

Abstract
Chronic, daytime sleepiness is a major, disabling symptom for many patients with traumatic brain injury (TBI), but thus far, its etiology is not well understood. Extensive loss of the hypothalamic neurons that produce the wake-promoting neuropeptide hypocretin (orexin) causes the severe sleepiness of narcolepsy, and partial loss of these cells may contribute to the sleepiness of Parkinson’s disease and other disorders. We have found that the number of hypocretin neurons is significantly reduced in patients with severe TBI. This observation highlights the often overlooked hypothalamic injury in TBI and provides new insights into the causes of chronic sleepiness in patients with TBI.
Introduction
Approximately 1.4 million people sustain traumatic brain injury (TBI) each year in the United States.1 Coma and obtundation are common during the acute period, but even after 6 months recovery, 43% of patients have symptoms of sleep disorders.2,3 In a prospective study of 65 TBI patients, 1 in 4 patients reported excessive daytime sleepiness (EDS) irrespective of the location or severity of the head trauma.2 Another study of 87 patients revealed similar results, with sleepiness in 25% after TBI.3
Though chronic sleepiness is common after head trauma, the cause remains unclear. Injury to ascending arousal pathways from the brainstem may contribute, but recent findings suggest a key role for the neuropeptides hypocretin-1 and -2 (also known as orexin-A and -B).2,4,5 Hypocretins are made by a small cluster of neurons in the lateral hypothalamus that play an essential role in promoting wakefulness. Selective loss of the hypocretin-producing neurons causes sleepiness and cataplexy in over 90% of patients with narcolepsy,5–9 and identical symptoms occur in mice genetically lacking hypocretins.10 Injury to the hypothalamus is common in patients with TBI,11 but its clinical impact remains unclear. We recently found that TBI patients have a marked reduction in CSF hypocretin-1 levels in the first days after injury.4 Even after 6 months recovery, many TBI patients with EDS have persistently low hypocretin-1 levels.2 Thus, we hypothesized that posttraumatic sleepiness is caused, at least in part, by injury to the wake-promoting hypocretin neurons.
Methods
Subjects
We collected brains at autopsy from 4 patients who died 7 to 42 (mean 19) days after severe TBI (Supplemental Table). Causes of death were multi-organ failure (n=2), elevated intracranial pressure (n=1), and brain death (n=1). We obtained 4 additional brains from control subjects with no clinical evidence of TBI or other neurological disorders. Those subjects died from ruptured aortic aneurysm (n=1), liver failure (n=1), drowning (n=1), and myocardial infarction (n=1). First-degree relatives gave written informed consent for autopsy and tissue research. This study was approved by the human subjects committees of Kantonale Ethikkommission Zurich and Beth Israel Deaconess Medical Center.
Tissue Processing
We fixed whole hypothalami in 10% buffered formalin and stored them at 4° C. After the tissue equilibrated for 2 to 3 days in phosphate-buffered saline (PBS) with 20% sucrose, 10% formalin, and 0.1% diethyl pyrocarbonate (DEPC) (pH 7.4), we coronally sectioned the right and left halves of the hypothalamus at 40 µm in a 1:24 series. We stored sections at −20 °C in a cryoprotectant solution.9
Immunohistochemistry
We immunostained sections for hypocretin-1 (orexin A) as described previously.9 Briefly, after treatment with 3% hydrogen peroxide and blocking in 3% normal horse serum, we incubated sections overnight in goat anti-hypocretin-1 antiserum (1:5,000; Santa Cruz Biotechnology) and then in biotinylated donkey anti-goat secondary antiserum at 1:500. We then exposed the sections to avidin-biotin complex and diaminobenzidine to produce light brown labelling of hypocretin-containing neurons. The specificity of the antiserum was tested and confirmed in hypothalamic tissue from hypocretin-knockout mice.12
To test whether other neurons in the lateral hypothalamus are lost with TBI, we immunostained adjacent sections for melanin concentrating hormone (MCH; rabbit primary antiserum, 1:10,000; a kind gift from Elefteria Maratos-Flier).
Finally, to test whether gliosis accompanies hypocretin neuron loss, we double-labelled sections for hypocretin-1 (1:5000) and glial fibrillary acidic protein (GFAP), a marker of astrocytes. Hypocretin was labelled using goat anti-hypocretin-1 antiserum (1:5,000), biotinylated donkey-anti goat IgG (Jackson Laboratories), and Alexa 488-conjugated streptavidin (Invitrogen). GFAP was labelled using rabbit anti-GFAP primary antiserum (1:2000; Millipore) and Cy3-conjugated donkey-anti rabbit IgG secondary antiserum (Jackson ImmunoResearch Laboratories).
Cell counts
After immunostaining, we examined sections at 200x with a Zeiss Axiophot microscope. We digitally superimposed a counting grid on every section and counted all hypocretin- and MCH-immunoreactive neurons across a 1:24 series of the entire hypothalamus. To prevent miscounting, we used a fine-scale counting grid that encompassed no more than 10 neurons per grid sector. Only clearly-immunolabeled neurons with neuronal morphology, outgoing axons and dendrites, and a nuclear void were counted. To estimate accurately the numbers of hypocretin and MCH neurons in each section, we performed Abercrombie corrections based on the size of the cell nuclei in each subject.13 We then calculated the total numbers of hypocretin and MCH neurons by multiplying the cell counts per section by the section sampling frequency (1:24). We also counted GFAP-immunolabeled astrocytes in 30 randomly selected 400 by 400 µm sites within the lateral and posterior hypothalamus. Cell counts were compared between groups using Student’s t-tests.
Results
In this pilot study, the TBI patients were younger (51 ± 19 years) than the controls (69 ± 18 years), and all subjects were male except for one woman in the control group. Postmortem delays were similar in both groups (TBI: 12±3 hours, controls: 13±3 hours) (Supplemental Table).
The number of hypocretin neurons in TBI subjects was 27% less than in controls (23,655±5,035 vs. 32,318±3,060 neurons, p = 0.001) (Figure 1). Among the TBI patients, the number of hypocretin neurons differed moderately between the left and right sides of the hypothalamus and between individuals, reflecting the asymmetry and heterogeneity of TBI. Many hypocretin neurons in TBI patients were small and pyknotic, and the density of hypocretin fibers appeared reduced. Though TBI brains contained fewer hypocretin neurons, the neurons were distributed in a normal pattern, thus demonstrating the reliability and completeness of the cell counts (Figure 2). In all subjects, the complete hypocretin neuron field was examined (Supplemental Figure A). In controls, the total number of hypocretin neurons and their distribution across the lateral and posterior hypothalamus were similar to prior reports.7,8,14,15
Figure 1.

Overview of hypocretin and MCH cell counts. Patients with traumatic brain injury (TBI) have fewer hypocretin neurons. On average, TBI patients have a total of 23,655 hypocretin neurons, whereas controls have 32,318 neurons (p = 0.001). The numbers of hypocretin neurons on the left and right sides of the hypothalamus differ more in TBI patients, probably reflecting asymmetry of the trauma. TBI patients also have fewer MCH neurons than controls (39,540 in total vs. 57,176, p = 0.06).
Figure 2.

TBI patients and controls have similar caudal-rostral distributions of hypocretin neurons. The graphs show the average number of hypocretin neurons per section (mean values across all cases). Overall, TBI patients have fewer neurons, especially in the center of the hypocretin field. Level a: fornix touches the dorsal edge of the mammillary bodies; level b: fornix next to the paraventricular nucleus.
Hypothalami from TBI patients also contained intense perivascular hypocretin immunoreactivity (Figure 3). This amorphous staining was present around small vessels in the posterior and lateral hypothalamus but not elsewhere. The perivascular staining may represent phagocytized hypocretin peptide following damage to the hypocretin neurons.
Figure 3.
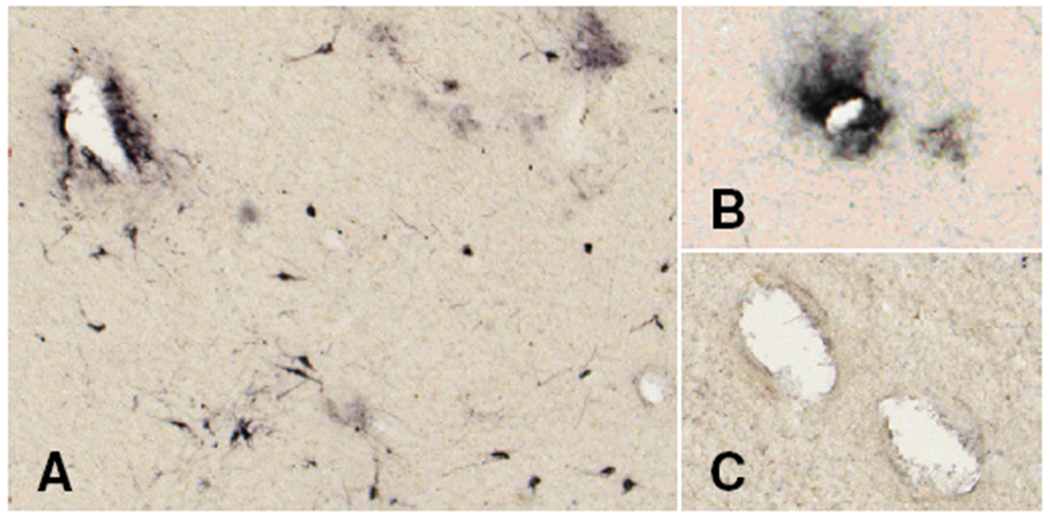
TBI patients have dense perivascular hypocretin immunoreactivity within the hypothalamic hypocretin field (A, B), but not in adjacent areas (C).
TBI patients also had 31% fewer MCH neurons than controls (39,540±10,900 vs. 57,176±24,016), but this reduction was not quite statistically significant (p = 0.06), probably because of variable numbers of MCH neurons. If one excludes the female control subject with surprisingly low numbers of MCH neurons (control subject 2), then the TBI subjects had 29% fewer hypocretin neurons than controls (33,342±2,785; p=0.002) and 40% fewer MCH neurons (66,434±18,730; p=0.003).
The density of astrocytes as assessed by GFAP staining was significantly higher (21±12 vs. 7±2 per mm2, p<0.001) in TBI patients than in controls (Supplemental Figure B). The pattern of gliosis differed across the TBI patients. Double immunofluorescence staining showed that patients with more severe loss of hypocretin neurons had more GFAP-labeled astrocytes (r=−0.46, p=0.01).
Discussion
In this pilot study, we found that patients with severe TBI have a significant loss of hypocretin-producing neurons. This injury was not limited to the hypocretin neurons as there was a partial loss of neurons producing MCH and patchy gliosis throughout the hypothalamus. These findings demonstrate that TBI can injure the hypothalamus and suggest that reduced hypocretin signaling may contribute to the persistent sleepiness often seen in TBI patients.
Descriptions of injury in TBI often focus on the midbrain and subthalamus,16 but hypothalamic injury is common. One early study showed damage to the hypothalamus in 42% of deceased TBI patients.11 Other reports describe pituitary and hypothalamic dysfunction,17,18 though to our knowledge, ours is the first to describe injury to specific types of hypothalamic neurons. In our patients, the loss of hypocretin neurons, coupled with gliosis and intense perivascular hypocretin-immunoreactive debris, strongly suggests that severe TBI can directly or indirectly kill hypocretin neurons. Perhaps, the hypocretin neurons are injured by the same shearing forces that often damage the midbrain, but vasospasm, or elevated intracranial pressure leading to compression or reduced perfusion of the hypothalamus may also contribute.11,17,18
Chronic excessive daytime sleepiness is a major disabling symptom for many TBI patients, but so far, its etiology is not well understood.2 We believe that partial loss of hypocretin neurons may contribute to chronic sleepiness after TBI. Clearly, extensive loss of hypocretin neurons is sufficient to cause the sleepiness of narcolepsy with cataplexy,5–9 and some hypothesize that a partial loss of these cells causes the generally milder sleepiness of narcolepsy without cataplexy and Parkinson’s disease.14,15,19 This assumption is supported by our recent finding of reduced CSF hypocretin-1 levels in sleepy TBI patients who survived their injury.2 Although the association between lumbar CSF hypocretin levels and the number of intact hypocretin neurons is still poorly understood, we hypothesize that reduced CSF levels of hypocretin in TBI patients may reflect partial loss of the hypocretin neurons and perhaps some reduction in the activity of the surviving cells. Still, it is likely that reduced hypocretin signalling is not the only factor to cause sleepiness after TBI. About one-third of TBI patients have brainstem injuries, often near the midbrain tegmentum, that may disrupt ascending monoaminergic or cholinergic wake-promoting pathways.20 In addition, some TBI patients have hypersomnolence, with excessively large amounts of sleep each day. Hypersomnolence is common with hypothalamic and rostral brainstem injuries, but is uncommon with selective loss of the hypocretin neurons.21 Loss of the MCH neurons may also contribute to the pathophysiology of posttraumatic sleep-wake disturbances as these cells are hypothesized to regulate REM sleep.22,23 Overall, it seems likely that the persistent sleepiness in some TBI patients is due to a combination of reduced hypocretin signaling and injury to other sleep-wake regulating systems.
This study has some limitations that should be addressed in future research. We examined a small number of brains, and the control subjects were not fully matched. However, our TBI patients had significantly fewer hypocretin neurons even though they were younger than the controls. In addition, we did not examine other wake-promoting systems, and the extent of injury to other arousal pathways should be further investigated in larger human and animal studies. Last, we studied patients with fatal TBI, which probably introduced some selection bias. It is possible that non-fatal TBI is associated with a less severe loss of hypocretin neurons.
TBI often results in chronic sleepiness, and the observation that wake-promoting hypocretin neurons are lost with TBI provides new insights into the underlying pathophysiology that may lead to novel treatment options. For example, stimulants such as modafinil that are helpful in narcolepsy and other hypocretin-deficient disorders should be studied systematically in affected TBI patients.24 Even better, hypocretin agonists could become an elegant and tailored treatment option not only for sleepy individuals with narcolepsy but also for those with TBI.
Supplementary Material
Anatomical distributions of hypocretin neurons in all TBI patients and controls. Each graph displays the total number of hypocretin neurons (y axis) per section (x axis). Each panel shows cell counts arranged from caudal to rostral.
Photomicrographs depicting the relationship between gliosis (GFAP staining of astrocytes) and hypocretin neurons. (A) Diaminobenzidine staining of a typical astrocyte in a control brain. Astrocyte density is much higher in the lateral hypothalamus of a TBI brain (B1) than in the same region of a control patient (C1). Double immunofluorescence staining shows that along with the reduction in the number of hypocretin neurons (green), there is an increase in red GFAP-labelled astrocytes (arrow) and fibers in TBI (B2) compared to control subjects (C2). Bar: 30 µm.
Clinical characteristics and cell counts of traumatic brain injury (TBI) patients and controls (Co). Severity of TBI was assessed clinically using the Glasgow Coma Scale (GCS) and by CT using the Marshall criteria.2 All patients were comatose from the time of injury, but no patient suffered a cardiac or respiratory arrest at the time of injury. None had facial or basilar skull fractures. TBI patient 2 had a minor subarachnoid hemorrhage.
Acknowledgements
This study was supported by the Foerderungskredit of the University of Zurich, Switzerland and NIH grant MH062589.
References
- 1.Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544–548. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Baumann CR, Werth E, Stocker R, et al. Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain. 2007;130:1873–1883. doi: 10.1093/brain/awm109. [DOI] [PubMed] [Google Scholar]
- 3.Castriotta RJ, Wilde MC, Lai JM, et al. Prevalence and consequences of sleep disorders in traumatic brain injury. J Clin Sleep Med. 2007;3:349–356. [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann CR, Stocker R, Imhof HG, et al. Hypocretin-1 (orexin A) deficiency in acute traumatic brain injury. Neurology. 2005;65:147–149. doi: 10.1212/01.wnl.0000167605.02541.f2. [DOI] [PubMed] [Google Scholar]
- 5.España RA, Scammell TE. Sleep neurobiology for the clinician. Sleep. 2004;27:811–820. [PubMed] [Google Scholar]
- 6.Adamantidis AR, Zhang F, Aravanis AM, et al. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 8.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crocker A, España RA, Papadopoulou M, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–1188. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 11.Crompton MR. Hypothalamic lesions following closed head injury. Brain. 1971;94:165–172. doi: 10.1093/brain/94.1.165. [DOI] [PubMed] [Google Scholar]
- 12.Baumann CR, Clark EL, Pedersen NP, Hecht JL, Scammell TE. Do enteric neurons make hypocretin? Regul Pept. 2008;147:1–3. doi: 10.1016/j.regpep.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guillery RW. On counting and counting errors. J Comp Neurol. 2002;447:1–7. doi: 10.1002/cne.10221. [DOI] [PubMed] [Google Scholar]
- 14.Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson's disease. Brain. 2007;130:1577–1585. doi: 10.1093/brain/awm090. [DOI] [PubMed] [Google Scholar]
- 15.Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain. 2007;130:1586–1595. doi: 10.1093/brain/awm097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ropper AH, Miller DC. Acute traumatic midbrain hemorrhage. Ann Neurol. 1985;18:80–86. doi: 10.1002/ana.410180114. [DOI] [PubMed] [Google Scholar]
- 17.Karavitaki N, Wass J, Henderson Slater JD, Wade D. A case of post-traumatic isolated ACTH deficiency with spontaneous recovery 9 months after the event. J Neurol Neurosurg Psychiatry. 2006;77:276–277. doi: 10.1136/jnnp.2005.070482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, et al. Hypopituitarism. Lancet. 2007;369:1461–1470. doi: 10.1016/S0140-6736(07)60673-4. [DOI] [PubMed] [Google Scholar]
- 19.Scammell TE. The frustrating and mostly fruitless search for an autoimmune cause of narcolepsy. Sleep. 2006;29:601–602. doi: 10.1093/sleep/29.5.601. [DOI] [PubMed] [Google Scholar]
- 20.Crompton MR. Brainstem lesions due to closed head injury. Lancet. 1971;1:669–673. doi: 10.1016/s0140-6736(71)92680-8. [DOI] [PubMed] [Google Scholar]
- 21.Scammell TE. Secondary Narcolepsy. In: Culebras A, editor. Sleep Disorders and Neurologic Diseases. 2nd edition. New York: Informa Healthcare; 2007. pp. 117–134. [Google Scholar]
- 22.Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009;106:2418–2422. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamantidis A, de Lecea L. Physiological arousal: a role for hypothalamic systems. Cell Mol Life Sci. 2008;65:1475–1488. doi: 10.1007/s00018-008-7521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scammell TE. Modafinil: Mechanisms of Action. In: Bassetti CL, Billiard M, Mignot E, editors. Narcolepsy and Hypersomnia. New York: Informa Healthcare; 2007. pp. 547–559. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Anatomical distributions of hypocretin neurons in all TBI patients and controls. Each graph displays the total number of hypocretin neurons (y axis) per section (x axis). Each panel shows cell counts arranged from caudal to rostral.
Photomicrographs depicting the relationship between gliosis (GFAP staining of astrocytes) and hypocretin neurons. (A) Diaminobenzidine staining of a typical astrocyte in a control brain. Astrocyte density is much higher in the lateral hypothalamus of a TBI brain (B1) than in the same region of a control patient (C1). Double immunofluorescence staining shows that along with the reduction in the number of hypocretin neurons (green), there is an increase in red GFAP-labelled astrocytes (arrow) and fibers in TBI (B2) compared to control subjects (C2). Bar: 30 µm.
Clinical characteristics and cell counts of traumatic brain injury (TBI) patients and controls (Co). Severity of TBI was assessed clinically using the Glasgow Coma Scale (GCS) and by CT using the Marshall criteria.2 All patients were comatose from the time of injury, but no patient suffered a cardiac or respiratory arrest at the time of injury. None had facial or basilar skull fractures. TBI patient 2 had a minor subarachnoid hemorrhage.