

Abstract
Receptor associated protein (RAP80), a nuclear protein containing two ubiquitin-interacting motifs (UIMs), was recently found to be associated with Breast cancer-1 (BRCA1) and to translocate to ionizing radiation-induced foci (IRIF). In this study, we demonstrate that the BRCT mutant of BRCA1, R1699W, which is associated with increased risk of breast cancer, is unable to interact with RAP80. Previously, we demonstrated that ataxia-telangiectasia mutated protein kinase (ATM) can phosphorylate RAP80 in vitro at Ser205, but whether this site is a target of ATM in whole cells was not established. To address this question, we generated an anti-RAP80Ser205P antibody that specifically recognizes RAP80 phosphorylated at Ser205. Our data demonstrate that RAP80 becomes phosphorylated at Ser205 in cells exposed to ionizing irradiation and that RAP80Ser205P translocates to IRIF. We show that this phosphorylation is mediated by ATM and does not require a functional BRCA1. The phosphorylation occurs within 5 min after irradiation, long before the translocation of RAP80 to IRIF. In addition, we demonstrate that ultraviolet (UV) irradiation induces translocation of RAP80 to DNA damage foci that co-localize with γ-H2AX. We further show that this translocation is also dependent on the UIMs of RAP80 and that the UV-induced phosphorylation of RAP80 at Ser205 is mediated by ATR, not ATM. These findings suggest that RAP80 has a more general role in different types of DNA damage responses.
INTRODUCTION
Genotoxic stress can induce different types of DNA damage, among which double strand breaks (DSB) are the most detrimental (1). To maintain their genomic integrity organisms have developed a sophisticated system to regulate DNA repair and cell cycle checkpoints. Receptor-associated protein 80 (RAP80) or ubiquitin-interacting motif containing 1 (UIMC1) is a nuclear protein containing two functional ubiquitin-interacting motifs (UIMs) at its amino terminus (2, 3). Recently, we and others demonstrated that RAP80 plays a critical role in DNA damage response signaling (4–7). These studies showed that RAP80 translocates to ionizing radiation-induced foci (IRIF) after IR and that the UIMs are essential for this re-localization. It was further shown that RAP80 forms a complex with BRCA1 and that this association is dependent on the BRCA1 C-terminal (BRCT) repeats of BRCA1. BRCA1 plays a critical role in DNA repair and activation of cell cycle checkpoints, and genetic alterations in the BRCA1 gene have been implicated in several cancers (8–10). RAP80 depletion disrupts the translocation of BRCA1 to IRIF and causes defects in G2/M checkpoint activation after IR (5–7). In addition, knockdown of RAP80 expression by small interfering RNA (siRNA) reduces DSB-induced homology-directed recombination (HDR) and increases the sensitivity of cells to IR-induced cytotoxicity (4, 5).
The ataxia-telangiectasia mutated (ATM) and ATM- and RAD3-related (ATR) kinases, members of phosphatidyl inositol 3-kinase-like kinase (PIKK) family, play a key role in several DNA damage repair response pathways (11). ATM is primarily activated by DSBs induced by ionizing radiation (IR) and various chemicals. Once activated, ATM phosphorylates a variety of proteins with different roles in damage response signaling pathways, including proteins involved in the control of cell cycle checkpoints (12–15). Deletion of or mutations in ATM causes defective activation of cell cycle checkpoints and less efficient DSB repair (16). ATR responds to stalled replication forks or other forms of DNA damage, such as UV photoproducts (17, 18). ATR null mice are embryonic lethal due to loss of genomic integrity suggesting a critical role for ATR in embryonic development (19), while deletion of ATR in cells causes loss of DNA damage checkpoint responses and cell death (20).
RAP80 was reported to be a target of ATM phosphorylation (4–7). We showed that ATM phosphorylates RAP80 at Ser205 and Ser402 in vitro (4), but whether these sites are phosphorylated by ATM in whole cells and are also targets of phosphorylation by ATR was not established. In this study, we report that RAP80 becomes phosphorylated at Ser205 in IR-treated cells. This phosphorylation was dependent on ATM and independent of BRCA1, and occurred at a time that preceded the translocation of RAP80 to IRIF by more than 60 min. Activation of ATR by UV treatment can catalyze the phosphorylation of some of the same substrates as IR-activated ATM (21–23). We demonstrate that after UV irradiation RAP80 translocates to damage foci and co-localizes with γ-H2AX. We further show that UV treatment also induces phosphorylation of RAP80 at Ser205 and provide evidence indicating that this phosphorylation is mediated by ATR. These findings suggest that RAP80 plays a more general role and is important in several types of DNA damage responses.
MATERIALS AND METHODS
Plasmids
pLXIN and pEGFP were purchased from BD Biosciences. pLXIN-3×FLAG-RAP80, pEGFP-RAP80, the mutant pEGFP-RAP80(S205G), and pEGFP-RAP80ΔUIM were described previously (2, 4). pLXIN-3×FLAG-RAP80 mutant, S205G, was generated by subcloning the RAP80(S205G) coding region of pEGFP-RAP80(S205G) into the EcoRI/BamHI sites of pLXIN-3×FLAG vector. The pcDNA3-Myc-BRCA1 (24) was kindly provided by Dr. Jane E. Visvader (Walter and Eliza Hall Institute of Medical Research and Bone Marrow Research Laboratories, Melbourne, Australia). The pcDNA3-Myc-BRCA1 mutants R1699W and pLXIN-3×FLAG-RAP80 mutants T373S, T373A, and F376W were generated using a Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The sequence of each insert was verified by DNA sequencing.
Generation of RAP80Ser205P-specific antibody
An antibody against the RAP80 phosphopeptide 199CGSWDQSpSQPVFEN was raised in rabbits injected with keyhole-limpet hemocyanin-conjugated phosphopeptide.
Cell culture and transfection
Normal human fibroblasts (GM05757), Ataxia-telangiectasia (A-T) cells (GM05823), and Seckel Cells (GM18366) (Coriell, Camden, NJ) were routinely maintained in Minimum Essential Medium (MEM) containing 15% fetal calf serum (FBS) and antibiotics. HEK293T cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and antibiotics. HEK293T cells were transfected with Polyfect reagent (Qiagen, Valencia, CA) as indicated by the manufacturer. The breast cancer cell lines MCF-7 and HCC1937 were maintained in RPMI1640 medium containing 10% FBS and antibiotics. For RAP80 knockdown, MCF-7 cells were transfected with control or RAP80 siRNA (Invitrogen, Carlsbad, CA) following the manufacturer’s suggestions. MCF-7 cells were seeded at 105/ml in antibiotics-free RPMI1640 medium containing 10% FBS. On the second day, siRNAs were mixed with Lipofectamine 2000 (Invitrogen) in antibiotics- and serum-free medium for 20 min and then added to the cells at a final concentration of 20 nM. After 72 h incubation cells were irradiated and then fixed for immunofluorescence or harvested for Western blot analysis.
Confocal microscopy
MCF-7 cells were transiently transfected with wild type pEGFP-RAP80 plasmid DNA and 48 h later treated with ionizing irradiation or UV at the dose indicated. At different time intervals after irradiation cells were fixed for 20 min in 4% paraformaldehyde, and subsequently treated for 7 min with 0.2% Triton X-100. Cells were washed in PBS and then incubated for 15 min in Superblock Blocking Buffer (Pierce, Rockford, IL). Cells were subsequently incubated for 2 h with anti-γ-H2AX antibody (Upstate Biotech., Temecula, CA), and finally for 40 min with anti-mouse Alexa 595 (Molecular Probes, Eugene, OR). Endogenous RAP80Ser205P was detected with the RAP80Ser205P antibody and an anti-rabbit Alexa 488 antibody (Molecular Probes). FLAG-RAP80 in MCF-7-RAP80 cells (4) was detected with an anti-FLAG M2 antibody (Sigma, St. Louis, MO). Nuclei were stained with 4′,6-diamidino-2-phenyindole (DAPI). Cells were then covered with 80% glycerol and fluorescence was observed in a Zeiss LSM 510 NLO confocal microscope (Zeiss, Thornwood, NY).
Co-immunoprecipitation assay
HEK293T cells were transfected with wild type pLXIN-3×FLAG-RAP80, pcDNA3-Myc-BRCA1, or their mutants as indicated, and 48 h later harvested and lysed for 1 h in RIPA buffer (Upstate Biotech.) containing protease and phosphatase inhibitor cocktails I and II (Sigma). The cell lysates were centrifuged at 14,000 ×g at 4 °C for 10 min. The supernatants were then incubated with anti-FLAG resin (Sigma) overnight to isolate RAP80 protein complexes. The beads were then washed three times with RIPA buffer. The bound protein complexes were then solubilized in sample buffer and analyzed by Western blot analysis using anti-FLAG M2 (Sigma) and anti-Myc (gift from Dr. Yue Xiong, UNC, Chapel Hill, NC) antibodies. In the case of phosphatase treatment, immunoprecipitated proteins were treated with λ-phosphatase (400 units/50 μl; New England Biolab.) for 30 min at 30°C before they were examined by Western blot analysis.
RESULTS
ATM phosphorylates RAP80 at Ser205
Previously, we found that ATM phosphorylates RAP80 at Ser205 and Ser402 in vitro (4). To investigate whether RAP80 is phosphorylated by ATM in whole cells, we generated a rabbit polyclonal antibody against a RAP80 phosphopeptide (from C199 to N212) phosphorylated at Ser205 (hereafter referred to as anti-RAP80Ser205P). To test the specificity of the antibody, HEK293T cells were transfected with wild type pLXIN-3×FLAG-RAP80 plasmid or its mutant, S205G and subsequently treated with IR. Protein lysates were then examined by Western blot analysis with the anti-RAP80Ser205P antibody. As shown in Figure 1A (left panel), the antibody did not recognize any protein in non-irradiated cells (lane 1) and reacted weakly with a 100 kD protein, presumably endogenous RAP80, in IR-treated non-transfected cells (lane 2). The anti-RAP80Ser205P antibody recognized low levels of phosphorylated FLAG-RAP80 in non-irradiated HEK293T cells transfected with pLXIN-3×FLAG-RAP80 (lane 3). This level was greatly enhanced after IR treatment (lane 4). The antibody recognized another lower band in lane 4, representing a C-terminal truncated FLAG-RAP80. The signal was specific for RAP80 phosphorylated on Ser205, because like mock-transfected cells, the antibody recognized only one weak band, representing endogenous phosphorylated RAP80, in IR-treated HEK293T cells expressing the RAP80 S205G mutant (lane 2 and 6). To further confirm the specificity of the antibody, proteins from irradiated U2OS cells were immunoprecipitated with anti-RAP80 antibody, subsequently treated with or without λ-phosphatase, and then examined by Western blot analysis with anti-RAP80Ser205P. As shown in (Fig. 1A, right panel), the recognition of RAP80Ser205P by the anti-RAP80Ser205P antibody was lost after phosphatase treatment. These results indicate that the anti-RAP80Ser205P antibody specifically recognizes RAP80 phosphorylated at Ser205 and demonstrate that RAP80 becomes phosphorylated on Ser205 in vivo in an IR-dependent manner.
Figure 1. ATM phosphorylates RAP80 at Ser205 in vivo.

A. Left panel: HEK293T cells were transfected with wild type pLXIN-3×FLAG-RAP80 or its mutant S205G, and 48 h later γ-irradiated (10 Gy). Cells were collected 3 h after irradiation and lysates examined by Western blot analysis with antibodies against RAP80Ser205P, FLAG, RAP80, and actin. Right panel: U2OS cells were γ-irradiated (10 Gy) and 30 min later cell lysates prepared. RAP80 protein complexes were immunoprecipitated with an anti-RAP80 antibody and subsequently treated with and without λ-phosphatase. Complexes were then examined by Western blot analysis with antibodies against RAP80Ser205P and RAP80. B. MCF-7 cells were treated with control or RAP80 siRNA, and 3 days later irradiated (10 Gy). Three hours later cells were fixed and stained with anti-RAP80Ser205P and anti-γ-H2AX antibodies. Nuclei were identified by DAPI staining. Localization of RAP80Ser205P and γ-H2AX was examined by confocal microscopy. Protein lysates from a parallel set of cells were examined by Western blot analysis with anti-RAP80 antibody to analyze the efficiency of the RAP80 knockdown (right panel). C. GM05757 and GM05823 cells were exposed to IR (10 Gy). Three h later cell lysates were prepared and proteins examined by Western blot analysis with anti-RAP80Ser205P and anti-RAP80 antibodies. D. GM05757 and GM05823 cells were exposed to IR (10 Gy). Cells were fixed and stained 3 h after irradiation as described under B.
To determine whether RAP80Ser205P translocated to DNA damage foci after IR, MCF-7 cells were treated with control or RAP80 siRNA and 3 days later irradiated Subsequently, cells were stained with anti-RAP80Ser205P and anti-γ-H2AX antibodies. Fig. 1B demonstrates that RAP80 expression was down-regulated by more than 90% in cells transfected with RAP80 siRNA. Confocal microscopy showed that 3 hours after 10-Gy irradiation 100% of the control cells (n>300) contained foci identified either by staining with the anti-γ-H2AX or the anti-RAP80Ser205P antibody. Most of the RAP80Ser205P foci co-localized with those of γ-H2AX. The induction of RAP80Ser205P foci was greatly abolished in cells in which RAP80 expression was knocked down by siRNAs while it did not affect the formation of γ-H2AX foci (Fig. 1B). These data are consistent with our conclusion that the antibody specifically recognizes RAP80Ser205P and that after IR RAP80Ser205P localizes to DNA damage foci.
Next, we determined whether the IR-induced phosphorylation of RAP80 at Ser205 was dependent on ATM. Normal human fibroblasts (GM05757) and Ataxia-Telangiectasia (A-T) cells (GM05823), which are deficient in ATM kinase activity, were treated with IR and protein lysates subsequently examined by Western blot analysis with anti-RAP80Ser205P antibody. The results showed that RAP80Ser205P was only detected in IR-treated normal fibroblasts (Fig. 1C) in agreement with the conclusion that phosphorylation of RAP80 at Ser205 is mediated by ATM. This conclusion was supported by confocal microscopy. RAP80Ser205P was detected in foci, that co-localized with those of γ-H2AX, in IR-treated normal fibroblasts but not in A-T cells (Fig. 1D).
RAP80 phosphorylation by ATM occurs before its translocation
To study the time course of RAP80 phosphorylation by ATM, MCF-7 and U2OS cells were irradiated and at different time intervals protein lysates were isolated and examined by Western blot analysis with the anti-RAP80Ser205P antibody. Our data showed that in both cell lines RAP80 became phosphorylated as early as 5 min after IR treatment (Fig. 2A). The upper band of RAP80 signal represents a non-specific signal, since it is not affected by RAP80 siRNA treatment (Sup Fig. 1). Previously, we reported that after IR treatment RAP80 translocates to DNA damage foci at times later than 60 min (4). Thus, phosphorylation of RAP80 by ATM occurs much earlier than its translocation to IRIF. To examine this further, MCF-7-RAP80 cells, which stably express FLAG-RAP80 (4), were irradiated and at different time intervals examined by confocal microscopy with anti-FLAG and anti-RAP80Ser205P antibodies. Consistent with Western blot analysis, RAP80 became phosphorylated at Ser205 within 10 min after IR. Initially, RAP80Ser205 was distributed within the nucleus in a rather homogeneous pattern but became localized to nuclear foci 60 min after IR (Fig. 2B). As expected, at 120 min after IR the patterns of foci formation obtained after anti-FLAG and anti-RAP80Ser205P staining largely overlapped. Similar results were obtained when the phosphorylation of endogenous RAP80 was examined in MCF-7 cells with the anti-RAP80Ser205P antibody. As shown in Fig. 2C, RAP80 was clearly phosphorylated at Ser205 within 15 min after IR; however, very few cells formed RAP80Ser205P foci that co-localize with γ-H2AX (2%, n>300). After 3 h, RAP80Ser205P co-localized with γ-H2AX to IRIF in all cells (100%, n>300). These results demonstrate that RAP80 phosphorylation by ATM happens very quickly and long before its translocation to IRIF.
Figure 2. Phosphorylation of RAP80 by ATM occurs long before its translocation to IRIF.

A. MCF-7 and U2OS cells were exposed to IR (10 Gy) and at time points indicated cells were collected and protein lysates prepared. The lysates were examine by Western blot analysis with anti-RAP80Ser205P and anti-RAP80 antibodies. B. MCF-7-RAP80 cells were exposed to 10 Gy γ-irradiation and fixed at the indicated time points. Cells were then stained with anti-FLAG and anti-RAP80Ser205P antibodies and the localization of FLAG-RAP80 and RAP80Ser205P was examined by confocal microscopy. C. MCF-7 cells were exposed to IR (10 Gy), fixed at the time points indicated, and the subcellular distribution of RAP80Ser205P and γ-H2AX examined by staining with DAPI, and anti-RAP80Ser205P and anti-γ-H2AX antibodies.
RAP80Ser205 phosphorylation by ATM is BRCA1-independent
Previous studies demonstrated that RAP80 is in a protein complex with BRCA1 (4–7). The phosphorylation of some proteins have been reported to depend on the presence of a functional BRCA1 whereas that of others occur in a BRCA1-independent manner (25). To determine whether a functional BRCA1 was required for RAP80 phosphorylation by ATM, we analyzed RAP80Ser205 phosphorylation in HCC1937 cells, which contain a mutant BRCA1. Fig. 3 shows that as in MCF-7 cells, RAP80 was phosphorylated in IR-treated HCC1937 cells indicating that a functional BRCA1 is not required for RAP80 phosphorylation by ATM.
Figure 3. RAP80 phosphorylation by ATM is independent of a functional BRCA1.
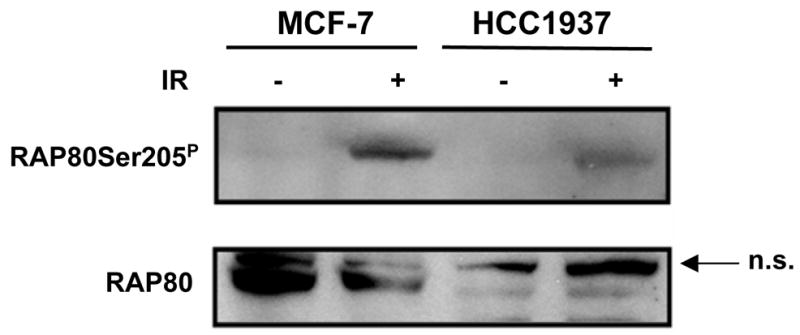
MCF-7 and HCC1937 were treated with 10 Gy γ-irradiation. Three hours later cells were collected and examined by Western blot analysis using anti-RAP80 and anti-RAP80Ser205P antibodies.
The R1699W BRCT mutation disrupts the interaction of BRCA1 with RAP80
Recent studies reported that RAP80 is required for the translocation of BRCA1 to DNA damage foci and that the BRCT motifs of BRCA1 are required for the interaction of RAP80 with BRCA1 (4–7). The structural integrity of the BRCT tandem repeats is essential for the interaction of BRCA1 with phosphorylated protein targets (26, 27). The missense mutation R1699W abolishes the binding of phosphopeptides and is associated with an elevated risk for hereditary breast/ovarian cancer (28–30). Co-immunoprecipitation analysis showed that this mutation abolished the interaction of BRCA1 with RAP80 (Fig. 4A) in agreement with the observation that the BRCT is essential for this association.
Figure 4. The cancer predisposing mutation R1699W disrupts the association of BRCA1 with RAP80.

HEK293T cells were transfected with pLXIN-3×FLAG-RAP80 and pcDNA3-Myc-BRCA1 or mutant pcDNA3-Myc-BRCA1(R1699W) (A) as indicated. Or cells were transfected with pcDNA3-Myc-BRCA1 and pLXIN-3×FLAG-RAP80 or one of the RAP80 mutants, pLXIN-3×FLAG-RAP80(T373S), (T373A), or (F376W) (B) as indicated. Forty-eight h after transfection, cell lysates were prepared and FLAG-RAP80 protein complexes isolated with an anti-FLAG resin. Complexes were then examined by Western blot analysis with antibodies against Myc and FLAG.
BRCT was identified as a phosphopeptide binding motif that specifically recognizes a pSer(Thr)-X-X-Phe motif (31, 32). Sequence analysis showed that RAP80 has one potential pSer(Thr)-X-X-Phe motif (373TKDF376) in a region (between a.a. 204–404) previously identified as being important for the association of RAP80 with BRCA1 (4). To determine whether the 373TKDF376 motif plays a role in this interaction, we made several point mutations T373A, T373S, and F376W in 3×FLAG-RAP80 and examined whether these mutations affected the interaction of RAP80 with BRCA1. The data showed that none of the mutations had an effect on this interaction (Fig. 4B), suggesting that this motif is not essential.
RAP80 translocates to the DNA damage foci after UV treatment and is phosphorylated by ATR
To determine whether RAP80 might be involved in DNA damage responses induced by UV treatment, we transfected MCF-7 cells with pEGFP-RAP80 and examined its subcellular localization after UV irradiation. Cells were treated with a moderate dose of UV (10J/M2) that affects cell survival to a similar extent as 4 Gy IR (5, 33). Our data demonstrated that after UV treatment RAP80 translocated to DNA damage foci that co-localized with those of γ-H2AX (Fig. 5A) (22). In addition, we found that UV treatment induced phosphorylation of RAP80 at Ser205 and translocation of RAP80Ser205P to DNA damage foci (Fig. 5B). It has been suggested that UV-induced phosphorylation of H2AX and γ-H2AX foci formation are triggered by DSBs induced by blocked replication forks in S-phase cells (34). However, subsequent studies showed phosphorylation of H2AX in non-S phase cells triggered by single-stranded DNA intermediates (33). Our observations showing that over 70% of the cells contained foci that were positive for both γ-H2AX and RAP80, a percentage that is much higher than that of the S-phase cell population, are consistent with this. These data suggest that RAP80 is involved in DNA damage responses other than those triggered by DSBs. Because UV treatment activates ATR instead of ATM, it is likely that UV-induced RAP80 phosphorylation is mediated by ATR. However, a recent study reported that ATR can phosphorylate and activate ATM after UV treatment (21). To rule out the possibility that UV-induced phosphorylation of RAP80 is mediated by ATM rather than ATR, we treated A-T and control cells with UV irradiation and examined RAP80 phosphorylation by Western blot analysis with anti-RAP80Ser205P antibody. The results in Fig. 5C show that RAP80 was phosphorylated at Ser205 in both control and A-T cells consistent with the conclusion that ATM is not required for RAP80 phosphorylation after UV treatment. To further establish that the phosphorylation of RAP80 after UV treatment was mediated by ATR, we examined the effect of UV or IR on RAP80Ser205 phosphorylation in Seckel cells which have an impaired ATR function (22). The results in Fig. 5D show that IR exposure of these cells induces phosphorylation of both H2AX and RAP80 whereas after UV treatment does not. These results suggest that after UV treatment ATR is responsible for the phosphorylation of H2AX and RAP80. As shown for IR (4), EGFP-RAP80ΔUIM lacking the UIMs, failed to translocate to damage foci after UV treatment (Fig. 6A) indicating that the UIMs are also critical for translocation of RAP80 in response to UV-induced DNA breaks. Moreover, UV treatment did not affect the association of RAP80 with BRCA1 (Fig. 6B).
Figure 5. UV irradiation induces translocation and phosphorylation of RAP80.

A. MCF-7 cells were transfected with pEGFP-RAP80 and 48 h later treated with UV (10 J/M2). After 3 h incubation, cells were fixed and subsequently stained with anti-γ-H2AX ntibody and DAPI. Localization of EGFP-RAP80 and γ-H2AX was examined by confocal microscopy. B. MCF-7 cells were UV-irradiated (10J/M2), and 2 h later fixed and stained with DAPI, and anti-RAP80Ser205P and anti-γ-H2AX antibodies. C. GM05757 and GM05823 cells were exposed to UV irradiation (10J/M2). Three h later cell lysates were prepared and examined by Western blot analysis using anti-RAP80 and anti-RAP80Ser205P antibodies. D. Seckel cells were exposed to IR (10 Gy) or UV (10 J/M2), and 3 h later stained with anti-RAP80Ser205P and anti-γ-H2AX antibodies. Nuclei were identified by DAPI staining. Localization of RAP80Ser205P and γ-H2AX was examined by confocal microscopy.
Figure 6. UIMs of RAP80 are required for its translocation to DNA damage foci after UV irradiation.

A. MCF-7 cells were transfected with pEGFP-RAP80 or pEGFP-RAP80ΔUIM and then treated as described under 5A. B. UV irradiation does not affect the association of RAP80 and BRCA1. HEK293T cells were transfected with pLXIN-3×FLAG-RAP80 and pcDNA3-Myc-BRCA1 and 48 h later exposed to UV-irradiation (10 J/M2). Three h later protein lysates were prepared and FLAG-RAP80 protein complexes isolated with an anti-FLAG resin. The complexes were then examined by Western blot analysis with antibodies against Myc and FLAG.
DISCUSSION
The human genome is under constant attack by radiation and different genotoxic chemicals. These exposures induce various DNA lesions that subsequently generate DNA damage responses to maintain genome integrity (11, 16, 35, 36). This involves a large number of proteins that are implicated in DNA damage repair, the regulation of cell cycle checkpoints and transcription, and apoptosis when damage is not properly repaired. We, and others, recently reported that RAP80 translocates to IRIF after IR treatment and plays an important role in DNA damage responses (4–7). RAP80 interacts with BRCA1 and is critical for efficient repair through DNA damage-induced homology-directed recombination and in cell cycle checkpoint control.
ATM plays a key role in the activation of cell cycle checkpoints after IR-induced DSBs. Loss of ATM, as in A-T cells, results in defects in the control of all checkpoints (16). In addition to ATM itself, ATM phosphorylates many DNA damage response proteins, including BRCA1 and H2AX. RAP80 was recently identified as a novel substrate of ATM and after IR became phosphorylated at several sites (4–7). Ser205 was identified as one of the ATM phosphorylation sites in vitro (4). In the current study, we show that exposure of cells to IR induces phosphorylation of RAP80 at Ser205 and that subsequently, phosphorylated RAP80Ser205P translocates to DNA damage foci where it co-localizes with γ-H2AX (Fig. 1). This phosphorylation was demonstrated to depend on the activation of ATM. Both ATM and DNAPK have been reported to be able to phosphorylate H2AX, and in A-T cells, it is DNAPK that phosphorylates H2AX after IR (37). Our results show that RAP80 is not phosphorylated at Ser205 in A-T cells after IR (Fig. 1C, D), indicating RAP80Ser205 is not a target of DNAPK.
We further demonstrate that the phosphorylation of RAP80 at Ser205 occurs rapidly and long before RAP80Ser205P translocates to DNA damage foci. The delayed translocation to IRIF suggests a role for RAP80 at a later stage of DNA repair whereas its rapid phosphorylation by ATM suggests that RAP80 might have a role in the regulation of an early event in the DNA damage response as well, such as control of cell cycle checkpoints. The latter hypothesis is consistent with findings showing that depletion of RAP80 causes defects in the control of the G2/M checkpoint after IR (5–7). Besides Ser205 and Ser402, several other ATM phosphorylation sites were identified in RAP80 (5–7). Interestingly, none of the sites is highly conserved across species (5). This raises several interesting questions: what is the function of RAP80 phosphorylation? Are all phosphorylation sites important for G2/M checkpoint control or is phosphorylation of RAP80 at different sites associated with different functions? Although RAP80 phosphorylation by ATM does not affect its association with BRCA1 or its translocation to IRIF (4), the phosphorylation might change the conformation of RAP80 and affect the interaction of RAP80 or RAP80/BRCA1 complex with other proteins. Subsequently this might affect the activity of the complex and the roles it plays in DNA damage response signaling. In support of this hypothesis, phosphorylation of BRCA1 at distinct sites has been reported to be linked to the regulation of different checkpoints (38). Whether phosphorylation of RAP80 at distinct sites is linked to different functions needs further investigation.
Previous studies reported that a functional BRCA1 is required for ATM- and ATR-dependent phosphorylation of several proteins, including p53, c-Jun, NBS1, CtIP, and Chk2 (25). These proteins are required for checkpoint activation and/or apoptosis, and as observed for RAP80, appear to be part of a BRCA1 complex even before the induction of DNA damage. However, we show that the phosphorylation of RAP80 is independent of a functional BRCA1 (Fig. 3).
In this study, we further demonstrate that RAP80 translocates to DNA damage foci after UV irradiation and that also this migration is dependent on the UIMs of RAP80 (Fig. 4). We show that RAP80 is phosphorylated at Ser205 and provide evidence that this phosphorylation is mediated by ATR and not ATM. Unlike IR, which induces DSBs directly, UV treatment mainly induces cyclobutane pyrimidine dimers (CPD) and pyrimidine (6-4) pyrimidone photoproducts (6-4PP) (39). Although UV-induced phosphorylation of H2AX and γ-H2AX foci formation can be triggered by DSBs induced by blocked replication forks in S-phase cells (34), recent studies demonstrated a cell cycle-independent induction of H2AX phosphorylation and γ-H2AX foci formation after UV triggered by single strand DNA repair intermediates (21, 22, 33). Our observations showing that after UV most cells contained foci that were positive for both γ-H2AX and RAP80, are consistent with this and suggest a role for RAP80 in DNA damage responses triggered by other types of DNA lesions, in addition to those induced by DSBs. This was supported by studies demonstrating that RAP80 depletion caused increased sensitivity to UV irradiation (5). Thus, RAP80 appears to play a role in several types of DNA damage responses.
BRCT motifs are found in a number of proteins with functions in DNA repair responses (29). Mutations in the BRCT motifs of BRCA1 have been linked to elevated risk for breast and ovarian cancer (29, 30). Structural analysis showed that many of the cancer-related mutations in the BRCT repeats disrupt the structure of the pSer(Thr)-X-X-Phe binding pocket, abolish the interaction of BRCT with its partners, and prevents BRCA1 from translocating to DNA damage foci (26–29). The BRCT motifs of BRCA1 were shown to be essential for its association with RAP80 and this interaction is required for the translocation of BRCA1 to DNA damage foci (4–7). In agreement with these findings, we demonstrated that the BRCT missense mutation R1699W abolished the interaction of BRCA1 with RAP80 (Fig. 4A). Although RAP80 has one potential pSer(Thr)-X-X-Phe motif within the region essential for its association with BRCA1 (4), this motif is not required for this interaction (Fig. 4B) suggesting that RAP80 and BRCA1 may interact indirectly by binding an intermediary protein. This concept was supported by recent studies showing that this interaction is mediated by CCDC98 (40, 41).
In summary, our study extends previous observations and demonstrates that RAP80 becomes phosphorylated at Ser205 in an ATM-dependent manner in IR-treated cells. This phosphorylation occurs long before RAP80 translocates to IRIF and is independent of BRCA1. UV irradiation also induces phosphorylation of RAP80 at Ser205 and its translocation to DNA damage foci. This phosphorylation requires ATR, not ATM. We further show that the BRCT mutant R1699W does not interact with RAP80. Future studies have to determine what the functions are of the different phosphorylation sites of RAP80.
Supplementary Material
Acknowledgments
We would like to thank Drs. Richard Paules and Daniel Menendez for their comments on the manuscript. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- 1.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 2.Yan J, Kim YS, Yang XP, Albers M, Koegl M, Jetten AM. Ubiquitin-interaction motifs of RAP80 are critical in its regulation of estrogen receptor α. Nucl Acids Res. 2007;35:1673–86. doi: 10.1093/nar/gkl1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan Z, Kim YS, Jetten AM. RAP80, a novel nuclear protein that interacts with the retinoid-related testis-associated receptor. J Biol Chem. 2002;277:32379–88. doi: 10.1074/jbc.M203475200. [DOI] [PubMed] [Google Scholar]
- 4.Yan J, Kim YS, Yang XP, et al. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007;67:6647–56. doi: 10.1158/0008-5472.CAN-07-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang B, Matsuoka S, Ballif BA, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–8. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobhian B, Shao G, Lilli DR, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–5. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Willers H, Feng Z, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:708–18. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuang J, Zhang J, Willers H, et al. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006;66:1401–8. doi: 10.1158/0008-5472.CAN-05-3278. [DOI] [PubMed] [Google Scholar]
- 11.Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–7. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 13.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–6. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 14.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 15.Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–77. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–42. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 17.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 18.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 19.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 20.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 21.Stiff T, Walker SA, Cerosaletti K, et al. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. Embo J. 2006;25:5775–82. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 23.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sum EY, Peng B, Yu X, et al. The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J Biol Chem. 2002;277:7849–56. doi: 10.1074/jbc.M110603200. [DOI] [PubMed] [Google Scholar]
- 25.Foray N, Marot D, Gabriel A, et al. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. Embo J. 2003;22:2860–71. doi: 10.1093/emboj/cdg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolopoulos G, Pyrpassopoulos S, Thanassoulas A, et al. Thermal unfolding of human BRCA1 BRCT-domain variants. Biochim Biophys Acta. 2007;1774:772–80. doi: 10.1016/j.bbapap.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem. 2003;278:52914–8. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- 28.Au WW, Henderson BR. The BRCA1 RING and BRCT domains cooperate in targeting BRCA1 to ionizing radiation-induced nuclear foci. J Biol Chem. 2005;280:6993–7001. doi: 10.1074/jbc.M408879200. [DOI] [PubMed] [Google Scholar]
- 29.Glover JN. Insights into the molecular basis of human hereditary breast cancer from studies of the BRCA1 BRCT domain. Fam Cancer. 2006;5:89–93. doi: 10.1007/s10689-005-2579-z. [DOI] [PubMed] [Google Scholar]
- 30.Vallon-Christersson J, Cayanan C, Haraldsson K, et al. Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet. 2001;10:353–60. doi: 10.1093/hmg/10.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 32.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–9. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 33.Hanasoge S, Ljungman M. H2AX phosphorylation after UV-irradiation is triggered by DNA repair intermediates and is mediated by ATR. Carcinogenesis. 2007;28:2298–304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 34.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 35.d’Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004;18:1781–99. doi: 10.1101/gad.1214504. [DOI] [PubMed] [Google Scholar]
- 36.Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7:165–72. doi: 10.1016/j.carrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 38.Xu B, O’Donnell AH, Kim ST, Kastan MB. Phosphorylation of serine 1387 in Brca1 is specifically required for the ATM-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 2002;62:4588–91. [PubMed] [Google Scholar]
- 39.Mitchell DL. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem Photobiol. 1988;48:51–7. doi: 10.1111/j.1751-1097.1988.tb02785.x. [DOI] [PubMed] [Google Scholar]
- 40.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nature Struct Mol Biol. 2007;14:710–5. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Wu J, Yu X. CCDC98 targets BRCA1 to DNA damage sites. Nature Struct Mol Biol. 2007;14:716–20. doi: 10.1038/nsmb1279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.