

Abstract
Protein kinases are central targets for drug-based cancer treatment. To avoid functional impairment, the cell develops mechanisms of drug resistance, primarily based on adaptive mutations. Redesigning a drug to target a drug-resistant mutant kinase constitutes a therapeutic challenge. We approach the problem by redesigning the anticancer drug imatinib guided by local changes in interfacial de-wetting propensities of the C-Kit kinase target introduced by an imatinib-resistant mutation. The ligand is redesigned by sculpting the shifting hydration patterns of the target. The association with the modified ligand overcomes the mutation-driven destabilization of the induced fit. Consequently, the redesigned drug inhibits both mutant and wild-type kinase. The modeling effort is validated through molecular dynamics, test tube kinetic assays of downstream phosphorylation activity, high-throughput bacteriophage-display kinase screening, cellular proliferation assays, and cellular immunoblots. The inhibitor redesign reported delineates a molecular engineering paradigm to impair routes for drug resistance.
Introduction
Protein kinases have been identified as central targets in molecular cancer therapy (1–3). However, kinases are actually moving targets because the cell develops mechanisms of drug resistance, mainly mutations, that hamper ligand association (1). The development of drug-resistant mutations on targeted proteins poses a challenge to inhibitor design (1–3). The C-KIT kinase, a therapeutic target for treating gastrointestinal stromal tumors (GIST), is inhibited by imatinib (Gleevec, STI571; refs. 4, 5), but in malignancies like systemic mastocytosis or acute myeloid leukemia (AML), the kinase develops the activation loop mutation D816V (6, 7), promoting resistance to imatinib inhibition (2, 3). Here, we report on a rational redesign of imatinib that inhibits the imatinib-resistant mutant and wild-type kinase. The prototype is designed to be a better stabilizer of the active induced-fit conformation of the activation loop. The ligand redesign is guided by the mutation-induced alterations to the interfacial hydration pattern of the target. The rationale for enhancing the affinity towards the active conformation hinges on the fact that the mutation conferring drug resistance cannot be too structurally deleterious as to impair kinase function; otherwise, it would not prevail in the cell life cycle. Thus, drug resistance is dealt with by a perturbative ligand modification.
To promote a tighter grip on the activation loop of C-Kit kinase and overcome the destabilizing effect of the mutation, we identified changes in the local dehydration propensities at the ligand/protein interface that may be compensated by redesigning the parental drug. The prototype ligand is engineered according to the blueprint of residence times of water molecules that solvate the target interface (8). Molecular modeling led us to introduce a specific methylation of imatinib. We confirmed through in vitro assays the dual inhibitory effect of the prototype by probing the downstream phosphorylation activity of wild-type and imatinib-resistant kinase in the presence of the parental and prototype competitive ligands. The focused effect of the prototype over a vast cross-section of the human kinome was corroborated by high-throughput screening (9). We also conducted cell proliferation assays on lines that express wild-type and imatinib-resistant kinase to confirm the dual anticancer activity of the prototype.
Materials and Methods
Calculation of local dehydration propensities
We introduce a descriptor of hydration tightness for soluble proteins defined as the mean residence time of hydrating molecules within a domain around each residue on the protein surface. The local mean residence time (<τ>i) of hydrating molecules at residue i is defined with respect to a spherical domain D(i) of 6.2-Å radius (approximately the width of three water layers; ref. 10) centered at the α-carbon of residue i. The actual computation of residence times is given in the Supplementary Material. The mean residence times were obtained from classic trajectories generated by molecular dynamic simulations (Supplementary Material).
Backbone exposure for protein targets
The extent of backbone exposure at a particular residue was determined by counting the number of nonpolar side chain groups contained within a 6.2-Å radius sphere (approximately the thickness of three water layers) centered at the α-carbon. The extent of backbone shielding (η) in structured regions averaged over a nonredundant curated PDB database (1,662 proteins) is 14.2, with Gaussian dispersion of 8.2. Thus, a backbone site is regarded as exposed if the region is structurally disordered or if η < 6. The statistics vary for desolvation radius in the range of 6 Å < r < 7 Å, but the tails of the distribution identify the same sites of backbone exposure. The structural integrity of soluble proteins requires that most backbone amides and carbonyls be protected from hydration. Thus, residues with absent backbone coordinates in a PDB entry are regarded as exposed and thus are residues from natively disordered proteins.
Molecular dynamics/free-energy computations
Classic molecular dynamic simulations were done starting from the crystal structure of the C-KIT kinase (PDB.1T46, 1.60-Å resolution; ref. 5). Simulations were done for the wild-type kinase complexed with imatinib and for the in silico generated D816V mutant complexed with imatinib and WBZ_7. The Amber package was used to obtain 50 ns of molecular dynamics with explicit solvent in an NPT ensemble (11). Binding free energies for the kinase/inhibitor systems were calculated using the MM-PB(GB)SA method (Supplementary Material; ref. 11). The average binding free energy was obtained from the sum of the average gas-phase energies, the solvation free energies, and the entropy contributions. Mechanical energies were evaluated in a single molecular dynamic step using an infinite cutoff for nonbonded interactions. Solvation free energies were estimated as the sum of an electrostatic solvation energy plus a nonpolar solvation energy. Entropic contributions to the binding free energy were estimated by calculating the quasiharmonic entropy (12), which takes into the account both the configurational and vibrational entropy contributions arising from structural changes and creation of new modes upon complexation.
Synthesis of imatinib derivative WBZ_7
The synthesis of the imatinib derivative resulting from methylation at position 6 on the pyperidine ring recapitulates Novartis patent WO03027100A1, 2003(13), replacing a single reactant (N,N-dimethylformamide dimethylacetal) for N,N-dimethylformamide methyl-dimethylacetal in the first step of synthesis. The total synthesis and spectroscopic characterization is provided in the Supplementary Material.
Spectrophotometric kinetic assay
To determine the inhibitory efficacy of WBZ_7, kinetic assays of the inhibition of active (phosphorylated) wild-type C-Kit kinase and active variant D816V (Upstate, Millipore) were conducted. To measure the rate of downstream phosphorylation due to kinase activity in the presence of inhibitors, a spectrophotometric assay has been adopted in which the ADP production is coupled to the NADH oxidation and determined by absorbance reduction at 340 nm, as described in ref. (14). Details are provided in the Supplementary Material.
High-throughput screening
A primary high-throughput screening of WBZ_7 at 10 μmol/L was conducted by Ambit Biosciences (San Diego, CA) against a bacteriophage library displaying 240 human kinases, using imatinib screening as control. A rough estimation of the binding constant (Kd–1) for each assay was provided by the single-hit value in the primary screen at a single compound concentration. Kinase profiling was done using a bacteriophage library displaying fused human kinases that may attach at the ATP site to a fixed-ligand matrix, which, in turn, may be competitively displaced from binding by the tested compound (9).
Cell proliferation assays
Proliferation of GIST cancer cells (line ST882) expressing C-Kit kinase (15) was determined by Alamar Blue assay (Bio Source International) following a 48-h treatment with imatinib and WBZ_7. The treated murine pro-B cells Ba/F3 (American Type Culture Collection) expressing C-Kit D816V (16) were investigated using a tetrazolium-based assay (17), adopting the same generic set-up described above. Human HMC-1 mast cells (ref. 18; gift from J.H. Butterfield, Mayo Clinic) expressing the C-Kit D816V mutant were monitored through a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt assay after 48 h of treatment. Details are provided in the Supplementary Material.
Western blots
HMC-1 and Ba/F3 cells were incubated untreated and treated with WBZ_7 or imatinib (0.1, 1, and 10 μmol/L) for 12 h. After treatment, cell pellets were lysed, and protein mixtures were separated though gel electrophoresis (SDS-PAGE). Membranes were subsequently probed with specific antibodies. Details are given in the Supplementary Material
Results
The structural basis for imatinib inhibition of C-Kit is revealed by examining the kinase-ligand interface (PDB.1T46; ref. 5). The ligand promotes an induced fit in the activation loop, thus binding to the active conformation. Nevertheless, there exists a sticky element in the active conformation, which is not interactive with imatinib. Residues F811 and A814 mark sites of weakness in the hydration shell of the target (Fig. 1A). These residues are paired by the solvent-accessible F811-A814 backbone hydrogen bond, which is prone to becoming dehydrated (10), as evidenced by the short residence times of local hydrating molecules in the crankshaft-like active conformation (Fig. 1A–C). The removal of surrounding water upon association enhances the backbone amide-carbonyl electrostatic interaction (10). Solvent accessibility is defined by a low number of surrounding nonpolar groups within residue microenvironments (Materials and Methods). Imatinib association contributes only partially to water removal from the pair F811-A814 whose de-wetting propensity is most affected by the D816V mutation (Fig. 1A).
Figure 1.
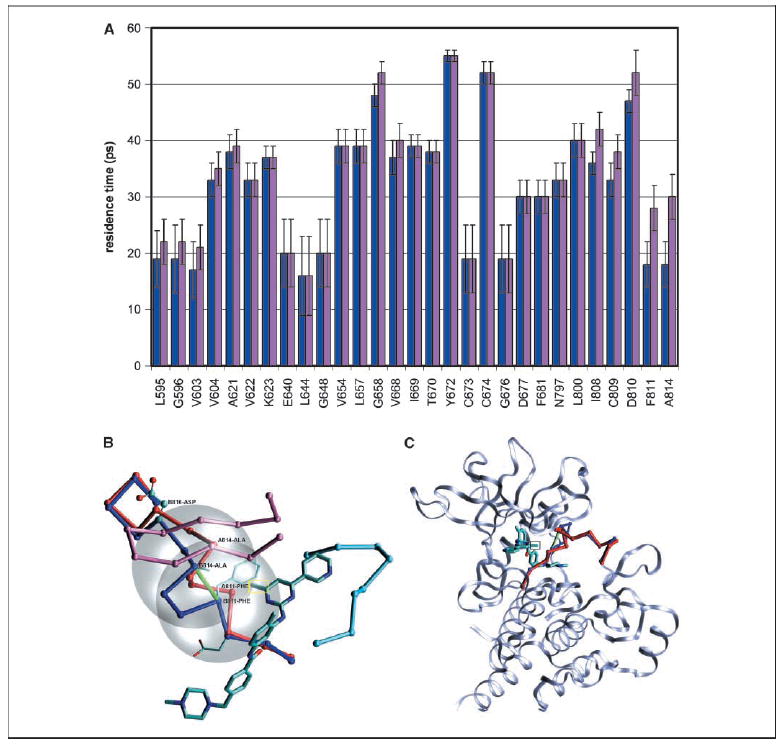
A, mean residence times of water molecules solvating the uncomplexed C-KIT wild-type (blue) and D816V mutant (pink) kinase domain. The mutation was introduced in silico, and the structures examined were obtained after 50 ns molecular dynamic equilibration of the uncomplexed kinases (Materials and Methods, Supplementary Material). Residue numbering follows PDB entry 1T46. The one-letter amino acid code was adopted for clarity. Columns, mean; bars, variances. Local dehydration propensities are signaled by relatively short residence times and represent weaknesses in the hydration shell of the target protein. Only residues in contact with the ligand in PDB complex 1T46 are indicated for clarity. A contact is defined by the presence of ligand atoms within a 6.2-Å sphere centered at the α-carbon. The changes in dehydration propensity introduced by the D816V mutation are most pronounced for residues F811 and A814. B, location of imatinib inhibitor with designed appended methyl group (yellow rectangle) relative to the activation loop of C-KIT kinase in the active (blue, chain B) conformation within the ligand-kinase complex PDB.1T46. The superimposed deactivating conformation (red, chain A) after full alignment of the autoinhibited form (PDB.1T45) with the active structure (PDB.1T46). Besides the activation loop (residues 808–820), the other two functional loops are also shown to give a better perspective of the inhibitor location within the ATP pocket: the P-loop (magenta, residues 594–604) and catalytic loop (light blue, residues 670–677). In addition, the side chains of the catalytic triad DFG (residues 810–812) are displayed for the active conformation. The chain conformation is indicated by virtual bonds joining α-carbons. Partially exposed F811-A814 backbone hydrogen bond involving a de-wetting hotspot in the active conformation (green). The two desolvation domains are given by 6.2-Å radius spheres (gray) centered at the α-carbons of the paired residues. By increasing the dehydration of F811 and A814, the highlighted methylation enhances the stabilization of the active loop upon ligand association to the active kinase. C, ribbon structure of C-Kit kinase target in complex with proposed imatinib modification, detailing the activation loop backbone (blue) and targeted de-wetting hotspot (green). Aligned inactive loop conformation (red).
Hydrating molecules with low residence times constitute our blueprint for ligand re-engineering because they signal a propensity for association. Because the 811-814 hydrogen bond is altogether absent in the inactive (autoinhibited) conformation of the loop (PDB.1T45), we were able to selectively enhance the affinity for the active conformation, by redesigning imatinib to increase its favorable exogenous dehydration. This higher level of favorable interactivity required a modulation of the target microenvironment through the incorporation of a methyl group at position 6 in the pyrimidine ring (13), yielding the compound WBZ_7 (Fig. 2; Materials and Methods).
Figure 2.
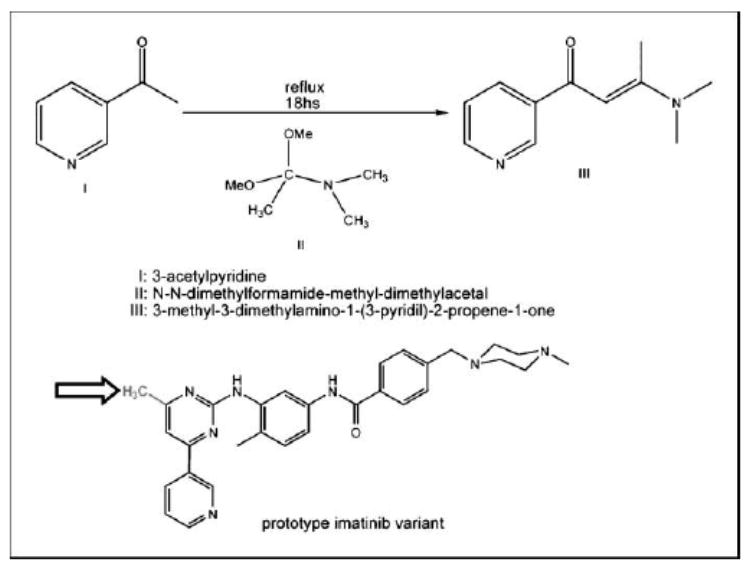
The initial step in the synthesis of the imatinib derivative WBZ_7 (total synthesis in the Supplementary Material). Structure of WBZ_7, highlighting the methyl group (arrow) that substitutes the original hydrogen at position 6 in the pyperidine ring of imatinib.
The ligand redesign was first validated through molecular dynamic simulations of the wild-type kinase-imatinib association and D816V mutant association with imatinib and WBZ_7 (Materials and Methods; refs. 11, 12). The calculated binding free energies are –17.0, –4.5, and –16.1 kcal mol–1, for wild-type kinase-imatinib association and D816V mutant complexation with imatinib and WBZ_7, respectively. These values show the same tendency in terms of inhibitor affinity found in the in vitro kinetic analysis reported below. The energetic contributions are –57.69, –59.15,and –59.24kcalmol–1,andtheentropiccontributions(–TΔS) (–TΔS) are 40.7, 54.7, and 43.2 kcal mol–1, respectively. These results indicate that the decrease in imatinib affinity caused by target mutation and the compensatory affinity increase introduced by WBZ_7 are mainly entropic effects. Imatinib resistance arises from the entropy increase (TΔS ˜ 14 kcal mol–1) of the uncomplexed mutant with respect to the uncomplexed wild type. This difference translates into the free energy difference between the respective imatinib associations. The entropy increase is due to the hydrophilic → hydrophobic destabilizing amino acid substitution in the (solvent exposed) activation loop. WBZ_7 restores the affinity for the mutant kinase by increasing the entropy of the complex (TΔS ˜ 12 kcal mol–1) with respect to the imatinib mutant counterpart, accounting for the difference in binding affinity. The added methyl in WBZ_7 becomes a surrogate for V816 in promoting water removal from the F811-A814 de-wetting hotspot (Fig. 1), thus enhancing loop flexibility and hence restoring ligand affinity.
The inhibiting efficacy of the prototype was tested in spectrophotometric kinetic assays (Materials and Methods; Fig. 3A; ref. 14), measuring the downstream phosphorylation rates of active wild type and D816V mutant at various inhibitor concentrations. Thus, by taking full advantage of the potential for interactivity with the activation loop in the activated conformation, we were able to redesign imatinib into WBZ_7, a nanomolar inhibitor (Kd ≈ 39 ± 7 nmol/L) of the imatinib-resistant D816V mutant as well as of the wild-type kinase (Kd ≈ 21 ± 5 nmol/L). The effect of the prototype compound should be contrasted with that of imatinib on the same targets: Kd ≈ 11 ± 2 μmol/L for the imatinib-resistant D816V mutant and Kd ≈ 25 ± 5 nmol/L for the wild-type C-Kit kinase. The latter powerful inhibition is entirely expected, given the therapeutic value of imatinib for treating GIST tumors by targeting wild-type C-Kit (4, 5). On the other hand, the affinity of WBZ_7 for the imatinib-resistant kinase is about 300 times higher. A tighter grip on the activation loop stabilizes its active conformation to such an extent that it counteracts the negative-design feature (Fig. 1A) introduced by the drug-resistant mutation.
Figure 3.
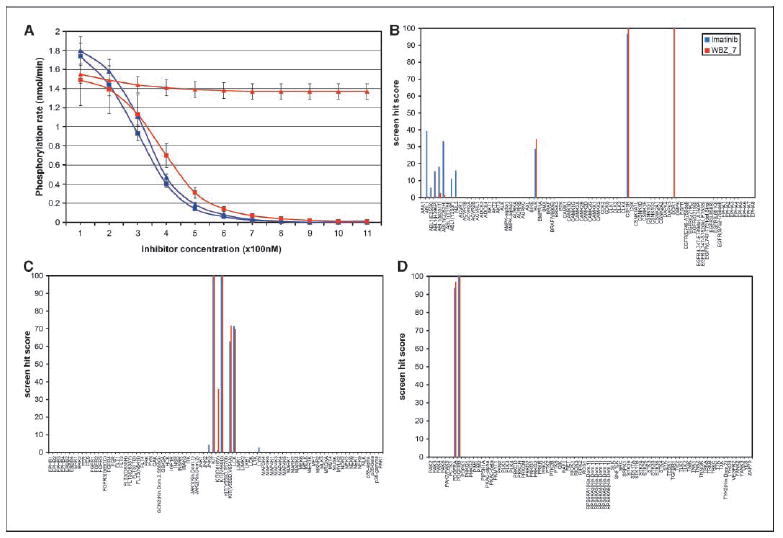
A, downstream phosphorylation rates from spectrophotometric kinetic assay of active C-Kit kinase and active imatinib-resistant D816V mutant (Upstate, Millipore). Both kinases are inhibited by WBZ_7 (squares), whereas only the wild-type C-Kit is significantly inhibited by imatinib (triangles). Phosphorylation rate plots are displayed for wild-type kinase (blue) and for the D816V variant (red). Bars, dispersion over five runs for each kinetic assay (Materials and Methods). Each assay run consists of 11 measurements of maximum phosphorylation rate at 100-nmol/L intervals in increasing inhibitor concentration for each of the four kinase/inhibitor pairs. Notice that in contrast with imatinib, WBZ_7 is an inhibitor of the imatinib-resistant mutated as well as of the wild-type kinase. Fluctuations in initial enzyme concentrations are likely sources of experimental error particularly apparent at low inhibitor concentrations (Km(app) ⪡ [ATP]). B to D, high-throughput screening of WBZ_7 (red) and imatinib (blue, control) over 240 human kinases displayed in a T7-bacteriophage library (Ambit Biosciences, Materials and Methods). Hit values are reported as % bound kinase.
A Michaelis-Menten scheme with ATP-competitive inhibition and throughout-saturating peptide-substrate concentration yields an accurate fit in all cases shown in Fig. 3A. Fluctuations in initial enzyme concentration yield rate-value dispersions (Materials and Methods; Fig. 3A) that are pronounced at low inhibitor concentration (because Km(app) ⪡ [ATP]) and translate into the SD reported for the Kd values. The plots generated with Kd/dispersions lie within the confidence bands for the rate/[I] plots (Fig. 3A).
To complete the in vitro analysis, WBZ_7 at 10 μmol/L was screened for affinity against a T7-bacteriophage library displaying 240 human kinases (Fig. 3B–D) as described previously (ref. 9; Materials and Methods), using imatinib screening as control. The dual affinity of WBZ_7 on C-Kit kinase and on the D816V mutant is noteworthy and contrasts with the lack of affinity of imatinib for the mutant. WBZ_7 is also more specific than the parental compound, as it has a low affinity for the Abelson (ABL) kinase, the primary imatinib target, and its variants (14). This selectivity is expected because WBZ_7 was designed to enhance the ligand interactivity towards the active conformation of the activation loop, whereas imatinib binds the constitutively active Bcr-ABL kinase in the inactive conformation (14). Also noteworthy is the WBZ_7 inhibition of discoidin receptor 1 kinase (DDR1; Fig. 3B). This finding suggests additional anticancer activity for WBZ_7 because DDR1 is up-regulated by p53, and DDR1 inhibition dramatically increases p53-mediated apoptosis through a positive feedback loop (19).
The anticancer activity of WBZ_7 was tested in vivo by selecting cell lines that express the wild-type and drug-resistant kinase (Materials and Methods; refs. 15 – 17 , 20 ). The proliferation of C-Kit–positive GIST cells (15) treated with WBZ_7 was significantly decreased (IC50 ≈ 72 ± 4 nmol/L) in a quantitative dose-dependent manner similar to imatinib (IC50 ≈ 65 ± 5 nmol/L; Fig. 4A). By contrast, the inhibitory effect of imatinib on murine Ba/F3 cells expressing the C-Kit (D816V) mutation (16) is very low at the same bulk physiologic doses (IC50 ≈ 19.0 ± 0.5 μmol/ L), whereas WBZ_7 significantly impairs the proliferation of these cells (IC50 ≈ 95 ± 5 nmol/L). The mast-cell line HMC-1 (18) expressing the D816V mutant was also treated with WBZ_7 and imatinib. Accordingly, a significant effect (38% decrease) on proliferation was observed for WBZ_7 at the 10 μmol/L bulk concentration, in sharp contrast with imatinib, which only impaired cell growth by 7%.
Figure 4.
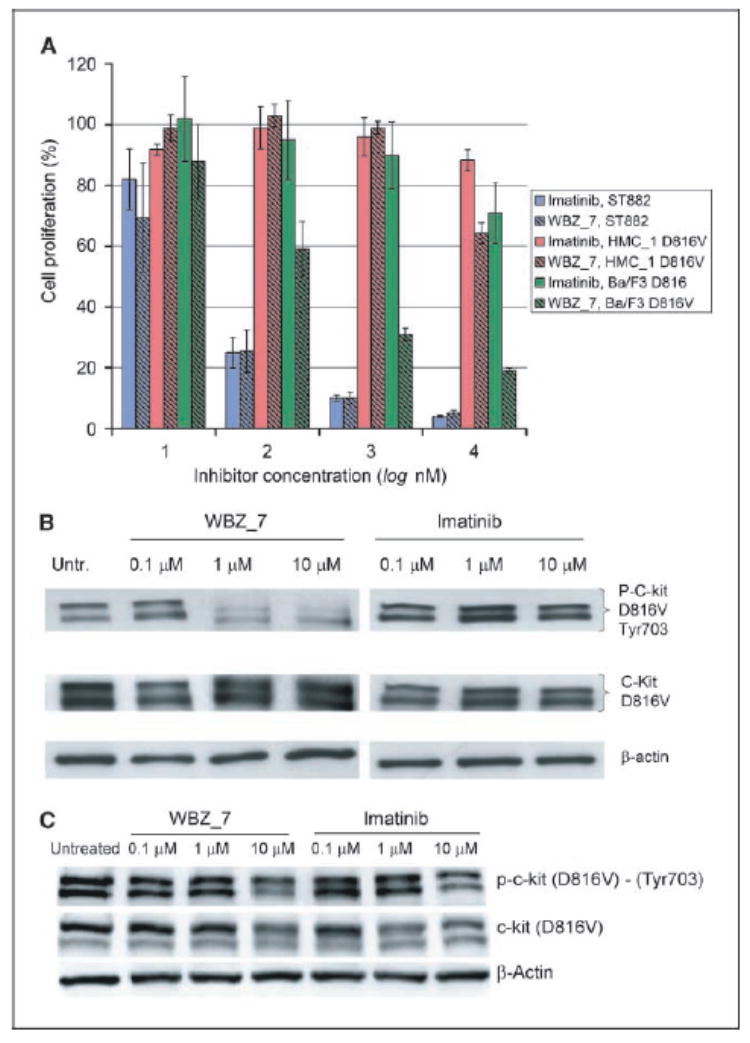
A, cell proliferation assay of antitumor activity for WBZ_7 (hatched columns) and imatinib (solid columns) on cell lines GIST-ST882 (blue), HMC-1 (red), and Ba/F3 murine pro-B (green). WBZ_7 inhibits proliferation of C-Kit–positive GIST cells and of C-Kit (D816V)–positive Ba/F3 murine pro-B and HMC-1 cells. The inhibitory effect of imatinib is essentially restricted to the GIST cells that express only the wild-type kinase. Cell proliferation, expressed as % proliferating cells relative to untreated cells, was determined by the spectrophotometric assay (Materials and Methods). Assays on batteries of 24 wells for each inhibitor concentration/cell type pair were repeated four times, and the value dispersions for each battery were averaged over the four batteries (columns). SD were obtained as root mean square deviations of measured populations from the arithmetic mean. The latter was obtained for each ligand concentration/cell type pair by averaging the four mean values obtained from the four repetitions of the batteries of 24 assays. B, Western blot assay of murine Ba/F3 cells untreated (Untr.) and treated with WBZ_7 and with imatinib at different inhibitor concentrations. Top, phosphorylated D816V mutant kinase; middle, total c-Kit D816V kinase; bottom, β-actin control. C, Western blot assay of HMC-1 cells untreated and treated with WBZ_7 and with imatinib at different inhibitor concentrations. Top, phosphorylated D816V kinase; middle, total C-Kit D816V kinase; bottom, β-actin control.
In searching for other appropriate human cell lines, we noted that mutations in C-Kit have been detected in some patients with AML; however, no human AML cell lines have been developed bearing the D816V mutation (21).
A Western blot (Materials and Methods) on treated Ba/F3 and HMC-1 cells expressing the drug-resistant mutation (Fig. 4B and C) was done to determine the inhibitory activity of WBZ_7 on kinase autophosphorylation. Densitometry revealed 91% inhibition of the D816V mutant by WBZ_7 at the 10 μmol/L bulk concentration in Ba/F3 cells. By contrast, imatinib inhibition of the mutant is 9% under the same conditions. For HMC-1 cells, WBZ_7 inhibition is 32%, in contrast with imatinib that inhibited the mutant by 12%. HMC-1 cells do not overexpress the kinase in response to the WBZ_7 inhibition of the active form, whereas Ba/F3 cells do, as evidenced by the stronger total mutant kinase (intermediate) band at 10 μmol/L inhibitor when compared with the band at lower inhibitor concentrations (Fig. 4B). The difference in the extent of inhibition of C-Kit (D816V) by WBZ_7 in the two cell lines is likely of pharmacokinetic origin and attributable to a reduced absorption of the drug into the human cells when compared with the murine cells, which in turn accounts for the higher expression levels of the mutant kinase found in the WBZ_7-treated murine cells.
Discussion
Drug resistance promoted by mutational modification constitutes a challenge for drug designers thus faced with a shifting target. More daunting is the problem of re-engineering an inhibitor to overcome the negative design introduced by mutations that confer resistance to the original inhibitor. We approached this problem in kinase targeting by redesigning the ligand according to changes in the target hydration pattern induced by the mutation. We rationally created a therapeutic agent that overcomes drug resistance. A combination of in silico, in vitro, and in vivo assays validates our design strategy, likely to inspire a new generation of molecular therapies for shifting targets arising from drug-resistant patterns.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: NIH grant R01-GM072614, National Science Foundation grant EIA-0216467, John and Ann Doerr Fund for Computational Biomedicine, and an unrestricted grant from Eli Lilly.
We thank Dr. J.H. Butterfield for the generous gift of the human mast cells.
References
- 1.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–6. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 2.Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamem-brane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–81. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 3.Shah N, Lee FY, Luo R, et al. (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108:286–91. doi: 10.1182/blood-2005-10-3969. [DOI] [PubMed] [Google Scholar]
- 4.Attoub S, Rivat C, Rodrigues S, et al. The c-kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res. 2002;62:4879–83. [PubMed] [Google Scholar]
- 5.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–63. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 6.Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the protooncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–44. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagata H, Worobec AS, Oh CH, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92:10560–4. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Y, Rossky PJ. Surface topography dependence of biomolecular hydrophobic hydration. Nature. 1998;392:696–9. doi: 10.1038/33653. [DOI] [PubMed] [Google Scholar]
- 9.Fabian MA, Biggs WH, Treiber DK, et al. A small molecule kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 10.Fernández A. Keeping dry and crossing membranes. Nat Biotechnol. 2004;22:1081–4. doi: 10.1038/nbt0904-1081. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Morin P, Wang W, Kollman PA. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of Efavirenz by docking and MM-PBSA. J Am Chem Soc. 2001;123:5221–30. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
- 12.Andricioaei I, Karplus MJ. On the calculation of entropy from covariance matrices of the atomic fluctuations. Chem Phys. 2001;115:6289–92. [Google Scholar]
- 13.Li JJ, Johnson DS, Sliskovic DR, Roth BD. Contemporary drug synthesis. New Jersey: Wiley-Interscience. 2004:32–3. [Google Scholar]
- 14.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 15.DeMatteo RP. The GIST of targeted cancer therapy: a tumor (gastrointestinal stromal tumor), a mutated gene (c-kit), and a molecular inhibitor (STI571) Ann Surg Oncol. 2002;9:831–9. doi: 10.1007/BF02557518. [DOI] [PubMed] [Google Scholar]
- 16.Corbin AS, Griswold IJ, La Rosee P, et al. Sensitivity of oncogenic KIT mutants to the kinase inhibitors MLN518 and PD180970. Blood. 2004;104:3754–7. doi: 10.1182/blood-2004-06-2189. [DOI] [PubMed] [Google Scholar]
- 17.La Rosee P, Corbin AS, Stoffregen EP, Deininger MW, Druker BJ. Activity of the Bcr-Abl kinase inhibitor PD180970 against clinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesylate (Gleevec, STI571) Cancer Res . 2002;62:7149–53. [PubMed] [Google Scholar]
- 18.Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345–55. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- 19.Ongusaha PP, Kim JI, Fang L, et al. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback. EMBO J. 2003;22:1289–301. doi: 10.1093/emboj/cdg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998;17:6250–62. doi: 10.1093/emboj/17.21.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scappini B, Onida F, Kantarjian HM, et al. Effects of signal transduction inhibitor 571 in acute myelogenous leukemia cells. Clin Cancer Res. 2001;7:3884–93. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).