

Abstract
Purpose
Recent studies have implicated caveolin-1 (cav-1), in the regulation of transforming growth factor-β (TGF-β) downstream signaling. Given the crucial role of TGF-β in the pathogenesis of Systemic Sclerosis (SSc) we examined here whether cav-1 is also involved in the pathogenesis of tissue fibrosis in SSc. We analyzed the expression of cav-1 in affected SSc tissues, studied the effects of lack of expression of cav-1 in vitro and in vivo, and analyzed the effects of restoration of cav-1 function on the fibrotic phenotype of SSc fibroblasts in vitro.
Methods
Cav-1 expression in tissues was analyzed by immunofluorescence and confocal microscopy. Extent of tissue fibrosis in cav-1 knockout mice was assessed by histology/histochemistry and quantified by hydroxyproline assays. Cav-1 null and SSc fibroblast phenotype and protein production were analyzed by real time PCR, immunofluorescence, Western blots and Multiplexed ELISA. The effects of cav-1 function restoration in SSc fibroblasts in vitro were also examined employing a cell permeable recombinant cav-1 peptide.
Results
Cav-1 is markedly decreased in affected lungs and skin from SSc patients. Cav-1 knockout mice develop pulmonary and skin fibrosis. Cav-1 downregulation is maintained in cultured SSc fibroblasts and restoration of cav-1 function in vitro normalizes their phenotype and abrogates TGF-β stimulation through inhibition of Smad3 activation.
Conclusions
Caveolin-1 appears to participate in the pathogenesis of tissue fibrosis in SSc. Restoration of cav-1 function by treatment with a cell permeable peptide corresponding to the cav-1 scaffolding domain may be a novel therapeutic approach in SSc.
Introduction
Systemic sclerosis (SSc) is characterized by excessive deposition of collagen and other connective tissue macromolecules in skin and multiple internal organs, prominent and often severe alterations in the microvasculature, and humoral and cellular immunologic abnormalities (1). The excessive collagen deposition in SSc is due to overproduction of this protein by fibroblasts (2-4). Indeed, it is the persistent activation of the genes encoding various collagens in SSc fibroblasts that distinguishes controlled repair, such as that occurring during normal wound healing, from the uncontrolled fibrosis that is the hallmark of SSc (5). Numerous alterations in the expression of cytokines and growth factors with potent effects on fibroblast collagen synthesis, various endothelial cell functions, and T cell responses have been demonstrated in SSc (6-8). Transforming growth factor β (TGF-β) is a growth factor that plays a crucial role in tissue fibrosis (9-12) and it has been implicated in the pathogenesis of SSc (1,12-17). An important effect of TGF-β is the stimulation of the expression of genes encoding various collagens and other matrix proteins and the processing and tissue deposition of interstitial collagens (9-11). Although SSc fibroblasts display increased TGF-β signaling (18,19), the mechanisms responsible are not completely known. A recent study confirmed the importance of the increased TGF-β pathway activation in SSc pathogenesis. In this study the conditional, fibroblast-specific gene expression of a constitutively active TGF-β Receptor-1 (TGFβR-1) recapitulated the fibrotic process occurring in the skin, the vasculature and possibly the lung of SSc patients (20).
Caveolin 1 (cav-1) is the most important member of a family of membrane proteins that are the major coating proteins of caveolae. Caveolae are 50- to 100-nm flask-shaped invaginations which represent a morphologically identifiable subset of lipid rafts (21). The spatial organization of cell receptors in lipid rafts can modulate the subsequent transmission of the specific signal (22,23). Indeed, TGF-β 1 receptors are internalized both by cav-1 associated lipid rafts and by early endosome antigen 1 non-lipid raft 1 pathways. Non-lipid raft associated internalization increases TGF-β signaling, whereas, caveolin-associated internalization increases TGF-β receptor degradation, thus, effectively decreasing or abolishing TGF-β signaling (24,25). The increased TGF-β R-1 degradation is mediated by an interaction between the receptor and the scaffolding domain of cav-1; the decreased availability of the activated TGFβR-1 diminishes the phosphorylation of Smad2/3 and disrupts its interaction with Smad4 and its subsequent nuclear translocation (26).
The possible participation of cav-1 in the pathogenesis of fibrotic diseases was first suggested by Tourkina et al. (27) who demonstrated that cav-1 knock-down in vitro resulted in a 5-fold increase in COL1A2 gene expression by normal human lung fibroblasts, whereas, increased cav-1 expression caused a reduction in collagen production. A recent study (28) described a marked reduction of cav-1 expression in lung tissues and in lung fibroblasts from patients with idiopathic pulmonary fibrosis compared to cells from normal lungs. These authors also demonstrated that induction of cav-1 expression markedly ameliorated bleomycin-induced pulmonary fibrosis and suppressed TGF-β1-induced stimulation of extracellular matrix production in cultured fibroblasts.
Based on these observations, we raised the question of whether cav-1 is involved in the pathogenesis of tissue fibrosis in SSc. Here, we present evidence supporting the hypothesis that cav-1 plays a role in the pathogenesis of tissue fibrosis in SSc. Furthermore, our results suggest that an increase of cav-1 function employing a cell membrane permeable cav-1 scaffolding domain peptide may be a novel therapeutic approach to limit the progression of tissue fibrosis in SSc.
Methods
Patients
Full thickness skin biopsies were surgically obtained from three patients with SSc of recent onset (<18 months from the appearance of clinically detectable skin induration) and three normal controls with IRB approval. The SSc patients satisfied the criteria for the classification of SSc (29) and had the diffuse cutaneous clinical subset of the disease as defined by LeRoy et al. (30). Skin biopsies were obtained from the leading edge of the lesion on the forearms. The tissues remaining after diagnostic histopathology were used for fibroblast isolation and for immunohistology. Paraffin embedded sections from surgical open lung biopsies from two patients with SSc associated pulmonary fibrosis and from one autopsy from a patient with SSc pulmonary hypertension were obtained. The histopathological examination of skin showed the typical changes of SSc. The two lung biopsies displayed the typical features of non-specific interstitial pneumonitis. The lungs from the autopsy case showed typical histopathological findings of pulmonary arterial hypertension.
Animals and histopathology studies
Cav-1 deficient mice were generated as previously described are in the C57Bl/6 genetic background (31). Six same gender mice per group were sacrificed at 3 months of age. After hair removal with a commercial depilatory, full thickness skin samples were surgically excised from the interscapular region of the animals, stretched and pinned in histopathology cassettes and fixed in phosphate buffered formalin for 24 h. The lungs were removed and the left lung was processed by perfusion of the left pulmonary artery with paraformaldehyde to attain fixation of the parenchyma avoiding alveolar collapse or atelectasis as described previously (32). The entire right lung was hydrolyzed and used for hydroxyproline assays. Paraffin embedded skin and lungs were stained with hematoxylin/eosin and Masson's trichrome according to standard procedures.
Hydroxyproline assays
The collagen content in the lungs and skin of the same animals sacrificed for the histopathology studies was determined by hydroxyproline assays. Skin samples obtained from an area adjacent to that used for histopathology with a 4mm tissue punch, and the right lung were weighted and hydrolyzed in 6M HCl overnight at 110°C for determination of their hydroxyproline content, as described (33).
Confocal microscopy studies
Cav-1 protein was analyzed by immunofluorescence using a anti-cav-1 rabbit polyclonal antibody (Santa Cruz Biotechnology, Cambridge, MA). Rabbit IgG (Sigma, St Louis, MO) was used as isotype control. For Smad localization studies, cells were fixed and permeabilized as described (34). Anti Smad3 antibodies (Upstate, Temecula, CA) were used at a 1:200 dilution. Secondary antibodies used in the studies were affinity purified sheep FAB' anti rabbit IgG conjugated with different fluorochromes (Sigma). Paraffin embedded sections from skin and lung were processed as described (35). The primary antibody incubation step was performed in a dark humidified chamber over-night at 4 °C and then incubated with the secondary antibody (1:200). The unbound antibodies were removed from the section following each incubation with 3 changes of PBS for 2 min each. Tissue sections or fixed cells were then counterstained with DAPI and analyzed using a Zeiss LSM 510 META Confocal Laser Scanning Microscope System and software. The two channels were recorded simultaneously if no cross-talk could be detected. The breakthrough of the DAPI signal into the red and the green channels was recorded separately and subtracted from the DAPI blue channel. Each image was scanned 8 times to separate signal from noise. Panels were assembled using Photoshop software without any RGB modification.
Cell culture
Fibroblasts from normal and SSc dermis were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, antibiotics and glutamine (complete medium) until confluent. Fibroblasts from cav-1 knockout or WT mice were obtained by subculturing full thickness skin biopsies from age and sex matched animals. Both human and mouse fibroblasts were used at passages 4 to 10.
RNA isolation and quantitative real time PCR
Fibroblasts were harvested with trypsin/EDTA, washed in PBS and then processed for RNA extraction (RNAeasy kit; Quiagen, Valencia, CA) according to the manufacturer's protocol, including a genomic DNA digestion step. Total RNA (2 μg) was reverse-transcribed using Super-script-II reverse transcriptase (Invitrogen). Real-time PCR was performed using SYBR green Master mix chemistry employing a standard amplification protocol on a Bio-Rad MyiQ real-time PCR system (Bio-Rad, Hercules, CA). Reactions were conducted as described (36). To confirm the amplification specificity, the PCR products were subjected to melting temperature dissociation curve analysis. No amplification and no template controls were examined in parallel. The differences in the number of mRNA copies in each PCR reaction were corrected for endogenous control transcript levels; the control experiment levels were set as 100 and all the other values expressed as a multiple thereof.
Primer sequences
All primers were designed using Primer Express (Applied Biosystem) and validated on NCBI database for specificity (Table 1).
Table 1. Oligonucleotides employed for Sybr-green quantitative RT-PCR.
| HUMAN | ||
| β -ACT | TTGCCGACAGGATGCAGAA | GCCGATCCACACGGAGTACTT |
| COL1A1 | CCTCAAGGGCTCCAACGAG | TCAATCACTGTCTTGCCCCA |
| CAV-1 | CGACCCTAAACACCTCAACGA | TCCCTTCTGGTTCTGTCA |
| CTGF | GTGTGCACTGCCAAAGATGGT | TTGGAAGGACTCACCGCTG |
| α-SMA | TGTATGTGGCTATCCAGGCG | AGAGTCCAGCACGATGCCAG |
| MOUSE | ||
| 18S | GAAACGGCTACCACATCCAAGGAA | ACAATACAGGACTCTTTCGAGCCCTGTAA |
| col1a1 | CTGGCCCAGGCAGTCAGAT | GGGTTTGCTACAACATGGGCTA |
| mmp3 | GTGTGCACTGCCAAAGATGGT | TTGGAAGGACTCACCGCTG |
| α-SMA | GTGGCCCCTGAAGAGCATC | AATCTGGGTCATTTTCTCCCG |
ELISA
SearchLight® proteome arrays (Pierce Biotechnology, Woburn, MA) were performed to measure the levels of IL-6 and MMP-3 in supernatants from mouse fibroblasts and the levels of MMP-3 and PAI in tissue culture supernatants form human fibroblasts as described (36). Briefly, samples were diluted 1:5, 1:50 or 1:1000 before a one h incubation on the array plates, which had been pre-spotted with capture antibodies specific for each protein. Plates were decanted and washed three times before adding a mixture of biotinylated detection antibodies to each well. After incubating with detection antibodies for 30 min, plates were washed three times and incubated for 30 min with streptavidin-horseradish peroxidase. Plates were again washed before adding SuperSignal® Femto Chemiluminescent substrate. The plates were immediately imaged using the SearchLight imaging system and data were analyzed using ArrayVision software.
Western Blot analysis
Intracellular proteins in whole cell lysates were obtained by lysing the confluent cells in 100 mm dishes as described (36). Fifty μg of protein were used per each sample. Primary antibodies used in the different experiments were: cav-1 polyclonal rabbit antibody (Santa Cruz) at a 1:1000 dilution, collagen I polyclonal rabbit antibody (Rockland, Gilbertsville, PA) at a 1:3000 dilution in 5% milk TBST overnight at 4 °C; rabbit anti connective tissue growth factor (CTGF) polyclonal antibodies (ABCAM, Cambridge, MA), and rabbit anti-phospho-Smad 2/3 polyclonal antibodies (Chemicon International, Temecula, CA). Anti rabbit HRP conjugated antibodies at a 1:3000 dilution were used as secondary antibodies. To verify equal loading and transfer the membranes employed for intracellular protein blots were stripped using Pierce RESTORE™ stripping buffer and re-probed with rabbit anti human actin antibodies (Rockland) at a 1:1000 dilution in 5% milk TBST overnight at 4 °C.
Treatment of cells with cell permeable peptides
It has been demonstrated that cav-1 regulates TGF-β/Smad signaling through an interaction between the TGFR-1 receptor and a peptide corresponding to the scaffolding domain of cav-1 (DGIWKASFTTFTVTKYWFY) (26). Confluent cells between passages 4 and 9 were treated for 24 h with either penetratin peptide, a 16-amino acid cell permeable peptide corresponding to the homeodomain of the Drosophila transcription factor antennapedia (RQPKIWFPNRRKPWKK) or with a peptide consisting of the fusion of the antennapedia peptide and the peptide corresponding to the scaffolding domain of cav-1 (32). This fusion peptide, referred to as cav-1p, was used at 2.5 and 5 μM final concentrations. Fibroblasts were incubated in duplicate for each experiment for 24 h with or without 10 ng/ml TGF-β. Following 24 h incubation with or without TGF-β, one experimental plate was harvested and processed for RNA extraction for real time PCR and the other was processed to obtain cell lysates for Western blots. Tissue culture supernatants were collected to analyze collagen production by Western blot.
Statistics
The resulting data were subjected to two-tailed t-test for statistical significance. A p value of <0.05 was considered significant.
Results
Decreased levels of Cav-1 in affected SSc lung
Affected tissues from patients with SSc-related pulmonary fibrosis and PAH displayed reduced cav-1 immunofluorescence compared to normal lungs (Figure 1). The reduction in fluorescence staining for cav-1 was observed in the thickened alveolar septa typical of SSc alveolitis (Figure 1B). This reduction was even more apparent in the neointima of pulmonary arteries in lungs from SSc patients affected by PAH (Figure 1C) and in the interstitium of a lung section from a region where the normal lung architecture has been replaced by fibrotic tissue (Figure 1D). In contrast, intense cav-1 immunofluorescence was observed in histopathologically non-affected areas of SSc lung tissues from the same patients (Figure 1E). Quantitative analysis of the integrated density of fluorescence in three different fields per each type of lung involvement represented in panels A-E was performed employing Image J software. The ratio between cav-1 immunofluorescence staining (green fluorescence) and the number of cells in the sample as assessed by DAPI staining of their nuclei was 1.95 in normal lung and 2.1 in histopathologically non-affected SSc lung biopsies, compared to 0.79 in the alveolitis sections, 0.60 in the PAH sections, and 0.66 in the late fibrotic lesions sections. These data are shown in Figure 1F. Hence, the three types of SSc lung involvement analyzed showed, respectively, 59%, 69% and 66% decrease in relative cav-1 immunofluorescence levels compared to either normal lung tissue or to histopathologically non-affected areas of SSc lung.
Figure 1. Confocal microscopy imaging of cav-1 in SSc lung.
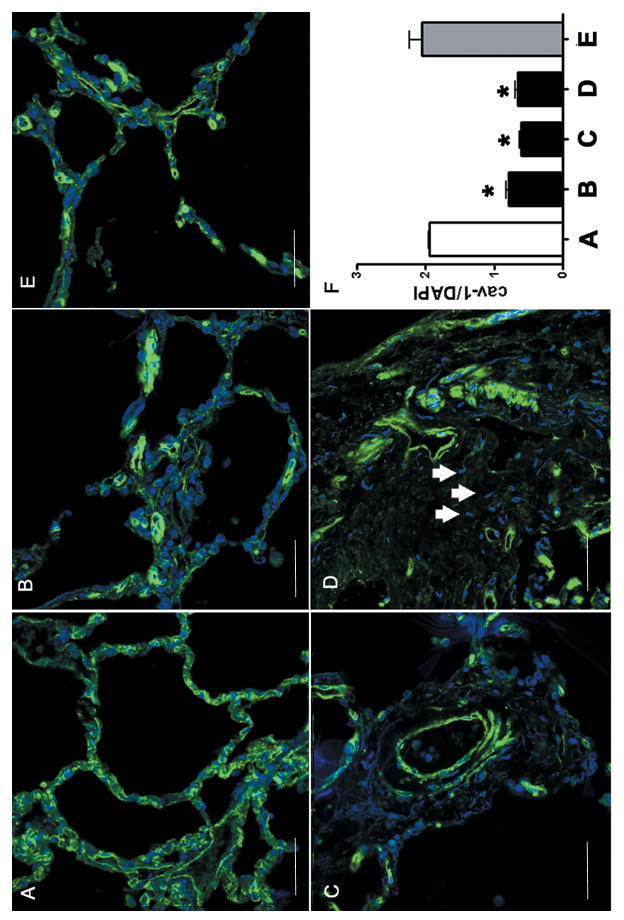
A: Normal lung immunostained for cav-1. Note the homogeneous green staining for cav-1 on the surface of each alveolar cell of the alveolar wall with an evident cell membrane pattern. The nuclei of all cells are stained with DAPI and appear blue. B: SSc lung immunostained for cav-1. Note that in contrast with A, there is non- homogeneous staining of cav-1 with markedly reduced levels of immunofluorescence in the involved alveolar septa as indicated by numerous cells (blue nuclei) without any detectable cav-1 staining in their surface. C: Pulmonary artery in the lung form a patient affected by SSc-related pulmonary artery hypertension. Note the reduced cav-1 immunofluorescent signal in most cells (blue nuclei) of the vessel wall. D: A fibrotic area of a lung from a SSc patient. Note the absence of cav-1 staining in many stromal cells (arrows). The images of each panel are representative of tissue samples from three different patients. E: Histopathologically non-affected area of lung from the same SSc patient shown in panels B and D. F: Bar graph of the ratio between cav-1 and DAPI integrated density of fluorescence (IDF) in panels A-E. (N=3; * = p<0.05 vs. A).
Cav-1 levels are decreased in SSc skin and in SSc dermal fibroblasts
The expression of cav-1 observed by confocal microscopy was markedly reduced in SSc affected skin (Figures 2A-D). We then compared by quantitative analysis of the integrated density of fluorescence the levels of cav-1 in three normal and three SSc skin biopsies. The results indicated that the cav-1/DAPI immunofluorescence ratio in the SSc affected skin was, in average, less than 50% than that observed in normal skin (Figure 2E). To confirm these results we studied in vitro cultured fibroblasts derived from SSc lesional skin because these cells recapitulate the most important molecular alterations responsible for the fibrotic phenotype observed in SSc skin in vivo. In these studies we compared the amount of cav-1 protein between SSc and normal dermal fibroblasts by immunofluorescence analysis (Figure 2F) and Western blot (Figure 2G). Both studies showed a dramatic decrease of cav-1 in SSc dermal fibroblasts compared to normal dermal fibroblasts.
Figure 2. A-D: Confocal microscopy imaging of cav-1 in SSc skin.
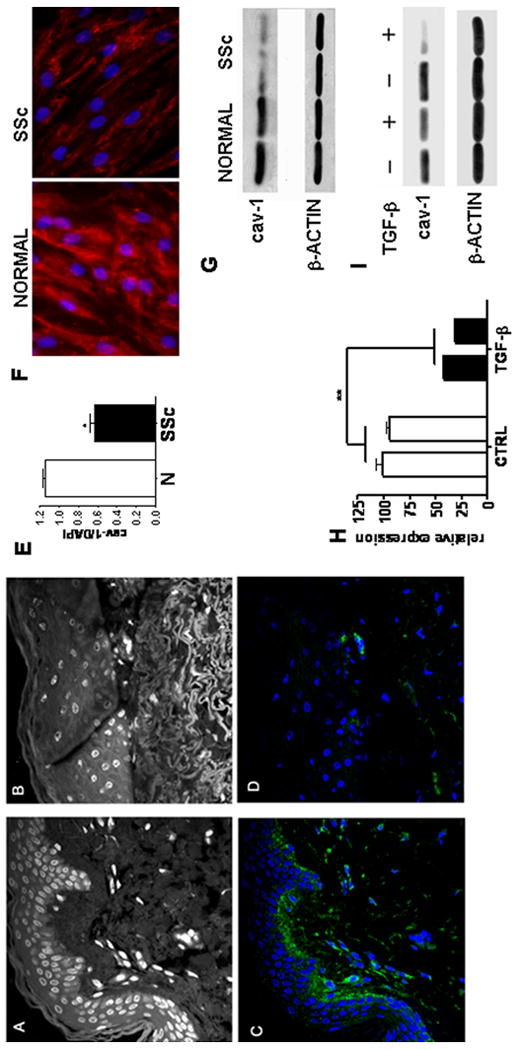
Non specific auto fluorescence of the DAPI channel is shown to display skin anatomy of (A) normal skin biopsy and (B) SSc skin biopsy. Note the tightly packed collagen fibrils in the lower dermis of the SSc biopsy. C: Confocal microscopy of the same section shown in A stained for cav-1 (green) and DAPI (Blue). Note the presence of intense cav-1 immunostaining surrounding almost every cell in the dermis. D: Same section shown in B stained for cav-1 (green) and DAPI (blue). Note that most cells (blue nuclei) do not show any detectable cav-1 staining (green) in their surface. The images shown in each panel are representative of tissue samples from three different patients. E: Quantitative analysis of fluorescence. Bar graph of the ratio between cav-1 and DAPI integrated density of fluorescence (IDF) in panels C and D. (*= p<0.05). F-G: Cav-1 expression in SSc fibroblasts in vitro. F: Immunofluorescence analysis of cav-1 expression (red) in confluent normal and SSc fibroblasts (both at passage 5). The figure shows a representative result from experiments performed with two normal and two SSc cell lines each in triplicate. Nuclei are stained with DAPI (Blue). G: Cell lysates from two SSc dermal fibroblast cell lines and two normal dermal fibroblast cell lines were analyzed by Western blot for cav-1. Antibodies directed against β-actin were used as control for equal protein loading. H-I: Cav-1 expression following TGF-β stimulation. H: mRNA levels for cav-1 were analyzed in two normal human fibroblast lines by real-time PCR following 24 h incubation with 10ng/ml TGF-β. Each bar represents the average ± SD of three different PCR. Cav-1 mRNA levels of the first line were arbitrarily set to 100 and all the other values calculated as multiples thereof. ** = p<0.01. I: Cell lysates from the same two human dermal fibroblast lines analyzed by Western blot for cav-1 following the same conditions described in panel H.
TGF-β downregulates cav-1 expression in vitro
To investigate whether the potent profibrotic growth factor, TGF-β may be involved in the observed reduction of cav-1 in affected SSc tissues we examined the effects of TGF-β on cav-1 expression in cultured human normal dermal fibroblasts. As shown in Figures 2H, and 2I, we observed a profound decrease in cav-1 mRNA and protein levels following 24 h incubation with 10 ng/ml human recombinant TGF-β.
Cav-1 knockout mice display pulmonary and dermal fibrosis and increased collagen content
To investigate whether the observed reduction of cav-1 in affected SSc lung and skin biopsies in vivo and in SSc cultured fibroblasts in vitro could play a role in the pathogenesis of tissue fibrosis we analyzed by histopathology the skin and lungs of mice lacking cav-1 expression. We observed that cav-1 null mice lungs had increased staining in the interstitium, indicating an increase of the collagen content in the lungs. The histopathologic analysis of paraffin embedded sections of skin biopsies from the same mice showed an increase in the thickness of the dermis which was accompanied by an increase in collagen staining (Figure 3A). Hydroxyproline assays on lung and skin from six 12-week old cav-1 knockout mice and six matched wild type (WT) littermates showed that the collagen content of cav-1 null mice lungs was 2.5 fold greater than that of WT animals (average cav-1 null = 0.86 μg/mg; WT = 0.35 μg/mg; p<0.001) and that skin of cav-1 null mice contained 2.4 fold more collagen that skin from WT mice (average cav-1 null = 6.8 μg/mg; WT = 2.8 μg/mg; p=0.002) (Figure 3B).
Figure 3. A-B: Cav-1 knockout mice display increased collagen content in their lungs and skin.
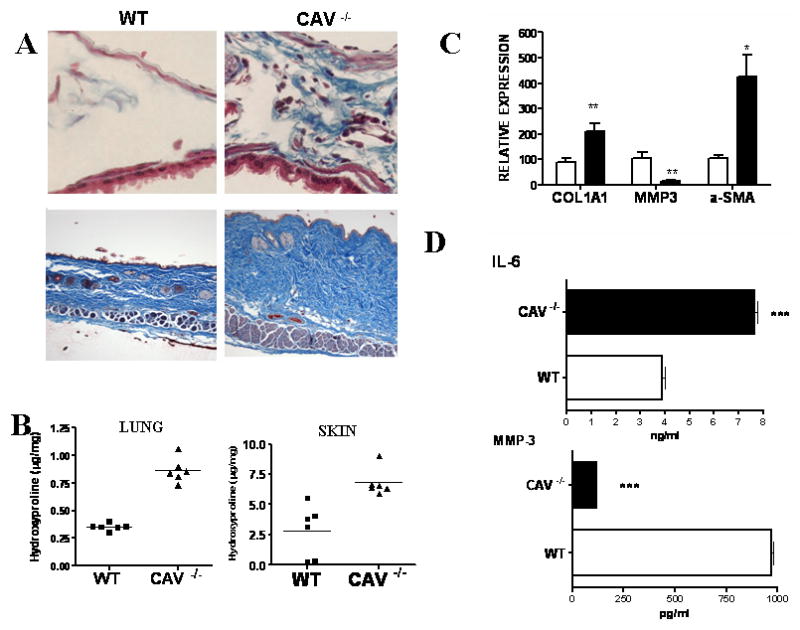
A. Masson's Trichrome staining of collagen in the lung interstitium (upper panel) and in the skin (lower panel) of wild type (WT) and cav-1-/- lung (CAV -/-) (upper panel: 400× lower panel: 200×). B: Hydroxyproline assays of lungs from 6 cav-1-/- and 6 WT 12-week-old mice. Each symbol (squares for WT and triangles for cav-1-/-) represents one animal. Results are shown as μg of hydroxyproline /mg of wet weight of the tissue. Paired t-test analysis showed p<0.001 for both. C-D: Cav-1-/- fibroblasts display a profibrotic phenotype. C: mRNA levels for COL1a1, MMP-3 and α-SMA were analyzed by quantitative real time PCR in WT (white bars) and cav-1-/- (black bars) fibroblasts. Each bar represents average ±SD of three different PCR. The mRNA levels of the WT line were arbitrarily set to 100 and all the other values calculated as multiples thereof. The transcript levels were corrected for 18S RNA levels. * = p < 0.05; ** = p < 0.005. D: Tissue culture supernatants from confluent WT or cav-1-/- fibroblasts were analyzed for IL-6 and MMP-3 by multiplex ELISA using Pierce Searchlight Multiplexed Proteome Arrays. Culture media from three separate WT and cav-1-/- cell lines were analyzed in duplicate; *** = p< 0.0001.
Cav-1 null fibroblasts display a profibrotic phenotype in vitro
To determine whether the skin fibroblasts obtained from cav-1 null mice recapitulate in vitro the molecular events associated with a profibrotic phenotype, we examined collagen, α-smooth muscle actin (α-SMA) and metalloproteinase (MMP) gene expression in cav-1 null fibroblasts by real time PCR. We observed a 2-fold increase in col1a1 transcript levels, a 3.5 fold increase in α-SMA expression and an 80% reduction in MMP-3 mRNA (Figure 3C). Furthermore, we also found an increase in IL-6 and a reduction in mmp-3 protein levels, as determined by ELISA of tissue culture supernatants (Figure 3D).
A cav-1 cell permeable peptide downregulates SSc fibroblast collagen production and suppresses the upregulation of TGF-β inducible genes in vitro
We next set out to examine whether cav-1p, a cell permeable cav-1 scaffolding domain peptide, previously shown to be able to inhibit TGF-β pathway activation and to mimic cav-1 function in a rat model of PAH (26,32), could exert an antifibrotic effect on SSc fibroblasts. We analyzed the effects of the cav-1p on SSc fibroblasts either under control conditions or following stimulation with TGF-β. We found that treatment of SSc dermal fibroblasts with cav-1p caused a reduction in their collagen expression both at the basal level and following TGF-β stimulation (Figure 4A). Furthermore, gene expression analysis of CTGF, a potent profibrotic TGF-β inducible gene, showed that treatment with cav-1p reduced its basal level of expression and abrogated the stimulation normally induced by TGF-β (Figure 4A). The results shown in Figure 4A were confirmed at the protein level for both proteins. Western blots for type I collagen indicated that incubation with 2.5 or 5 μM cav-1p caused a profound reduction in basal collagen production (Figure 4B). Furthermore, we also observed that 5 μM cav-1p abrogated TGF-β induced type I collagen stimulation (Figure 4B). These effects were not caused by cellular toxicity induced by the cav-1p as cell viability assays did not show any differences between treated and untreated cells at 18 and 24 h time points (Figure 4C). Western blots for CTGF of the cell lysates from SSc fibroblasts incubated with TGF-β for 24 or 48 h confirmed at the protein level the inhibitory effects of 24h cav-1p treatment observed at the mRNA level (Figure 4D). PCR analysis and immunofluorescence studies of SSc fibroblasts indicated that the basal expression of α-SMA transcripts and α-SMA containing stress fibers were reduced by treatment with 10 μM cav-1p and that treatment with the peptide inhibited the TGF-β induced increased levels of α-SMA mRNA and stress fibers (Figures 5A and 5B). The cav-1 peptide also reduced the effects of TGF-β on the production of plasminogen activator inhibitor 1 (PAI1) and increased the production of MMP-3 regardless of TGF-β stimulation, as demonstrated by ELISA of the tissue culture supernatants (Figure 5C).
Figure 4. Effects of cav-1p on SSc fibroblast profibrotic phenotype.
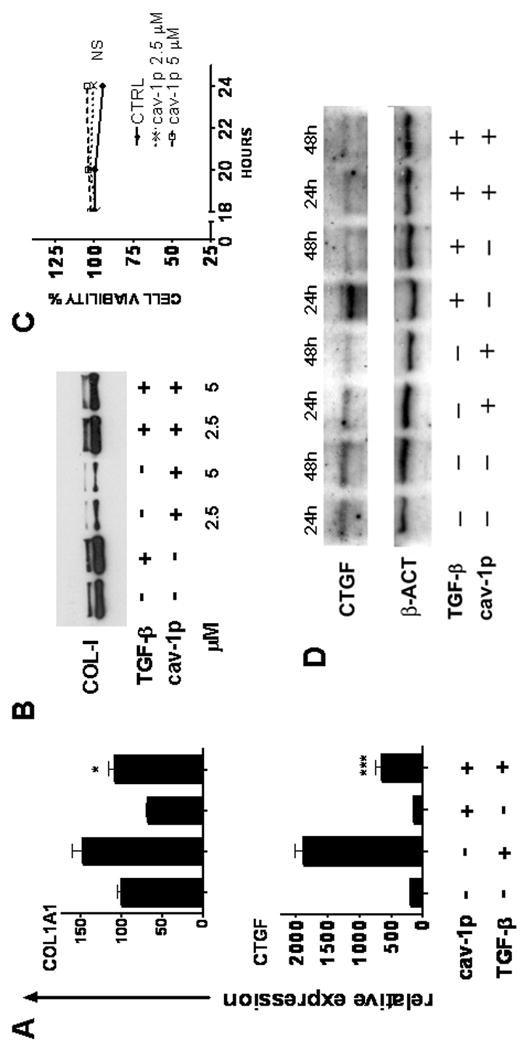
A: Real time PCR analysis of COL1A1 and CTGF mRNA from SSc dermal fibroblasts treated for 24 h with 10 ng/ml of human recombinant TGF-β in the presence of either 5 μM cav-1p (cav-1p) or 5 μM control peptide (penetratin-AP). mRNA levels for each gene under control conditions (penetratin without TGF-β) were set to 100. Other values were then calculated as a multiple thereof. Bars represent the average of three different PCR reactions. The results shown are representative of three different experiments. * = p<0.05; *** = p< 0.001. B: Western blot for collagen I in tissue culture supernatants from SSc fibroblasts treated for 24 h with cav-1p or penetratin alone (Control) with and without TGF-β. C: Cell viability of confluent dermal fibroblast monolayers assessed by WST-1 assay following 18, 20 or 24 h treatment with cav-1p, or penetratin alone (CTRL). NS: Not statistically significant. D: Western blot for CTGF in cell lysates from SSc fibroblasts following 24 h and 48 h TGF-β incubation in the presence of either control peptides (-) or cav-1p (+).
Figure 5.
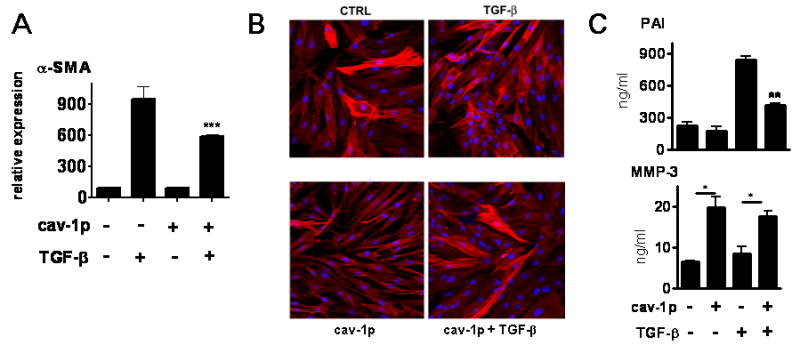
A: Real time PCR analysis of mRNA levels of α-SMA from SSc dermal fibroblasts treated for 24 h with 10 ng/ml of human recombinant TGF-β in the presence of either 5 μm cav-1p (cav-1p) or 5 μm control peptide (penetratin-AP). mRNA levels for each gene under control conditions (penetratin without TGF-β) were set to 100. Other values were then calculated as a multiple thereof. Bars represent average of three different PCR reactions. The results shown are representative of three different experiments. *** = p< 0.001. B: Immunofluorescence for α-SMA in SSc cells in the same experimental conditions described in A. Note the decreased number of fibroblasts expressing stress fibers in the presence of cav-1p both at basal level and following TGF-β stimulation. Original magnification 200×. C: ELISA of tissue culture supernatants for PAI and MMP-3. The results shown are the average of two different cell lines, each assayed in duplicate at three different dilutions. * =p<0.05; ** p= 0.01.
A cav-1 cell permeable peptide inhibits TGF-β pathway activation through suppression of Smad3 phosphorylation
To gain a mechanistic insight in the observed inhibitory effects of the cav-1p on TGF-β pathway activation, we analyzed Smad3 phosphorylation in SSc fibroblasts following TGF-β stimulation. We observed that cav-1p inhibited the TGF-β induced phosphorylation of Smad3 as early as 5 min following TGF-β stimulation. The inhibition was more pronounced at 15, 30 and 60 min following TGF-β (Figure 6A). To examine whether cav-1p also reduced the translocation of activated Smad3/4 complexes from the cytoplasm to the nucleus we analyzed by immunofluorescence the cellular localization of Smad following incubation of SSc dermal fibroblasts with TGF-β. We observed that the expected nuclear localization of Smad3 at 60 min following TGF-β stimulation was almost completely abrogated in the presence of 5 μM cav-1p (Figure 6B).
Figure 6.
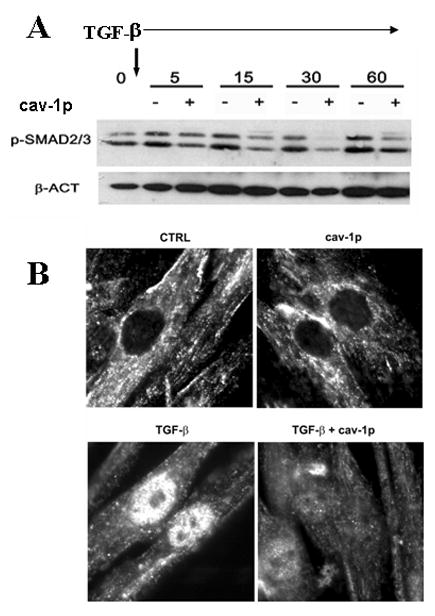
A: Cell lysates from SSc fibroblasts prepared 5, 15, 30 and 60 min following TGF-β stimulation (10ng/ml) in the presence or absence of cav-1 peptides were examined by Western blot using an antibody specific for phosphorylated Smad2/3. To document equal loading and transfer, the same membrane was stripped and reblotted with β-actin (β-ACT) antibodies B: Intracellular immunolocalization of total Smad3 in SSc fibroblasts following 60 min incubation with cav-1p or control peptide (CTRL) in the presence or not of TGF-β (10ng/ml). Note the marked nuclear accumulation of Smad3 following TGF-β treatment and the almost complete abrogation of this accumulation in cells treated with cav-1p. Original magnification 1000×.
Discussion
Since the discovery of the potent profibrotic and immunomodulatory activities of TGF-β, this growth factor has been recognized as one of the most important molecules in the pathogenesis of SSc and other fibroproliferative diseases (1,9-20). The classic pathway of TGF-β signal transduction involves the ligand-bound TβRII which recruits the TGF-βI receptor (TβRI) and then transphosphorylates specific serine and threonine residues in a short (30 amino acid) regulatory sequence known as the GS region (37,38). Signaling from the phosphorylated TβRI receptor to the nucleus then occurs through the Smad family of proteins (39). Smad2 or Smad3, two of the five receptor activated Smads (RSmads), bind to the activated TGF-β receptor complex and become phosphorylated, allowing the formation of a complex with the co-Smad, Smad4, a cytoplasmic protein which translocates the Smad complex through nuclear membrane pores into the nucleus. Once in the nucleus, Smad3/Smad4 complexes act as transcription factors, binding with the help of intranuclear transcriptional partners to specific DNA binding sites in the promoter regions of target genes and activating their expression. To further add to the complexity of the TGF-β activation and signaling cascades, it has been recognized that there are numerous other non-Smad mediated events involved in TGF-β functions and effects (40,41). Furthermore, it has also been recently demonstrated that other non-TGF-β dependent pathways may be necessary for the expression of the full SSc fibroblast phenotype. For example, Chen et al. (42) found that the overexpression of CTGF and α-SMA in SSc fibroblasts was independent of TGF-β signaling, as it could not be abolished by treatment of the cells with a specific inhibitor of ALK-5, the receptor involved in the canonical TGF-β activation cascade.
Another important mechanism of regulation of TGF-β activation involves the intracellular degradation of the activated TGF-β receptor complex. The important role of cav-1 in this regulation has recently been recognized (23-26) and it has been suggested that alterations in this process may participate in the initiation and progression of tissue fibrosis (27,28). Thus, the purpose of the present studies was to examine the role of cav-1 in the pathogenesis of tissue fibrosis in SSc. We demonstrate here that the levels of cav-1 in affected SSc lung and skin were substantially lower in comparison with normal tissues. Interestingly, our findings that cav-1 expression was not reduced in histopathologically non-affected areas of SSc lungs indicate that reduction of cav-1 expression may be a specific component of the actual pathologic process rather than the result of a generalized manifestation of SSc. Additionally, our observations that cav-1 is also low in lesional SSc skin indicates that regardless of the anatomic site, a reduction in cav-1 likely represents a marker of the pathologic process in SSc.
To explore the functional effects of decreased cav-1 expression in vivo, we employed cav-1 knockout mice (31) and performed a detailed histopathologic analysis of skin and lung tissues from these mice. In these studies we found that the lack of cav-1 expression was accompanied by skin and lung fibrosis supporting the notion that the observed decreased expression of cav-1 in SSc tissues may participate in the fibrotic process rather than being only an epiphenomenon. Furthermore, we found that cav-1 null fibroblasts displayed a profibrotic phenotype characterized by an increased expression of type I collagen, α-SMA and IL-6 and decreased production of MMP-3. In support of our observations are the results of a recent study (28) which showed that induced overexpression of cav-1 in vitro employing an adenoviral vector suppressed the expression of various genes encoding extracellular matrix molecules as well as TGF-β induced α-SMA upregulation in the MRC-5 fibroblast line.
To further examine the role of decreased cav-1 expression in the development of tissue fibrosis we explored the effects of increasing cav-1 function in SSc fibroblasts on the expression of genes encoding various key proteins participating in the process of pathologic fibrogenesis. The numerous and potentially serious difficulties encountered with the clinical application of virally-mediated gene therapy approaches (43-47) has stimulated the search for alternative methods of delivery of therapeutic proteins into cells. One of the most promising approaches is the utilization of novel cell permeable carriers capable of shuttling small peptides and proteins inside cells (48). One such carrier which has been utilized extensively is the small peptide, penetratin. Hence, we treated SSc fibroblasts in vitro with a cell permeable cav-1 scaffolding domain peptide (cav-1p), previously shown to be capable of increasing the intracellular bioavailability of cav-1 (26,32), and examined whether the reconstitution of intracellular cav-1 function resulted in a modulatory effect on the profibrotic phenotype of SSc fibroblasts. We found that the administration of cav-1p in vitro to SSc fibroblasts caused a significant reduction in the basal level of COL1A1 production, as well as a reduction in CTGF protein levels and α-SMA containing stress fibers. Of further interest, was the observation that the TGF-β induced upregulation of COL1A1, CTGF and α-SMA was strongly inhibited by treatment of SSc cells with cav-1p. Subsequently, we demonstrated that the inhibitory effects of cav-1p on TGF-β pathway activation were associated with the inhibition of Smad3 phosphorylation and of its nuclear translocation.
However, it should be noted that in the absence of TGF-β stimulation SSc fibroblasts did not show appreciable nuclear accumulation of Smad3. Although these results are in contrast with previously published data on Smad3 nuclear localization in SSc fibroblasts (19), they support the participation of non-Smad3-mediated pathways in the activation of the SSc fibroblast phenotype as suggested by Chen et al (42) and suggest that in the absence of exogenous TGF-β stimulation cav-1p could function in a TGF-β/Smad independent pathway.
In summary, the studies described here support strongly the hypothesis that decreased expression of cav-1 in SSc tissues plays a role in the development and progression of tissue fibrosis by mediating a sustained activation of the TGF-β pathway. Furthermore, we demonstrated that an in vitro intervention capable of increasing cav-1 function employing a cell permeable cav-1 peptide, reverted the profibrotic phenotype of SSc fibroblasts in vitro an that this effect was mediated by the suppression of the cascade of events induced by TGF-β receptor ligation as shown by the abrogation of Smad3 phosphorylation and nuclear translocation.
These results suggest that restoration of cav-1 function in vivo employing cell permeable cav-1 peptides may be effective to prevent the progression and limit the extension of tissue fibrosis in SSc. Future studies should be performed to fully assess the long-term safety and effectiveness of cav-1p administration in vivo as a novel therapeutic approach for SSc.
Acknowledgments
Supported by NIH grant RO1-AR 019616 to S.A.J. and grant R01 CA 080250 to M.P.L.
The excellent assistance of Susan V. Castro, Ph.D., in the preparation of the manuscript is gratefully acknowledged.
References
- 1.Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of Systemic Sclerosis. Ann Int Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 2.LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest. 1974;54:880–9. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jimenez SA, Bashey RI. Collagen synthesis by scleroderma fibroblasts in culture. Arthritis Rheum. 1977;20:902–3. doi: 10.1002/art.1780200323. [DOI] [PubMed] [Google Scholar]
- 4.Jimenez SA, Feldman G, Bashey RI, Bienkowski R, Rosenbloom J. Co-ordinate increase in the expression of type I and type III collagen genes in progressive systemic sclerosis fibroblasts. Biochem J. 1986;237:837–43. doi: 10.1042/bj2370837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jimenez SA, Hitraya E, Varga J. Pathogenesis of scleroderma. Collagen. Rheum Dis Clin North Am. 1996;22:647–74. doi: 10.1016/s0889-857x(05)70294-5. [DOI] [PubMed] [Google Scholar]
- 6.White B. Immunopathogenesis of systemic sclerosis. Rheum Dis Clin North Am. 1996;22:695–708. doi: 10.1016/s0889-857x(05)70296-9. [DOI] [PubMed] [Google Scholar]
- 7.Sakkas LI, Chikanza IC, Platsoucas CD. Mechanisms of Disease: the role of immune cells in the pathogenesis of systemic sclerosis. Nat Clin Pract Rheumatol. 2006;2:679–85. doi: 10.1038/ncprheum0346. [DOI] [PubMed] [Google Scholar]
- 8.Chizzolini C. Update on pathophysiology of scleroderma with special reference to immunoinflammatory events. Ann Med. 2007;39:42–53. doi: 10.1080/07853890601098152. [DOI] [PubMed] [Google Scholar]
- 9.Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–45. [PubMed] [Google Scholar]
- 10.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–71. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varga J, Jimenez SA. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem Biophys Res Commun. 1986;138:974–80. doi: 10.1016/s0006-291x(86)80591-5. [DOI] [PubMed] [Google Scholar]
- 12.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. 1987;247:597–604. doi: 10.1042/bj2470597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varga J, Abraham D. Systemic Sclerosis: A prototypic multi-system fibrotic disorder. J Clin Invest. 2007;117:557–67. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blobe GC, Schiemann EP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 15.Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 2005;26:587–95. doi: 10.1016/j.it.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Verrecchia F, Mauviel A, Farge D. Transforming growth factor-beta signaling through the Smad proteins: role in systemic sclerosis. Autoimmunity Rev. 2006;5:563–9. doi: 10.1016/j.autrev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Ihn H. Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis. J Dermatol Sci. 2008;49:103–13. doi: 10.1016/j.jdermsci.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Ihn H, Yamane K, Kubo M, Tamaki K. Blockade of endogenous transforming growth factor β signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor β receptors. Arthritis Rheum. 2001;44:474–80. doi: 10.1002/1529-0131(200102)44:2<474::AID-ANR67>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003;48:1964–78. doi: 10.1002/art.11157. [DOI] [PubMed] [Google Scholar]
- 20.Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56:334–44. doi: 10.1002/art.22328. [DOI] [PubMed] [Google Scholar]
- 21.Severs NJ. Caveolae: static inpocketings of the plasma membrane, dynamic vesicles or plain artifact? J Cell Sci. 1988;90:341–8. doi: 10.1242/jcs.90.3.341. [DOI] [PubMed] [Google Scholar]
- 22.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–31. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 23.Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol. 2006;23:49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang XL, Topley N, Ito T, Phillips A. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem. 2005;280:12239–45. doi: 10.1074/jbc.M413284200. [DOI] [PubMed] [Google Scholar]
- 25.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 26.Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276:6727–38. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- 27.Tourkina E, Gooz P, Pannu J, Bonner M, Scholz D, Hacker S, et al. Opposing effects of protein kinase C alpha and protein kinase C epsilon on collagen expression by human lung fibroblasts are mediated via MEK/ERK and caveolin-1 signaling. J Biol Chem. 2005;8:13879–87. doi: 10.1074/jbc.M412551200. [DOI] [PubMed] [Google Scholar]
- 28.Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AM. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subcommittee for scleroderma criteria of the American Rheumatism Association. Preliminary criteria for the classification of systemic sclerosis (Scleroderma) Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 30.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 31.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 32.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–20. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- 33.Stegemann H, Stalder KH. Determination of hydroxyproline. Clin Chim Acta. 1967;18:267–73. doi: 10.1016/0009-8981(67)90167-2. [DOI] [PubMed] [Google Scholar]
- 34.Sotgia F, Williams TM, Schubert W, Medina F, Minetti C, Pestell RG, et al. Caveolin-1 deficiency (-/-) conveys premalignant alterations in mammary epithelia, with abnormal lumen formation, growth factor independence, and cell invasiveness. Am J Pathol. 2006;168:292–309. doi: 10.2353/ajpath.2006.050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Galdo F, Maul GG, Jiménez SA, Artlett CM. Expression of allograft inflammatory factor 1 in tissues from patients with systemic sclerosis and in vitro differential expression of its isoforms in response to transforming growth factor beta. Arthritis Rheum. 2006;54:2616–25. doi: 10.1002/art.22010. [DOI] [PubMed] [Google Scholar]
- 36.Del Galdo F, Jiménez SA. T cells expressing allograft inflammatory factor 1 display increased chemotaxis and induce a profibrotic phenotype in normal fibroblasts in vitro. Arthritis Rheum. 2007;56:3478–88. doi: 10.1002/art.22877. [DOI] [PubMed] [Google Scholar]
- 37.Schmierer B, Hill CS. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 38.Massagué J, Gomis RR. The logic of TGFβ signaling. FEBS Lett. 2006;580:2811–20. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 39.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 40.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature. 2005;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 41.Rahimi RA, Leof EB. TGF-β signaling: a tale of two responses. J Cell Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Shi-wen X, Eastwood M, Black CM, Denton CP, Leask A, Abraham DJ. Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum. 2006;54:1309–16. doi: 10.1002/art.21725. [DOI] [PubMed] [Google Scholar]
- 43.Woods NB, Bottero V, Schmidt M, von Kalle C, Verma IM. Gene therapy: therapeutic gene causing lymphoma. Nature. 2006;440:1123. doi: 10.1038/4401123a. [DOI] [PubMed] [Google Scholar]
- 44.Couzin J, Kaiser J. Gene therapy. As Gelsinger case ends, gene therapy suffers another blow. Science. 2005;307:1028. doi: 10.1126/science.307.5712.1028b. [DOI] [PubMed] [Google Scholar]
- 45.Check E. Gene therapy put on hold as third child develops cancer. Nature. 2005;433:561. doi: 10.1038/433561a. [DOI] [PubMed] [Google Scholar]
- 46.Williams DA, Baum C. Gene therapy--new challenges ahead. Science. 2003;302:400–1. doi: 10.1126/science.1091258. [DOI] [PubMed] [Google Scholar]
- 47.Check E. Harmful potential of viral vectors fuels doubts over gene therapy. Nature. 2003;423:573–4. doi: 10.1038/423573a. [DOI] [PubMed] [Google Scholar]
- 48.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–49. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]