our earlier work studying the roles of the tethered ligand and extracellular loop 2 for PAR2 signaling, we used calcium signaling only as an index of receptor activation (Al-Ani et al., 1999, 2002). That work pointed to differences between the interactions of the extracellular receptor domains with the proteolytically revealed tethered ligand, compared with the comparable soluble synthetic PAR-activating peptides. We were able to conclude that several distinct sites on the extracellular domains of PAR2 can interact with TL amino acid sequences either as the intact TL or as synthetic peptides to trigger calcium signaling. We showed further that the first two amino acids of the revealed TL of PAR2 (SL—) play a major role in triggering calcium signaling (Al-Ani et al., 2004), but the impact of those two revealed amino acids on MAP kinase activation was not assessed. Our current work shows that when the SL sequence in the revealed TL is replaced by alanines (rPAR2-Ala37–38) or if the residues are simply switched in sequence (rPAR2-Leu37Ser38), the ability of the revealed tethered ligand to stimulate an elevation of intracellular calcium is lost, but the activation of MAP kinase is retained. This agonist-biased signaling by the trypsin-revealed TL, with a triggering of MAP kinase but not calcium, was also observed for the synthetic PAR-activating peptide, SLAAAA-NH2, but not for AAIGRL-NH2 or LSIGRL-NH2 that were inactive for both pathways. Thus, SLAAAA-NH2 is a biased agonist for PAR2 and its ability to interact with the receptor seems to differ from that of the same sequence when presented to the receptor as a trypsin-revealed tethered ligand, which can activate both calcium and MAP kinase signaling.
MAP Kinase Activation and Interaction with Arrestins.
Based on previous observations indicating a role for β-arrestin in regulating PAR2-stimulated MAP kinase signaling (Ge et al., 2003; Zoudilova et al., 2007; Defea, 2008), we hypothesized that all receptor variants that can activate MAP kinase in the absence of Ca2+ mobilization would do so via interaction with β-arrestins. It was thus surprising that the receptor mutants did not interact with β-arrestin-1 and -2 when triggered by trypsin (Fig. 8, C and D; Supplementary Information). Furthermore, although the magnitude of MAP kinase activation by the wt-rPAR2 was greater than for the TL-mutated receptors when transfected into wt-MEFs, all three receptors promoted similar levels of MAP kinase activation in the absence of β-arrestins (Figs. 8, B and C). These data suggest that trypsin-activated wt-rPAR2 can activate MAP kinase by both a β-arrestin-dependent and β-arrestin-independent mechanism, whereas the mutated receptors, upon trypsin activation, can trigger MAP kinase principally via a β-arrestin-independent mechanism. Notwithstanding, activation of all receptor variants by the soluble PAR-activating peptide seems to trigger MAP kinase by both the-arrestin-dependent and -independent pathways, suggesting that the tethered ligand mutations do not affect the overall signaling ability of these mutants, but rather the binding of the mutant tethered ligand domain promotes a distinct set of responses. This dual process for MAP kinase activation by PAR2 mirrors the ability of various β-adrenergic receptor-targeted ligands to activate MAP kinase by distinct mechanisms (Galandrin et al., 2008).
As reported previously, wt-PAR2 interacts with β-arrestin-1 and -2, and our BRET-based analysis of this interaction revealed that PAR2 is a class B GPCR that interacts stably with and is cointernalized with β-arrestin (Hamdan et al., 2005). This stable interaction is probably due to the presence of serine and threonine clusters in the C tail of PAR2 (residues 380–397), as has been reported for a number of other GPCRs (Oakley et al., 2001).
Trypsin versus PAR-Activating Peptide-Stimulated Receptor Internalization and the Ability of Receptors to Interact with Arrestins.
Because the interaction of PAR2 with β-arrestin is critical for PAR2 internalization (DeFea et al., 2000) and because the internalized receptor scaffold plays an important role in PAR2-mediated signaling and chemotaxis (Ge et al., 2003; Zoudilova et al., 2007), we anticipated differences in internalization between the trypsin-activated receptor constructs that did and did not interact with β-arrestins (presence or absence of BRET signals in Fig. 9). Indeed, whereas trypsin triggered internalization of the wild-type PAR2 to an extent comparable with that triggered by the PAR2-activating peptide (Fig. 10, ii and iii), there was only minimal trypsin-triggered internalization of the mutated receptors PAR2-Leu37Ser38 and PAR2-Ala37–38 (Fig. 10, v and viii). In contrast, morphometric analysis of receptor internalization (Fig. 10B) demonstrated that the PAR2-activating peptide caused an equivalent internalization of all receptor constructs (Figs. 9B and 10A, iii, vi, and ix). These observations correlated very well with the ability of SLIGRL-NH2, but not trypsin, to stimulate interactions between the TL-mutated receptors and β-arrestins (Fig. 9).
Signaling Pathways and Activation of MAP Kinase by Trypsin-Stimulated Receptors.
Given the β-arrestin independence of trypsin-mediated activation of MAP kinase by the mutated receptors, our focus turned to other signal pathways. Unlike other GPCR-triggered MAP kinase activation, the PAR2-stimulated process was not affected by either doxycycline or the EGF receptor kinase inhibitor AG1478/PD153035 (Figs. 4 to 6). Those data indicated that the predominant PAR2 mechanism for activating MAP kinase in the KNRK cell background does not involve either the MMP-mediated release of an EGF receptor agonist (Daub et al., 1996) or an MMP-independent transactivation of the EGF receptor. The inhibition of either Gq (GP2A) or Gi (PTX treatment) had no effect on MAP kinase stimulation by the trypsin-activated mutated receptors, PAR2-Ala37–38 or PAR2-Leu37Ser38 and had only a very modest effect (approximately 30% inhibition) on trypsin-mediated activation of MAP kinase by the wt-rPAR2. Likewise, the Src-targeted inhibitor PP1 had no effect on trypsin-triggered activation of MAP kinase by the two mutated receptors and only a small inhibitory effect (approximately 30%) on MAP kinase activation by the wild-type receptor (Figs. 4–6). Thus, in the KNRK cell background, the process of proteinase-mediated PAR2-stimulated MAP kinase activation seemed to be largely independent of pathways involving many of the common signal components (Gq, Gi, Src, EGFR). That said, the Rho kinase inhibitors Y27362 and H1152 had a major inhibitory effect on the ability of all PAR2 constructs to stimulate MAP kinase when activated by trypsin (Figs. 4–6). The role of Rho kinase in activation of MAP kinase by PAR2 is in keeping with the role of this kinase for the activation of MAP kinase and other responses by PAR1 (Vouret-Craviari et al., 1998; Seasholtz et al., 1999; Carbajal et al., 2000; McLaughlin et al., 2005) and PAR2 (Scott et al., 2003). Overexpression of constructs encoding Gα12 and Gα13 resulted in an increase in trypsin sensitivity of the cells to MAPK signaling, suggesting that the Rho kinase-dependent MAPK signal seen in these cells is Gα12/13-dependent (Fig. 7). Thus, in future work with intact tissue PAR2 targets (e.g., vasculature, neurons), it will be of considerable importance to evaluate the potential role of Rho kinase and Gα12/13-dependent signaling events.
We found that synthetic soluble PAR-activating peptides, like the distinct trypsin-revealed tethered ligands, are capable of agonist-biased signaling by PAR2. Specifically, SLIGRL-NH2 was able to trigger both an elevation of intracellular calcium and an activation of MAP kinase, whereas SLAAAA-NH2 triggers only MAP kinase activation (Fig. 11) without causing a calcium signal (Al-Ani et al., 2004). In contrast with the trypsin-revealed mutated tethered ligands with the sequences, AAIGRL— (PAR2-Ala37–38) and LSIGRL— (PAR2-Leu37Ser38), the analogous synthetic peptides with the sequences AAIGRL-NH2 and LSIGRL-NH2 were unable to activate either MAP kinase or calcium signaling. These results indicate that the tethered and soluble ligands most likely bind differentially and/or stabilize different conformations of the receptor leading to the activation of distinct subsets of signaling cascades. Further work will be needed to better understand the specific structural basis of this biased signaling.
In summary, we describe agonist-biased signaling by both a trypsin-revealed tethered ligand and a synthetic PAR2-activating peptide that can lead to the selective activation of MAP kinase versus calcium signaling by PAR2 as well as distinctions in recruitment of β-arrestin and receptor endocytosis. The ability of the PAR-activating peptide, SLAAAA-NH2 to signal selectively via MAP kinase versus calcium establishes in principle the possibility of developing signal-selective PAR2 agonists that may be of therapeutic utility.
Supplementary Material
Acknowledgments
We thank the live cell imaging facility directed by Dr. Giuseppina (Pina) Colarusso for assistance with the imaging experiments.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
These studies were supported in part the Canadian Institutes of Health Research [Grants MT-13759, MOP-53289] (to M.D.H.), [Grant FRN11215] (to M.B.); the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM066151] (to K.D.); and by a Canadian Association of Gastroenterology/Canadian Institutes of Health Research/Ortho-Jensen postdoctoral fellowship.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- PAR
- proteinase-activated receptor
- A23187
- calcimycin
- AG1478
- 4-(3′-chloroanilino)-6,7-dimethoxy-quinazoline
- BRET
- bioluminescence resonance energy transfer
- DKO
- double-knockout β-arrestin-deficient mouse embryo-derived fibroblasts
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal-regulated kinase
- GP2A
- N-cyclohexyl-1-(2,4-dichlorophenyl)-1,4-dihydro-6-methylindeno[1,2-c]pyrazole-3-carboxamide
- GPCR
- G-protein-coupled receptor
- H1152
- (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinolinyl)sulfonyl]-hexahydro-1H-1,4-diazepine dihydrochloride
- HEK
- human embryonic kidney
- KNRK
- normal rat kidney cell line transformed by Kirsten murine sarcoma virus
- MAP
- mitogen-activated protein
- MAPK
- mitogen-activated protein kinase
- MEF
- mouse embryo-derived fibroblasts
- MMP
- matrix metalloproteinase
- PAR2 or wt-rPAR2
- wild-type rat proteinase-activated receptor-2 having the trypsin-revealed tethered ligand sequence SLIGRL—
- PAR2-Ala37–38
- mutated rat PAR2 with a trypsin-revealed tethered ligand sequence AAIGRL—
- PAR2-Ala39–42
- mutated rat PAR2 with a trypsin-revealed tethered ligand sequence SLAAAA—
- PAR2-Leu37Ser38
- mutated rat PAR2 with a trypsin-revealed tethered ligand sequence LSIGRL—
- PBS
- phosphate-buffered saline
- PP1
- 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine
- PTX
- pertussis toxin
- r
- rat
- TBST
- Tris-buffered saline/Tween 20
- TL
- tethered ligand
- wt
- wild type
- YFP
- yellow fluorescent protein
- Y27362
- (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide, 2HCl.
References
- Al-Ani B, Hansen KK, Hollenberg MD. (2004) Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharmacol 65:149–156 [DOI] [PubMed] [Google Scholar]
- Al-Ani B, Saifeddine M, Kawabata A, Hollenberg MD. (1999) Proteinase activated receptor 2: Role of extracellular loop 2 for ligand-mediated activation. Br J Pharmacol 128:1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ani B, Wijesuriya SJ, Hollenberg MD. (2002) Proteinase-activated receptor 2: differential activation of the receptor by tethered ligand and soluble peptide analogs. J Pharmacol Exp Ther 302:1046–1054 [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG, et al. (1996) Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J 314:1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajal JM, Gratrix ML, Yu CH, Schaeffer RC., Jr (2000) ROCK mediates thrombin's endothelial barrier dysfunction. Am J Physiol Cell Physiol 279:C195–C204 [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. (2000) Thrombin signalling and protease-activated receptors. Nature 407:258–264 [DOI] [PubMed] [Google Scholar]
- Darmoul D, Gratio V, Devaud H, Laburthe M. (2004) Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem 279:20927–20934 [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379:557–560 [DOI] [PubMed] [Google Scholar]
- Defea K. (2008) Beta-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br J Pharmacol 153 (Suppl 1):S298–S309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. (2000) beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148:1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon S, Candé C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. (2003) Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol 28:339–346 [DOI] [PubMed] [Google Scholar]
- Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. (2005) Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 32:411–419 [DOI] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bonin H, Ogawa K, Galés C, Bouvier M. (2008) Conformational rearrangements and signaling cascades involved in ligand-biased mitogen-activated protein kinase signaling through the beta1-adrenergic receptor. Mol Pharmacol 74:162–172 [DOI] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bouvier M. (2007) The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci 28:423–430 [DOI] [PubMed] [Google Scholar]
- Ge L, Ly Y, Hollenberg M, DeFea K. (2003) A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem 278:34418–34426 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. (2005) High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen 10:463–475 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Rochdi MD, Breton B, Fessart D, Michaud DE, Charest PG, Laporte SA, Bouvier M. (2007) Unraveling G protein-coupled receptor endocytosis pathways using real-time monitoring of agonist-promoted interaction between beta-arrestins and AP-2. J Biol Chem 282:29089–29100 [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem 271:695–701 [DOI] [PubMed] [Google Scholar]
- Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. (1999) Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther 288:358–370 [PubMed] [Google Scholar]
- Kenakin T. (2007) Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci 28:407–415 [DOI] [PubMed] [Google Scholar]
- McCole DF, Keely SJ, Coffey RJ, Barrett KE. (2002) Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-alpha. J Biol Chem 277:42603–42612 [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. (2005) Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 280:25048–25259 [DOI] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. (1994) Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A 91:9208–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem 276:19452–19460 [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Hollenberg MD. (2008) Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol 153 (Suppl 1):S263–S282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott G, Leopardi S, Parker L, Babiarz L, Seiberg M, Han R. (2003) The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J Invest Dermatol 121:529–541 [DOI] [PubMed] [Google Scholar]
- Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. (1999) Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res 84:1186–1193 [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, et al. (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990–994 [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13 [DOI] [PubMed] [Google Scholar]
- van der Merwe JQ, Hollenberg MD, MacNaughton WK. (2008) EGF receptor transactivation and MAP kinase mediate proteinase-activated receptor-2-induced chloride secretion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 294:G441–451 [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, Boquet P, Pouysségur J, Van Obberghen-Schilling E. (1998) Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell 9:2639–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. (2003) Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A 100:10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. (2007) Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem 282:20634–20646 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}