

Abstract
Purpose
Activity of ornithine decarboxylase (ODC), the first enzyme in polyamine synthesis, is required for normal growth and is elevated in many cancers, including colorectal cancer (CRC). We examined associations of the +316 ODC1 single nucleotide polymorphism (SNP) with CRC-specific survival among CRC cases, and then investigated its functional significance in colon cancer cells.
Experimental Design
The study included 400 incident stage I-III CRC cases from the population-based UC Irvine Gene-Environment Study of Familial CRC (diagnosed 1994-1996 with follow-up through March 2008). The primary outcome was CRC-specific survival (CRC-SS) dependent on ODC1 (rs2302615) genotype (GG vs. GA/AA). In human colon cancer cell lines, ODC1 allele-specific binding of E-box transcription factors was determined via western blotting and chromatin immunoprecipitation (CHIP) assays. ODC1 allele-specific promoter activity was determined using promoter constructs in combination with vectors expressing either the transcriptional activator c-MYC or the repressor MAD1.
Results
Genotype-specific survival differences were observed among CRC cases: compared to cases with ODC1 GG genotype (HR=1.00, reference) the adjusted CRC-SS hazards ratio (HR) was 2.02 (1.17-3.50) for ODC1 GA/AA cases (P = 0.012). In colon cancer cells, the ODC1 SNP, flanked by two E-boxes, predicts ODC1 promoter activity. The E-box activator c-MYC and repressors MAD1 and MAD4 preferentially bind to ODC1 minor A-, compared to major G-, alleles in cultured cells.
Conclusions
These results have implications for conditional regulation of polyamine homeostasis and suggest a model in which the ODC1 SNP may be protective for colon adenoma recurrence and detrimental for survival after colon cancer diagnosis.
Keywords: Polyamines, E-box transcription factors, colorectal cancer, survival
Introduction
Polyamines are small ubiquitous molecules involved in various processes, including transcription, RNA stabilization, ion channel gating and others (1). Ornithine decarboxylase (ODC), the first enzyme in polyamine synthesis, is essential for normal development and tissue repair in mammals but is down-regulated in most adult tissues (2). Multiple abnormalities in the control of polyamine metabolism and transport result in increased polyamine levels that can promote tumorigenesis in several tissues (3). Polyamine metabolism is up-regulated in intestinal epithelial tissues of humans with familial adenomatous polyposis (FAP) (4), a syndrome associated with high risk of colon and other cancers. FAP is caused by mutations in the adenomatous polyposis coli (APC) tumor suppressor gene, and we have shown that wild-type APC signaling downregulates ODC1 expression in both human cells (5) and in a mouse model of FAP (6). Wild type APC expression leads to decreased expression of ODC, while mutant APC leads to increased expression of ODC. The mechanism of APC-dependent regulation of ODC involves E-box transcription factors, including the transcriptional activator c-MYC and the transcriptional repressor MAD1 (5, 7). c-MYC was shown by others to regulate ODC1 transcription (8). Several genes involved in polyamine metabolism are essential genes for optimal growth in most organisms, and are down-regulated in non-proliferating and/or adult cells and tissues (2). The polyamines influence specific cellular phenotypes, in part, by affecting patterns of gene expression, as reviewed elsewhere (9).
D, L-α-Diflouromethylornithine (DFMO), a selective ODC inhibitor, decreases APC-dependent intestinal tumorigenesis in mice (6). Oral DFMO administered daily to humans inhibits ODC enzyme activity and polyamine contents in a number of epithelial tissues (10-15). Recently, DFMO in combination with the non-steroidal anti-inflammatory drug (NSAID) sulindac, has been reported to markedly lower the adenoma recurrence rate among individuals with colonic adenomas when compared to placebos in a randomized clinical trial (16). We have associated a common SNP in the ODC1 promoter, 316 nucleotides 3′ of the transcription start site, with decreased risk of colon polyp recurrence especially in people who report using aspirin (7), and this finding has been confirmed by others (17, 18). This SNP falls between two consensus E-box binding elements. Although the risk of adenoma recurrence based on ODC1 genotype has been investigated previously, the effects of this SNP on clinical outcomes after CRC diagnosis are unknown.
In this report, we studied the association of the ODC1 +316 SNP to CRC-specific mortality among CRC cases from a population-based study. Subsequently, using experimental models involving cultured colon cancer cells we investigated the functional significance of the ODC1 SNP, as depicted in Figure 1. Previous work by our group and others has associated the ODC A-allele at +316 to reduced risk of colon polyp recurrence (7). Ornithine decarboxylase +316 genotype is prognostic for colorectal adenoma recurrence and predicts efficacy of aspirin chemoprevention (18). Visvanathan et al. (19) have reported results apparently at odds with our previous findings in colon carcinogenesis. Visvanathan and colleagues have associated the ODC1 A-allele at +316 with increased risk of prostate cancer. Our earlier functional studies, using promoter reporter methods (7) showed that the repressor MAD1 could suppress the expression of the ODC1 promoter in an allele-specific manner. However, no direct evidence for binding of E-box transcription factors to the ODC1 promoter in an allele-specific manner has yet been reported. Consequently, we tested the hypothesis that both transcriptional activators and repressors might bind selectively to the ODC1 promoter in an allele-specific manner. Such allele specific binding might explain the apparent discordant associations of this allele with both reduced risk of precancerous lesions, but increased risk of invasive cancer, based on differences in the expression of E-box transcriptional activators and repressors. For example, c-MYC is known to be expressed at low levels in normal intestinal epithelium, but at high levels in intestinal polyps, in a mouse model of intestinal carcinogenesis (20).
Fig. 1.
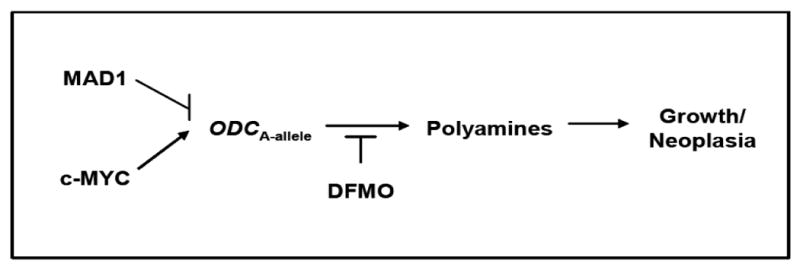
Schema depicting the proposed differential effects of polyamine regulation by MAD1 and c-MYC on the ornithine decarboxylase-1 (ODC-1) +316 minor A-allele. Effects of the ODC inhibitor DFMO (difluoromethylornithine) are also shown.
Materials and Methods
Epidemiologic Studies: ODC1 +316 SNP Associations with CRC-Specific Survival
Study population
We studied incident cases of invasive CRC with stage I-III disease at presentation enrolled in the University of California, Irvine Gene-Environment Study of Familial Colorectal Cancer (21, 22) during 1994-1996 with follow-up through March 2008. The parent study was designed to determine the incidence of HNPCC in a large, population-based cohort of colorectal cancer cases. Participants were identified through the population-based cancer registries of the Cancer Surveillance Program of Orange County/San Diego Imperial Organization for Cancer Control using the April 2008 data file. In the parent study (21), all subjects with CRC diagnosed at all ages in Orange County, CA, from 1994 to1996 were ascertained. All subjects diagnosed in San Diego and Imperial Counties, CA, at ages <65 y between 1994 and 1995 were also ascertained. Cases were then contacted if they were eligible for the study (alive at the time ascertained and having a contact address) and if their physicians did not deny permission to contact. At the time of study entry, cases signed a consent form allowing for blood draws and the release of medical information. This study was approved by the UC Irvine Institutional Review Board (#93-257). Clinical and demographic data including vital status and follow-up were obtained through linkage to the regional cancer registry databases as previously described (21-23). Tumor grade was recorded according to standard convention: grade I-well differentiated, grade II-moderately differentiated, grade III-poorly differentiated, grade IV-undifferentiated. Tumor, node, metastasis (TNM) staging determination was derived from existing AJCC codes where available and conversion of extent of disease codes, as previously reported (24). Family history of cancer in a first-degree relative was ascertained by self-reporting during a telephone interview conducted at enrollment (23, 25). Twenty-two cases with hereditary non-polyposis colon cancer (HNPCC), as defined by Amsterdam criteria, were identified and excluded from the analysis. The median time from CRC diagnosis until study entry (i.e., date of family history interview) was 18 months (95% CI 12-32 months).
DNA extraction and ODC1 +316 SNP genotyping
DNA was extracted from 2.0 mL red blood cell clot samples using the QIAGEN QIAamp DNA Midi or Mini Kits, (Qiagen) following the manufacturer's instructions. Genotyping of the ODC1 (National Center for Biotechnology Information SNP database ID rs2302615) +316 SNP was conducted using oligonucleotide primers designed to amplify a 172-bp fragment containing the polymorphic base at +316 (Applied Biosystems, Foster City, CA). Allele-specific TaqMan probes were synthesized with different 5′ labels (6-carboxyflourescein or VIC) and the same 3′ quencher dye (6-carboxytetramethylrhodamine) (26). Each PCR reaction (5 μL total) contained 10 ng of participant DNA, 30 pmol of each primer, 12.5 pmol of each TaqMan probe, and 1× TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA), as previously reported (7, 26).
Statistical analysis – population-based study
An estimated 1:1 ratio of ODC1 GG genotype to ODC1 GA/AA genotype was expected based on previous literature (7, 17, 18, 26). 440 of 481 DNA samples were successfully genotyped. 41 cases (8.5%) resulted in an undetermined ODC1 +316 genotype due to low DNA concentration and/or poor DNA quality, however no clinicopathologic differences were observed between the successfully genotyped and unsuccessfully genotyped cases. The final analytic cohort was restricted to 400 CRC cases with stage I-III disease. Comparisons of demographic, clinical, and pathologic variables among colon and rectal cases were done using Pearson chi-square statistic or Fisher's exact test for nominal variables and Student t-test for continuous variables. Colorectal cancer-specific survival was defined as mortality from CRC itself, and data censoring occurred in the following instances: alive at the end of follow-up, loss to follow-up, or death from any cause other than CRC. Overall survival (OS) was defined with mortality from any cause. Survival curves were constructed for colon and rectal cancer cases using the Kaplan-Meier method and analyzed with the log rank test for univariate analyses. Cox proportional hazards modeling was performed for all CRC cases, colon cancer cases, and rectal cancer cases using time since diagnosis to profile the adjusted risk of overall and CRC-specific death based on ODC1 genotype. The effects of ODC1 genotype on survival were analyzed in the Cox models with stratification for TNM stage at diagnosis and adjustment for the following covariates: age, gender, ethnicity, family history of CRC, tumor site within the colon, histologic subtype, treatment with surgery, radiation therapy, and chemotherapy. For covariates with categorical data, dummy coding was assigned (0 or 1) and each category was included in the multivariate models in comparison to the referent group. ODC1 genotype was analyzed using the dominant model (GG vs. GA/AA) using dummy variables with GA/AA coded as 1 and GG as the referent group. Subset analyses were performed using the additive model for ODC1 either as continuous variable 0, 1 and 2 test for trend, or using two dummy variables one for GA and one for AA with GG as the referent group. All analyses were conducted using SAS 9.2 statistical software (SAS Institute, Cary, NC). Statistical significance was assumed for a 2-tailed P value <0.05.
Experimental Studies: ODC1 +316 SNP Regulation in Colon Cancer Cells
Cell culture
The human colon cancer cell lines HT29 and HCT116 were maintained in McCoy's 5A medium (Invitrogen, Carlsbad, CA). All media used were supplemented with 10% FBS plus 1% penicillin/streptomycin solution (Invitrogen, Carlsbad, CA). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2.
Genotyping assay
DNA samples from HT29 and HCT116 cells were subjected to a PCR-RFLP procedure to detect the polymorphic PstI site. Sequences were amplified by PCR, using the following primers: 5′– TCTGCGCTTCCCCATGGGGCT-3′ and 5′-TTTCCCAACCCTTCG-3′. Each reaction contained − 1 μl DNA, 4 pmol of each primer, 12.5 μl 2× PCR PreMixes buffer “G” (EPICENTRE Biotechnologies, Madison, WI) and 0.5 unit of Taq DNA polymerase, in a final volume of 25 μl. The expected size of the PCR product was 351 bp. After amplification, 10-20 μl of the PCR product were digested with 10 units of PstI in 30 μl for 2 hours at 37°C. DNA from HT29 cells (GA), containing the PstI site, yielded two fragments of 156 and 195 bp.
Western blot analysis
Cells were harvested, lysed and proteins were separated on a 12.5% SDS-PAGE gel. Proteins were transferred by electrophoresis onto a Hybond-C membrane. The membrane was blocked with Blotto A (5% blocking grade dry milk in TTBS solution) and probed using 1:300 dilutions of primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) in Blotto A. Primary antibodies were incubated at 4°C overnight, followed by incubation with an appropriate HRP-tagged secondary antibody (1:1000 dilution) for 1 hour at room temperature. Chemiluminescent detection was conducted using ECL Western Detection reagent (Amersham Biosciences, Piscataway, NJ) and exposed on Biomax XAR film (Kodak).
Chromatin immunoprecipitation (CHIP)
CHIP assays were performed using a commercial kit, as recommended by the manufacturer (Upstate Biotech, Lake Placid, NY, USA). Briefly, cells were treated with 1% formaldehyde to crosslink DNA and proteins, and DNA-protein complexes were disrupted by sonication to lengths between 200 to 1000 bp. Lysates were diluted 10-fold with immunoprecipitation (IP) dilution buffer containing protease inhibitors. Antibodies for c-MYC, MAD1 and MAD4 (Santa Cruz Biotechnology, Santa Cruz, CA) were used to precipitate chromatin, while additional sample was left as a minus-antibody (-Ab) control. Samples were immunoprecipitated overnight at 4°C with rotation. Immune complexes were obtained by adding 60ul of salmon sperm DNA/protein A Agarose slurry and incubating for an hour at 4°C with rotation followed by gentle centrifugation (1000 rpm, 1 min). Protein A agarose pellets were washed with low salt buffer, high salt buffer, LiCl buffer and TE buffer. Then the complexes were eluted by adding 250 μl elution buffer (0.1M NaHCO3, 1% SDS) twice, and DNA-protein crosslinks were reversed with 0.2 M NaCl by heating at 65°C for 4 hours for all samples, including the input DNA and −Ab DNA controls. DNA was resuspended in 30ul of ddH2O. For visualization of PCR product and its size, standard PCR reactions were carried out. The sequences of ODC1 primers used for PCR were 5′- CCTGGGCGCTCTGAGGT-3′ (17mer) and 5′-AGGAAGCGGCGCCTCAA-3′ (17mer). Quantitative real-time PCR was performed using TaqMan gene expression assays kit (Applied Biosystems, Foster City, CA) on an ABI7700 sequence detection system. Details for the computation of relative binding can be found on the manufacturer's web site (http://www.appliedbiosystems.com/).
Transient transfections
Transient transfections were preformed using LipofectAMINE reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol, as detailed in the supplementary file. HCT116 and HT29 cells were transfected with 1 μg of pGL3-ODC1/A or pGL3-ODC1/G plasmids (7) along with 0.01 μg of Renilla-TK plasmid. The Renilla-TK plasmid was purchased from Promega (Madison, WI) and used as a transfection efficiency control in all promoter-reporter transfection experiments. For MYC experiments, ODC1 pGL3-plasmids were co-transfected with either pcDNA 3.0 or CMV-MYC expression vector (OriGene, Rockville, MD). For MAD1 experiments, the ODC1 plasmids were co-transfected with either pcDNA 3.1 or pcDNA-MAD1. For MYC and MAD1 co-transfection, ODC1 promoter reporter constructs were prepared which contain the first 1.6 Kb of the ODC1 gene cloned into a pGL3 vector. The constructs included E-box 1 (−485 to −480 bp) intact (wt E-box 1) or deleted (mut E-box 1). Additionally, both variants of the +316 ODC1 SNP were used, creating a total of 4 different constructs. After 6 hours of incubation, cells were supplemented with complete medium containing 20% FBS and left to grow overnight. The next day after transfection 20% FBS-containing complete medium was replaced with 10% FBS-containing medium. 48 hours after transfection, cells were washed with PBS and lysed in Passive Lysis Buffer from the Dual Luciferase Assay kit (Promega, Madison, WI). Dual luciferase activities were measured using a Turner Designs TD-20/20 luminometer, as described by the manufacturer, and presented as relative luciferase units (RLU). Experiments were preformed in triplicates and repeated at least 2 times.
Statistical analysis - experimental studies
For transient transfection experiments, two-sample t-tests were used (Microsoft Excel Microsoft Corp., Redmond, WA). The effect of MYC expression on ODC1 allele-specific promoter activity was examined in HT29 colon cancer cells using ODC1 promoter constructs differing by the presence of the first E-box element: (a) wild type (wt) E-box1+316 G, (b) mutant (mut) E-box1 + 316 G, (c) wt E-box1 + 316 A, and (d) mut E-box1 + 316 A. For each promoter construct, two-sample t-tests were used to compare promoter activity between cells co-transfected with pcDNA3.0 plasmid versus those transfected with the CMV-MYC expression vector. Similarly, to examine the effect of MAD1 expression on ODC1 allele-specific promoter activity, two-sample t-tests were used to compare the effect of promoter activity in promoter constructs co-transfected with pcDNA3.1 plasmid versus those transfected with pcDNA-MAD1 plasmid. Statistical significance was assumed for a 2-tailed P value <0.05.
Results
Epidemiologic Studies: Association of the ODC1 +316 SNP with CRC-Specific Survival among Colon and Rectal Cancer Cases
A total of 400 stage I-III CRC cases identified from the UC Irvine CRC gene-environment study were used in the case-only analysis. Median follow-up duration was 11 years, 1 month. There were 252 (63%) colon cancer cases, 143 (36%) rectal cancer cases, and 5 (1%) CRC cases of unspecified location. Clinicopathologic data for colorectal cancer cases are shown in Table 1. Timing of radiation therapy was available for 60 cases (55 rectal and 5 colon cancer cases) of the 63 receiving radiation therapy. Thirteen (21.7%) received neoadjuvant (i.e. pre-surgery) radiation, 45 (75.0%) received adjuvant (i.e., post-surgery) radiation, and 2 (3.3%) received combination neoadjuvant and adjuvant radiation therapy. ODC1 genotype distribution among all CRC cases was 52% GG, 41% GA, and 7% AA. There were no significant differences in ODC1 genotype distribution (GG vs. GA/AA) by age, gender, ethnicity, family history, stage, site within the colorectum, histology, or tumor grade (Table 1). When analyzed by three genotypes (GG, GA, AA), ODC1 genotype distribution by ethnicity revealed significant differences: Caucasian (351cases: 54% GG, 40% GA, 6% AA, minor-A allele frequency = 26%), African-American (4 cases: 50% GG, 50% GA, 0% AA, minor-A allele frequency = 25%), Hispanics (20 cases: 55% GG, 45% GA, 0% AA, minor-A allele frequency = 23%), and Asians (23 cases: 30% GG, 44% GA, 26% AA, minor-A allele frequency = 48%) (P = 0.012). However, within each race ODC1 genotype distribution was in Hardy-Weinberg equilibrium (Caucasians P = 0.51, African-Americans P = 0.51, Hispanics P = 0.19, Asians P = 0.54).
Table 1.
Descriptive analysis for colorectal cancer cases overall and based on ODC1 genotype
| All CRC Cases n = 400 |
ODC1 GG n = 208 |
ODC1 GA/AA n = 192 |
P-value | ||
|---|---|---|---|---|---|
| Median Age (with age range) | 56.6 (29-89) | 56.8 (29-89) | 56.4 (29-82) | 0.24 | |
| Gender | Male | 230 (58%) | 117 (56%) | 113 (59%) | 0.60 |
| Female | 170 (41%) | 91 (44%) | 79 (41%) | ||
| Ethnicity | Non-Hispanic Caucasian | 351 (88%) | 188 (90%) | 163 (85%) | 0.14 |
| African American | 4 (1%) | 2 (1%) | 2 (1%) | ||
| Caucasian-Hispanic | 20 (5%) | 11 (5%) | 9 (5%) | ||
| Asian | 23 (6%) | 7 (3%) | 16 (8%) | ||
| Other | 2 (<1%) | 0 (0%) | 2(1%) | ||
| Family History of CRC | Yes | 106 (26%) | 56 (27%) | 50 (26%) | 0.84 |
| No | 294 (74%) | 152 (73%) | 142 (74%) | ||
| Stage at Diagnosis | I | 146 (37%) | 72 (35%) | 74 (39%) | 0.49 |
| II | 128 (32%) | 72 (35%) | 56 (29%) | ||
| III | 126 (31%) | 64 (31%) | 62 (32%) | ||
| Colon/Rectum Site | Proximal and transverse | 120 (30%) | 65 (31%) | 55 (29%) | 0.90 |
| Descending | 22 (6%) | 12 (6%) | 10 (5%) | ||
| Sigmoid | 110 (28%) | 55(26%) | 55 (29%) | ||
| Rectosigmoid | 57 (14%) | 32 (15%) | 25 (13%) | ||
| Mid-Low Rectum | 86 (22%) | 41 (20%) | 45 (23%) | ||
| Colorectum, unspecified | 5 (1%) | 3 (1%) | 2 (1%) | ||
| Histologic Subtype | Adenocarcinoma Mucinous | 364 (91%) | 183 (88%) | 181 (94%) | 0.16 |
| adenocarcinoma | 26 (7%) | 18 (9%) | 8 (4%) | ||
| Carcinoma | 9 (2%) | 6 (3%) | 3 (2%) | ||
| NOS | 1 (<1%) | 1 (<1%) | 0 (0%) | ||
| Tumor Grade | I | 62 (17%) | 27 (14%) | 35 (20%) | 0.36 |
| II | 261 (71%) | 140 (73%) | 121 (68%) | ||
| III | 46 (13%) | 24 (13%) | 22 (12%) | ||
| No. missing | 31 | 17 | 14 | ||
| Surgical Treatment | Yes | 395 (99%) | 204 (98%) | 191 (99%) | 0.21 |
| No | 5 (1%) | 4 (2%) | 1 (1%) | ||
| Radiation Therapy | Yes | 63 (16%) | 29 (14%) | 34 (18%) | 0.30 |
| No | 337 (84%) | 179 (86%) | 158 (82%) | ||
| Chemotherapy | Yes | 170 (45%) | 84 (44%) | 86 (46%) | 0.56 |
| No | 208 (55%) | 109 (56%) | 99 (54%) | ||
| No. missing | 22 | 15 | 7 | ||
Of the 400 stage I-III CRC cases, 109 (27%) were deceased at the time of analysis. Forty-seven (43%) deaths occurred in cases carrying the ODC1 GG genotype, compared to 62 (57%) deaths in cases with the AA/AG genotypes. Cause of death was available for 76 of the 109 deceased CRC cases. Fifty-nine (77%) CRC cases died as a result of CRC. A statistically significant improvement in CRC-specific survival was observed among CRC cases homozygous for the ODC1 G-allele (10-year survival = 89%) compared to cases with at least one A-allele (ODC1 GA/AA) (10-year survival = 81%; P = 0.011). CRC-specific survival analysis by stage revealed that significantly different survival differences were not observed for AJCC stage I (P = 0.055) or II (P = 0.61) CRC. However, among cases with stage III CRC the ODC1 GG genotype was associated with improved 10-year CRC-specific survival: 75% compared to 60% for ODC1 GA/AA genotype cases; P = 0.024 (Fig. 2). This difference was significant among rectal (10-year OS = 88% for ODC1 GG vs. 75% for ODC1 GA/AA; P = 0.041) but not colon cancer cases (10-year OS = 89% for ODC1 GG vs. 84% for ODC1 GA/AA; P = 0.11).
Fig. 2.

Kaplan-Meier colorectal cancer (CRC) specific survival rate estimates for cases with stage III CRC, stratified by ornithine decarboxylase 1 (ODC1) +316 genotype. Includes cases from the University of California Irvine Gene-Environment Study of Familial CRC diagnosed during 1994-1996 with follow-up through March 2008; ODC1 GG (64 cases, 15 CRC-specific deaths), ODC1 GA/AA (62 cases, 25 CRC-specific deaths).
Among CRC cases, the CRC-specific survival estimates based on ODC1 genotype after stratification for TNM stage at diagnosis and adjustment for age (years), gender, ethnicity, family history of CRC, tumor site within the colon, histologic subtype, treatment with surgery, radiation therapy, and chemotherapy were a follows: ODC1 GG hazards ratio (HR) = 1.00 (referent), ODC1 GA/AA HR = 2.02 (P = 0.021) (Table 2). In the additive model, adjusted CRC-specific survival estimates were as follows: ODC1 GG hazards ratio (HR) = 1.00 (referent), ODC1 GA HR = 1.95, ODC1 AA HR = 2.25 (P-trend = 0.015). Subset multivariate CRC-specific survival analysis revealed that ODC1 genotype was a significant prognostic factor among rectal (ODC1 GA/AA HR=2.92, 95% CI 1.22-7.03 using ODC1 GG as a referent) but not colon cancer cases (ODC1 GA/AA HR=1.76, 95% CI 0.85-3.63 using ODC1 GG as a referent). No significant associations with ODC1 genotype and overall survival were detected in subset adjusted analyses of rectal, or colon cancer cases.
Table 2.
Multivariate overall survival and CRC-specific survival analysis for CRC cases based on ODC1 genotype.
| ODC1 Genotype | P-value | ||
|---|---|---|---|
| GG | GA/AA | ||
| Overall Mortality | |||
| Number of events | 47 | 62 | |
| Number at risk | 208 | 192 | |
| Unadjusted HR (95% CI) | 1 (Reference) | 1.57 (1.07-2.29) | 0.020 |
| Adjusted HR (95% CI)* | 1 (Reference) | 1.58 (1.07-2.34) | 0.021 |
| CRC-Specific Mortality | |||
| Number of events | 22 | 37 | |
| Number at risk | 208 | 192 | |
| Unadjusted HR (95% CI) | 1 (Reference) | 1.97 (1.16-3.34) | 0.012 |
| Adjusted HR (95% CI)* | 1 (Reference) | 2.02 (1.17-3.50) | 0.012 |
Includes stratification for stage (I, II, III) and adjustment for age (years), gender, ethnicity, family history of CRC, TNM stage at diagnosis, tumor site within the colorectum, histologic subtype, treatment with surgery, radiation therapy, and chemotherapy.
As noted above, the ODC1 +316 genotype distribution differed across ethnicity. The observed mortality risk, other than by chance, likely reflects differences based on ODC1 genotype, however the risk may be restricted to a particular ethnic group. Thus multivariate analyses were conducted among Caucasian colon cancer cases, to assess genotype-specific mortality risk within this single ethnic group. Among the 351 Caucasians, there were 52 CRC-related deaths. Multivariate CRC-specific survival analysis revealed that the ODC1 SNP was an independent predictor of CRC-specific survival among Caucasian CRC cases after adjustment for the aforementioned relevant clinical variables. Compared to cases with ODC1 GG genotype (HR=1.00, reference), the CRC-specific risk of death (HR) was 2.37 (1.33-4.23) for ODC1 GA/AA genotype.
Experimental Studies: ODC1 +316 SNP Regulation in Colon Cancer Cells
ODC1 allele-specific binding of E-box transcription factors
We evaluated the functional significance of the +316 ODC1 SNP, located between two E-boxes (E-box 2 and 3 as depicted in Fig. 3, A). Each cell line genotype influences a consensus Pstl restriction site in this region. Fig. 3, B shows that a polymerase chain reaction (PCR) product made from human colon HT29 cells was partially sensitive to PstI cutting, suggesting that these cells contained at least one ODC1 A-allele. A PCR product made from human colon HCT116 cells using the same primers was insensitive to PstI action, implying that these cells contained only ODC1 G-alleles. This result was confirmed by direct DNA sequencing (data not shown).
Fig. 3.
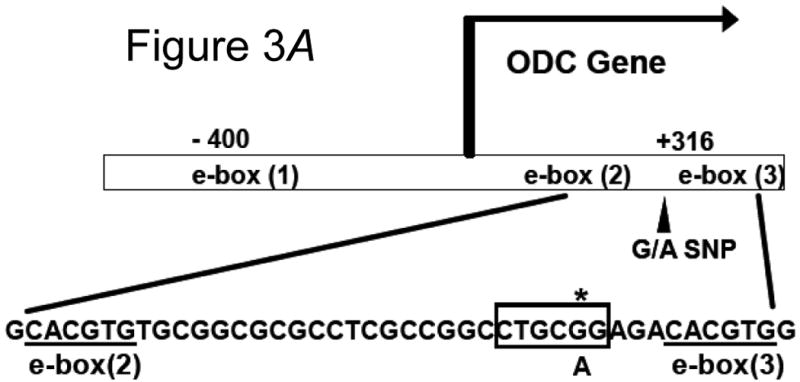
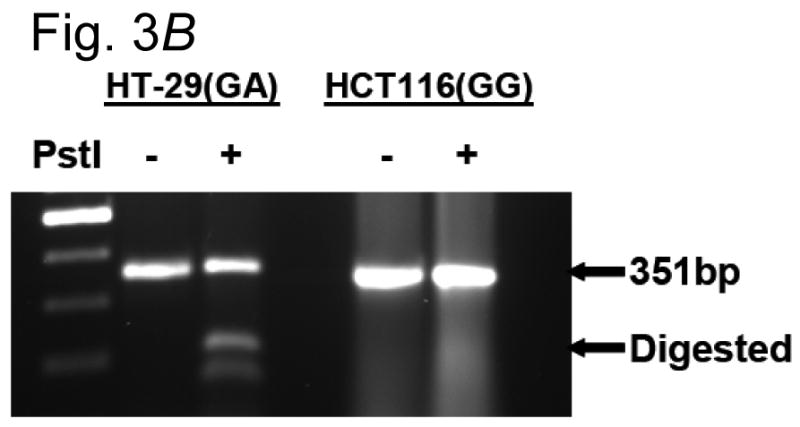
A) Location of the ODC1 promoter SNP. The SNP under investigation in this study is 316 nucleotides 3′ of the ODC1 transcription start site (marked with an asterisk). This SNP resides between two consensus E-boxes as shown by the underlined sequences, and affects a PstI restriction site, marked with a box. B) RFLP analysis of ODC1 SNP. DNA was obtained from two cell types, and the region surrounding the ODC1 SNP site was sequenced. Colon-derived HT29 cells were found to be heterozygous GA, while HCT116 cells were found to be homozygous GG, at the ODC1 SNP locus. A 350 bp PCR product of this region was obtained from each cell type and subjected to digestion with PstI. Evidence of an A-allele was indicated by restriction products smaller than 350 bp.
Expression of specific E-box binding proteins, including the transcriptional activator c-MYC and several transcriptional repressors in HT29 and HCT116 cells (e.g. MAD1 and MAD4), was established by Western blotting (Fig. 4, A). Chromatin immunoprecipitation (CHIP) analysis of the region surrounding +316 of the ODC1 promoter was conducted, using antibodies directed against these proteins. As shown in Fig. 4, B, ODC1 promoter-specific PCR products were synthesized from HT29 DNA obtained after immunoprecipitation of chromatin with antibodies directed against c-MYC, MAD1 or MAD4. PCR products synthesized from HCT116 DNA after similar chromatin immunoprecipitation were substantially reduced compared to those synthesized from HT29 DNA. Quantification of these results indicated that c-MYC, MAD1, and MAD4 binding to the ODC1 SNP region was 4-14 times greater in HT29 cells, which contained one ODC1-A allele, compared to HCT116 cells, which contained only ODC1-G alleles (Supplemental Table).
Fig. 4.
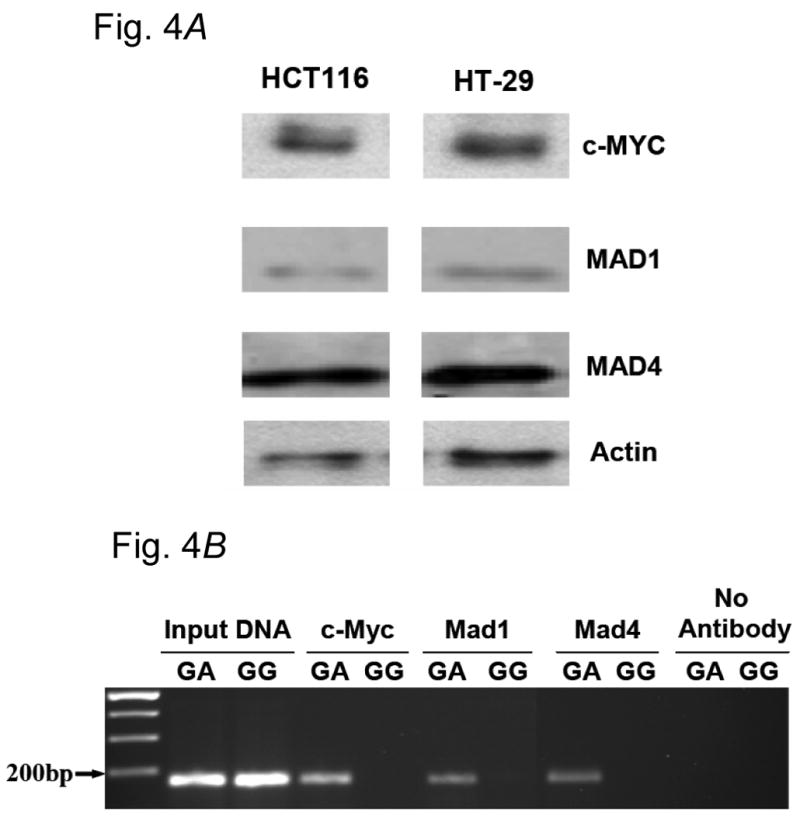
A) E-box protein expression in colon-derived cells. Expression of proteins to be evaluated for binding to the +316 ODC1 SNP was assessed by Western blot analysis. Extracts of both HT29 and HCT116 cells were evaluated for c-MYC, MAD1 and MAD4; β-actin was used as a loading control. B) Documentation of allele-specific transcription factor binding by chromatin immunoprecipitation (CHIP) analysis. CHIP analysis was conducted as described in Material and Methods. HT29 cells were a source of ODC1 A-alleles, as these cells are heterozygous GA at this site. HCT116 cells were used as a source of ODC1 G-alleles.
ODC1 allele-specific promoter activity
We tested the hypothesis that +316 ODC1 SNP influenced ODC1 expression in a manner dependant on the expression of E-box activators and repressors. Transient co-transfection of colon cancer-derived HT29 cells was accomplished with OD1C allele-specific promoter constructs in combination with vectors expressing either the transcriptional activator MYC or the repressor MAD1 (Fig. 5). The standard error bars shown reflect the variability in triplicate measurements within a single representative experiment, which has been replicated. The allele-specific promoter-reporters used in these experiments included all three E-boxes shown in Fig. 3, A. As shown in Fig. 5, A, MYC expression had the greatest stimulatory effect on promoters containing three consensus E-boxes and the ODC1-A allele (wt E-box1 +316 A, P = 0.0014). Deletion of the upstream E-box reduced promoter activity, but MYC expression continued to stimulate this activity (mut E-box1 +316 A, P = 0.0013). Substitution of a G for the A at the ODC1 +316 SNP position reduced the ability of c-MYC to stimulate promoter activity even with an intact 5′ flanking consensus E-box. Mutation of the 5′ flanking consensus E-box in combination with the ODC1-G allele further reduced promoter activity
Fig. 5.
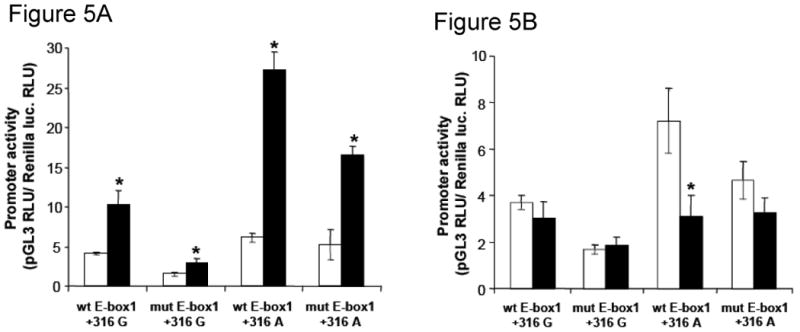
A) Effect of c-MYC expression on ODC1 allele-specific promoter activity in HT29 colon derived cells. Promoter activity was measured after transfection with ODC1 promoter reporter plasmids co-transfected with pcDNA 3.0 plasmid (□) or CMV-MYC expression vector (■). Promoter constructs differ by the presence of the first E-box element, located in −485 to −480 bp (“wt E-box1” for the wild-type sequence or “mut E-box1”, for a mutant sequence). The constructs differ also by the ODC1 +316 SNP (“+316 G” or “+316 A”). *, P ≤ 0.013 for each of the four comparisons relative to promoter activity with pcDNA 3.0 co-transfection. B) Effect of MAD1 expression on ODC1 allele-specific promoter activity in HT29 colon tumor derived cells. Promoter activity was measured after transfection with ODC1 promoter reporter plasmids co-transfected with pcDNA 3.1 plasmid (□) or with a pcDNA-MAD1 plasmid (■). Promoter constructs used were described in the legend for panel A of this figure. *, P = 0.027, statistical significance relative to promoter activity with pcDNA 3.1 co-transfection.
When MAD1, rather than MYC, was co-transfected with the ODC1 allele-specific promoter reporters (Fig. 5, B), the repressor was only able to reduce the activity of the ODC1 promoter which contained all three E-boxes and the wild-type +316 A-allele (P = 0.027). Deletion of the upstream E-box (mut E-box 1 +316A) significantly reduced the effect of MAD1 on ODC1 promoter activity. Substitution of G for A at the +316 position rendered promoters containing either two or three E-boxes unresponsive to MAD1 suppression.
Discussion
In our population-based analysis of colorectal cancer cases with more than eleven years follow-up duration, we observed that the +316 ODC1 SNP was associated with colorectal cancer specific survival (CRC-SS) among CRC cases, with a greater association observed among rectal cancer cases. A statistically significant increased risk of CRC-specific mortality was observed for ODC1 GA/AA cases among rectal cancer cases, after stratification for stage and adjustment for age, gender, ethnicity, family history of CRC, tumor site, histology, treatment with surgery, radiation therapy, and chemotherapy.
The experimental data presented here provide insights into potential biologic mechanisms underlying our clinical observations. In colon cancer epithelial cells, we have shown that the ODC1 +316 SNP is functionally significant, as evidenced by increased binding of E-box transcription factors to promoter elements containing A-, compared to G-, alleles. Both the activator c-MYC and the repressor MAD1 show greater effects on promoter activity in reporter elements containing A- versus G- alleles. These results suggest allele-specific regulation of ODC1 by E-box transcription factors. ODC protein enzyme activity is not apparently affected by the ODC1 +316 SNP genotype, which we believe influences ODC1 transcription.
In colon cells, we have shown that conditional expression of wild type APC, a gene expressed in normal colonic mucosa, suppresses MYC, and increases MAD1, expression (5). Further, we have reported that wild type APC can regulate ODC1 promoter activity in a manner dependent on the +316 SNP (7). Wild type APC is expressed in the apparently normal colonic mucosa of individuals not afflicted with FAP, while the majority of sporadic colon adenomas show evidence of mutated or deleted APC (27). MYC is expressed at low levels in normal intestinal mucosa but is increased in intestinal adenomas of APCMin/+ mice. Conditional knockout of intestinal epithelial MYC expression suppresses intestinal tumorigenesis in APCMin/+ mice (20). As described above, previous work by our group (7) and others (18) demonstrated a protective role for the ODC1 A-allele, especially in aspirin users, against recurrence of colon polyps in clinical prevention trials. However, in the population-based study presented here, the ODC1 A-allele was associated with poor survival. This apparent contradiction may be explained by the results shown here, which indicate that both E-box activators and repressors bind the ODC1 A-allele selectively. If the transformed epithelium begins to express E-box activators (such as c-MYC), then cancer progression may be more likely to occur in individuals with the ODC1 A-allele genotype. Our results for risk of colon cancer-specific mortality are consistent with those of others showing that risk of prostate cancer may be associated with the ODC1 A-allele among specific individuals as the result of gene environment interactions (19, 28). Such colon cancer progression could be due to enhanced polyamine synthesis, as has been demonstrated already for prostate cancer (15).
Our finding that a factor, such as the ODC1 SNP, may have both promoting and inhibiting effects on carcinogenesis is not unique. For example, transforming growth factor-beta (TGF-β) has diverse roles in carcinogenesis and cancer progression (29-31). TGF-β in untransformed cells inhibits cell proliferation and induces apoptosis. Yet, it is overexpressed in all human tumors and is associated with late cancer progression, specifically tumor invasion and metastasis. A single study by Matsubara et al. reporting ODC1 activity in human colorectal tumors demonstrated that high levels of ODC1 expression was significantly associated with improved survival (32). This suggests that, although ODC1 overexpression promotes the formation of human colorectal adenomas, it is possible that in established lesions, ODC1 overexpression causes enhanced proliferation and is associated with improved response to anti-proliferative treatments. However, that study did not include stratification by ODC1 genotype, so it is not known if these effects are independent of ODC1 genotype. Population differences between the Matsubara et al. study and ours could account for the differences. For example, the former study included few rectal cancers (5%), whereas our study cohort was comprised of a substantial proportion of rectal cancer cases (36%). This difference is not trivial, since the observed associations of the ODC1 SNP with CRC-SS were greater for rectal compared to colon cancer cases. The Matsubara et al. study was comprised of 63.5% females compared to our study which included 41% females. Furthermore, the distribution by race/ethnicity differs substantially between the two studies. Additionally, chance variation could explain why our findings differ from the prior analysis.
The observed associations of the ODC1 +316 SNP with CRC-specific mortality were greatest among rectal cancer cases. Among all CRC cases, particularly strong effects were observed for Caucasians. Similar to other reports, the ODC1 +316 SNP allele frequency differs considerably by ethnicity (28). When we limited the survival analysis to Caucasians only (i.e., the only ethnic group with adequate power for such analyses), the associations of the ODC1 +316 SNP were significant, and of greater magnitude than the estimates observed for the entire cohort.
The epidemiologic study shares limitations of other population-based analyses, including lack of data on comorbid conditions, performance status, and particular chemotherapeutic regimens utilized. Additionally, the tissue biopsy samples obtained from participants of the UC Irvine Gene-Environment Study of Familial Colorectal Cancer are paraffin-embedded specimens and therefore cannot be used for accurate assessment of tissue polyamine quantification by high performance liquid chromatography (HPLC). There is also the potential for selection bias, favoring a relatively healthy group of CRC survivors, since there was a median 18month delay from the time of CRC diagnosis until study enrollment. Other factors affecting polyamine metabolism that were not accounted for in the present study may explain our observations. For example, aspirin activates polyamine acetylation and export and works with the ODC1 A-allele to reduce cell and tissue polyamine contents (2, 7, 33). Our experimental findings are limited to the effects of the ODC1 +316 SNP in two colon cancer cell lines. Thus validation of our findings is warranted.
In summary, we have observed clinical consequences of the ODC1 +316 SNP on CRC-specific mortality among CRC cases. Additionally, we have further established the functional significance of the ODC1 +316 SNP in the c-MYC- and MAD1-dependent transcription of this gene in human colon cancer cells. Together, these experimental and epidemiologic findings suggest a role for the ODC1 SNP in progression of CRC distinct from its previously reported role in progression to colon adenomas. Provided that our results are validated in future research, these novel findings may contribute to our ability to assess risk of CRC progression and may direct patient-specific pharmacogenetic management, surveillance monitoring, and inform novel targeted approaches to secondary and tertiary CRC prevention.
Supplementary Material
Acknowledgments
We thank Tom O'Brien for the ODC1 allele specific promoter reporters.
Financial support: This work was supported by grants from the National Institutes of Health (USA), CA72008 (EWG), CA78134 (HAC), CA78285 (HAC), CA95060 (EWG), K23 CA133142 (Zell), and L30 CA130160 (Zell)
The collection of cancer incidence data used in this study was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885; the National Cancer Institute's Surveillance, Epidemiology and End Results Program under contract N01-PC-35136 awarded to the Northern California Cancer Center, contract N01-PC-35139 awarded to the University of Southern California, and contract N01-PC-54404 awarded to the Public Health Institute; and the Centers for Disease Control and Prevention's National Program of Cancer Registries, under agreement 1U58DP00807-01 awarded to the Public Health Institute. The ideas and opinions expressed herein are those of the author(s) and endorsement by the State of California, Department of Public Health the National Cancer Institute, and the Centers for Disease Control and Prevention or their Contractors and Subcontractors is not intended nor should be inferred.
Abbreviations
- ODC
ornithine decarboxylase
- SNP
single nucleotide polymorphism
Footnotes
Disclosure of Potential Conflict of Interest: Eugene W. Gerner has an ownership interest in Cancer Prevention Pharmaceuticals, LLC.
Statement of Translational Relevance: Excess polyamine formation has long been implicated in epithelial carcinogenesis, particularly colorectal carcinogenesis. Recently, a strategy involving inhibition of ornithine decarboxylase (ODC) activity (i.e., the rate-limiting enzyme of polyamine synthesis) has demonstrated remarkable efficacy in preventing recurrence of colorectal polyps in humans. Epidemiologic and experimental results from the present research demonstrate conditional regulation of polyamine homeostasis by genetic polymorphism in ODC1, and suggest a model in which the +316 ODC1 SNP may be protective for colon adenoma recurrence and detrimental for survival after diagnosis of non-metastatic colorectal cancer. This information may be useful for determining colon cancer prognosis. By identifying patients at increased risk for cancer progression/recurrence, early implementation of tertiary prevention management strategies could be instituted. Additionally, this research may help to identify high-risk but otherwise optimally-treated locoregional colorectal cancer patients that would benefit from tertiary cancer prevention clinical trials.
References
- 1.Wallace HM. The physiological role of the polyamines. Eur J Clin Invest. 2000;30:1–3. doi: 10.1046/j.1365-2362.2000.00585.x. [DOI] [PubMed] [Google Scholar]
- 2.Gerner EW, Meyskens FL. Polyamines and cancer: Old molecules, new understanding. Nature Reviews Cancer. 2004;4:781–92. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 3.Thomas T, Thomas TJ. Polyamine metabolism and cancer. JCell MolMed. 2003;7:113–26. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giardiello FM, Hamilton SR, Hylind LM, Yang VW, Tamez P, Casero RA. Ornithine decarboxylase and polyamines in familial adenomatous polyposis. Cancer Research. 1997;57:199–201. [PubMed] [Google Scholar]
- 5.Fultz KE, Gerner EW. APC-dependent regulation of ornithine decarboxylase in human colon tumor cells. MolCarcinog. 2002;34:10–8. doi: 10.1002/mc.10043. [DOI] [PubMed] [Google Scholar]
- 6.Erdman SH, Ignatenko NA, Powell MB, et al. APC-dependent changes in expression of genes influencing polyamine metabolism, and consequences for gastrointestinal carcinogenesis, in the Min mouse. Carcinogenesis. 1999;20:1709–13. doi: 10.1093/carcin/20.9.1709. [DOI] [PubMed] [Google Scholar]
- 7.Martinez ME, O'Brien TG, Fultz KE, et al. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7859–64. doi: 10.1073/pnas.1332465100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellofernandez C, Packham G, Cleveland JL. The Ornithine Decarboxylase Gene Is A Transcriptional Target of C-Myc. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7804–8. doi: 10.1073/pnas.90.16.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Childs AC, Mehta DJ, Gerner EW. Polyamine-dependent gene expression. Cell Mol Life Sci. 2003;60:1394–406. doi: 10.1007/s00018-003-2332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love RR, Carbone PP, Verma AK, et al. Randomized phase I chemoprevention dose-seeking study of alpha-difluoromethylornithine. J Natl Cancer Inst. 1993;85:732–7. doi: 10.1093/jnci/85.9.732. [DOI] [PubMed] [Google Scholar]
- 11.Gerner EW, Garewal HS, Emerson SS, Sampliner RE. Gastrointestinal Tissue Polyamine Contents of Patients with Barretts-Esophagus Treated with Alpha-Difluoromethylornithine. Cancer Epidemiology Biomarkers & Prevention. 1994;3:325–30. [PubMed] [Google Scholar]
- 12.Meyskens FL, Emerson SS, Pelot D, et al. Dose De-Escalation Chemoprevention Trial of Alpha-Difluoromethylornithine in Patients with Colon Polyps. Journal of the National Cancer Institute. 1994;86:1122–30. doi: 10.1093/jnci/86.15.1122. [DOI] [PubMed] [Google Scholar]
- 13.Meyskens FL, Gerner EW, Emerson S, et al. Effect of alpha-difluoromethylornithine on Rectal Mucosal levels of polyamines in a randomized, double-blinded trial for colon cancer prevention. Journal of the National Cancer Institute. 1998;90:1212–8. doi: 10.1093/jnci/90.16.1212. [DOI] [PubMed] [Google Scholar]
- 14.Simoneau AR, Gerner EW, Phung M, McLaren CE, Meyskens FL., Jr Alpha-difluoromethylornithine and polyamine levels in the human prostate: results of a phase IIa trial. J Natl Cancer Inst. 2001;93:57–9. doi: 10.1093/jnci/93.1.57. [DOI] [PubMed] [Google Scholar]
- 15.Simoneau AR, Gerner EW, Nagle R, et al. The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: results of a year-long phase IIb randomized placebo-controlled chemoprevention trial. Cancer Epidemiol Biomarkers Prev. 2008;17:292–9. doi: 10.1158/1055-9965.EPI-07-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyskens FL, McLaren CE, Pelot D, Fujikawa S, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prevention Research. 2008;1:32–8. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barry ELR, Baron JA, Bhat S, et al. Ornithine decarboxylase polymorphism modification of response to aspirin treatment for colorectal adenoma prevention. Journal of the National Cancer Institute. 2006;98:1494–500. doi: 10.1093/jnci/djj398. [DOI] [PubMed] [Google Scholar]
- 18.Hubner RA, Muir KR, Liu JF, Logan RF, Grainge MJ, Houlston RS. Ornithine decarboxylase G316A genotype is prognostic for colorectal adenoma recurrence and predicts efficacy of aspirin chemoprevention. Clin Cancer Res. 2008;14:2303–9. doi: 10.1158/1078-0432.CCR-07-4599. [DOI] [PubMed] [Google Scholar]
- 19.Visvanathan K, Helzlsouer KJ, Boorman DW, et al. Association among an ornithine decarboxylase polymorphism, androgen receptor gene (CAG) repeat length and prostate cancer risk. Journal of Urology. 2004;171:652–5. doi: 10.1097/01.ju.0000108384.74718.73. [DOI] [PubMed] [Google Scholar]
- 20.Ignatenko NA, Holubec H, Besselsen DG, et al. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol Ther. 2006;5:1658–64. doi: 10.4161/cbt.5.12.3376. [DOI] [PubMed] [Google Scholar]
- 21.Peel DJ, Ziogas A, Fox EA, et al. Characterization of hereditary nonpolyposis colorectal cancer families from a population-based series of cases. JNatlCancer Inst. 2000;92:1517–22. doi: 10.1093/jnci/92.18.1517. [DOI] [PubMed] [Google Scholar]
- 22.Zell JA, Ignatenko NA, Yerushalmi HF, et al. Risk and risk reduction involving arginine intake and meat consumption in colorectal tumorigenesis and survival. International Journal of Cancer. 2007;120:459–68. doi: 10.1002/ijc.22311. [DOI] [PubMed] [Google Scholar]
- 23.Zell JA, Honda J, Ziogas A, Anton-Culver H. Survival after Colorectal Cancer Diagnosis is Associated with Colorectal Cancer Family History. Cancer Epidemiol Biomarkers Prev. 2008;17:3134–40. doi: 10.1158/1055-9965.EPI-08-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le H, Ziogas A, Lipkin SM, Zell JA. Effects of socioeconomic status and treatment disparities in colorectal cancer survival. Cancer Epidemiol Biomarkers Prev. 2008;17:1950–62. doi: 10.1158/1055-9965.EPI-07-2774. [DOI] [PubMed] [Google Scholar]
- 25.Ziogas A, Anton-Culver H. Validation of family history data in cancer family registries. Am J Prev Med. 2003;24:190–8. doi: 10.1016/s0749-3797(02)00593-7. [DOI] [PubMed] [Google Scholar]
- 26.Guo YJ, Harris RB, Rosson D, Boorman D, O'Brien TG. Functional analysis of human ornithine decarboxylase alleles. Cancer Research. 2000;60:6314–7. [PubMed] [Google Scholar]
- 27.Iwamoto M, Ahnen DJ, Franklin WA, Maltzman TH. Expression of beta-catenin and full-length APC protein in normal and neoplastic colonic tissues. Carcinogenesis. 2000;21:1935–40. doi: 10.1093/carcin/21.11.1935. [DOI] [PubMed] [Google Scholar]
- 28.O'Brien TG, Guo Y, Visvanathan K, et al. Differences in ornithine decarboxylase and androgen receptor allele frequencies among ethnic groups. Molecular Carcinogenesis. 2004;41:120–3. doi: 10.1002/mc.20047. [DOI] [PubMed] [Google Scholar]
- 29.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nature Genetics. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 30.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochimica et Biophysica Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8621–3. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsubara N, Hietala OA, Gilmour SK, et al. Association between high levels of ornithine decarboxylase activity and favorable prognosis in human colorectal carcinoma. Clinical Cancer Research. 1995;1:665–71. [PubMed] [Google Scholar]
- 33.Babbar N, Gerner EW, Casero RA., Jr Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochem J. 2006;394:317–24. doi: 10.1042/BJ20051298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.