

Abstract
Signaling from the BCR and B cell activating factor receptor (BAFF-R or BR3) differentially regulates apoptosis within early transitional (T1) and late transitional (T2; CD21int-T2) B cells during selection processes to generate mature B lymphocytes. However, molecular mechanisms underlying the differential sensitivity of transitional B cells to apoptosis remain unclear. In this study, we demonstrate that BCR signaling induced more long-term c-Rel activation in T2 and mature than in T1 B cells leading to increased expression of anti-apoptotic genes as well as prosurvival BAFF-R and its downstream substrate p100 (NF-κB2). Sustained c-Rel activation required de novo c-Rel gene transcription and translation via Btk-dependent mechanisms. Like T1 cells, mature B cells from Btk- and c-Rel-deficient mice also failed to activate these genes. These findings suggest that the gain of survival potential within transitional B cells is dependent on the ability to produce a long-term c-Rel response, which plays a critical role in T2 B cell survival and differentiation in vivo by inducing anti-apoptotic genes, BAFF-R and NF-κB2, an essential component for BAFF-R survival signaling. Thus, acquisition of resistance to apoptosis during transitional B cell maturation is achieved by integration of BCR and BAFF-R signals.
Development of B lymphocytes in the bone marrow yields surface IgM+ immature B cells that immigrate to the spleen as early transitional (T1) B cells. Selection and maturation of transitional B cells continues in the periphery leading to the generation of at least mature follicular (Fo B)6 and marginal zone (MZ) B cells likely via late transitional T2-follicular (CD21int-T2) precursor stage (1–5). Peripheral differentiation is accompanied by increasing resistance to passive or BCR-induced apoptosis (1–5). In this regard, T1 cells are the most sensitive and have been shown to die in response to BCR cross-linking (1, 4, 6, 7). They are therefore the likely targets of negative selection, which weeds out self-reactive B cells (8–10). Prior studies also indicate that positive selection of transitional B cells contributes to generation of the mature B cell repertoire (2, 11, 12). Because this process involves selection of BCR specificities, it is believed to occur in late transitional or T2 cells that have reduced sensitivity to death in response to BCR cross-linking (4).
B cells gain a second form of survival advantage during positive selection by becoming responsive to B cell activating factor (BAFF) (4, 13–15). Loss of BAFF or BAFF-receptor 3 (BAFF-R or BR3) results in a severe B cell deficiency, particularly affecting T2 and mature B cells (16–18). Recent studies show that CD21int-T2 B cells express higher levels of BAFF-R and display enhanced responsiveness to BAFF relative to T1 B cells (4). BAFF-R activates several signaling pathways, including the canonical and noncanonical NF-κB pathways that increase B cell survival (13, 15, 19–21). Thus, differential survival competence in response to both BCR and BAFF-R engagement is a hallmark of the T1- to T2 transition. However, the mechanisms that underlie this “gain of survival potential” remain unclear.
To account for the hypersensitivity of T1 cells to BCR cross-linking, we and others have compared the signaling characteristics and biological responses of T1, T2, and mature Fo B cells (1, 3, 4, 6, 7, 15). The unifying feature of these studies is that T1 cells respond poorly to BCR cross-linking compared with T2 cells in many biochemical and biological assays. However, it remains unclear how this hypersensitivity is implemented to cause enhanced T1 cell death. In this regard, studies with B cells deficient in BCR signal transducer, Bruton’s tyrosine kinase (Btk) show that, like T1 cells, Btk-deficient B cells undergo apoptosis in response to BCR cross-linking (6, 7, 22). This similarity may be significant as, like wild-type (WT) T1 B cells, Btk-deficient B cells fail to induce Bcl-xL and A1 anti-apoptotic genes of the Bcl-2 family in response to BCR signaling (6, 7, 22). Impaired activation of these genes in T1 cells likely contributes to their extreme sensitivity to BCR-induced cell death, as evidenced by the relative resistance of B cells from Bim-deficient or Bcl-xL transgenic mice to apoptosis (23, 24). In this regard, the NF-κB family member c-Rel has been previously shown to regulate Bcl-xL and A1 gene expression in B cells (25). Consistent with an anti-apoptotic function of c-Rel, mature B cells deficient in c-Rel also display extreme sensitivity to BCR-induced cell death (25–28). Thus, B cells deficient for Btk and c-Rel behave similarly to T1 cells in their response to BCR cross-linking. We therefore considered the possibility that differential c-Rel activity is under Btk control and may at least in part explain the extreme sensitivity of T1 cells to BCR-induced death.
In this study, we show that sustained BCR-induced nuclear c-Rel activity depends on de novo c-Rel gene transcription and translation, and that this process occurs in T2 and mature Fo B but not in T1 cells. Btk deficiency blocks c-Rel induction in mature B cells, implicating Btk-dependent signaling as the basis for the differential regulation of c-Rel in T1 and T2 B cells. Importantly, c-Rel also regulates BCR-inducible expression of BAFF-R and substrate p100 of the noncanonical NF-κB pathway, thereby providing additional anti-apoptotic functions in T2 cells. We propose that gain of c-Rel inducibility at the T1 to T2 transition contributes to increased B cell resistance to apoptosis by two distinct mechanisms, by directly regulating the expression of anti-apoptotic genes and by up-regulating expression of BAFF-R and its substrates.
Materials and Methods
Mice
The generation of Btk-, c-Rel-deficient mice as well as B cell-specific bcl-2 transgenic mice has been previously described (29–31). C57BL/6 mice from The Jackson Laboratory were used as a source of WT B cell populations. Mice were treated humanely in accordance with federal and state government guidelines and their use was approved by the institutional animal committee.
B cell isolation and transitional B cell purification by FACS
Primary B lymphocytes from C57BL/6 mice were isolated using MiniMACS magnetic sorting by negative selection (CD43 depletion) to avoid inadvertent activation of B cells as previously described (3, 6). B cell purity was 90–95% based on flow cytometric (FCM) analysis following CD19 staining.
Splenic B cell subsets were purified based on a combination of schemes previously described (1, 3, 4, 6, 32, 33). Highly purified T1 and T2 B cells were obtained by MACS enrichment of AA4.1+ transitional B cells followed by FACS purification using Abs directed against CD23, CD24, and CD21. The purity of T1 (AA4.1+HSAhighCD21−CD23−) and T2 (AA4.1+HSAhighCD21+CD23+) B cell populations was 85–92 and 85–95%, respectively, as determined by FCM (see Fig. 2A). Using this sorting scheme, T2-preMZ (AA4.1low/−HSAhighCD21highCD23+) B cells were excluded from sorted T1 and T2 B cells. Thus, the T2 population analyzed in this study is the same as we and Meyer-Bahlburg et al. have recently reported (CD21intT2; Refs. 3 and 4). Mature Fo B cells were obtained by MACS-enrichment of the AA4.1− fraction with anti-CD43 conjugated microbeads (Miltenyi Biotec) followed by FACS purification using the same Ab combination as the T1 and T2 sorts. The purity of Fo B cells (AA4.1−HSAlowCD21+CD23+) was 95–100%, as determined by FCM. B cell subsets from Bcl-2 transgenic (Tg) mice were also purified using this scheme as shown in Fig. 3A. In experiments with Bcl-2 Tg B cell subsets (Fig. 3C), CD23+ cells were selected from AA4.1+ depleted splenocytes.
FIGURE 2.

BCR signaling induces cRel and anti-apoptotic genes preferentially in T2 and mature B cells. A, Purification of T1, T2 (CD21int-T2), and mature Fo B cell populations. Freshly isolated splenocytes from 4-wk-old C57BL/6 mice were depleted of RBCs. T1 and T2 B cells were obtained by MACS enrichment of AA4.1+ transitional B cells, followed by FACS purification using Abs directed against CD23, CD24 and CD21 to sort AA4.1+C23−CD24+CD21low (T1) and AA4.1+ CD23+CD24+CD21+ (T2) B cells. T2-PreMZ (AA4.1+C23+CD24+CD21high) B cell subset was excluded from sorted T1 and T2 B cells. Thus, the T1 and T2 B cells we have used here have previously been described by Hoek et al. and Meyer-Bahlburg (3, 4). The purity of T1 and for T2 was determined by postsort FCM analysis. Mature Fo B cells were MACS enriched from the AA4.1 negative fraction by CD43 depletion followed by FACS purification using the same Ab scheme as for T1 and T2 sorts; AA4.1−CD23+CD24lowCD21+ (Fo B). The purity of the mature Fo B cells was determined by postsort FCM analysis. B, Analysis of BCR-induced steady-state levels of c-Rel mRNA in FACS-purified T1, T2, and mature Fo B cells from C57BL6 mice. Cells were stimulated with 10 μg/ml anti-IgM F(ab′)2 for 4 h and analyzed for c-Rel mRNA by qRT-PCR. Relative fold levels of c-Rel was normalized to 18S and calibrated to the nonstimulated sample for each population. Data are mean with SD of five experiments using five to ten mice as a source of B cells for cell sorting. *, p = 0.0036; **, p = 0.0103. Levels of c-Rel protein in freshly isolated T1, T2, and mature B cells were similar (data not shown). C and D, Expression profile of NF-κB dependent A1 and Bcl-xL genes in transitional and mature Fo B cells. mRNA levels for A1 and Bcl-xL were determined by qRT-PCR in T1, T2 and mature Fo (M) B cells stimulated with 10 μg/ml anti-IgM F(ab′)2 for 4 h. Relative fold expression of each gene was normalized to 18S and calibrated to the nonstimulated sample for each population. Data are mean with SD of five experiments using five to ten mice as a source of B cells for FACS sorting as described in A. p values for A1 gene in anti-IgM treated samples; *, p = 0.051; **, p = 0.047. p values for Bcl-xL gene in anti-IgM treated samples; *, p = ≪0.000; **, p = 0.0024. D, Increased BCR-induced NF-κB DNA binding activity in T2 and mature Fo B cells relative to T1 cells. EMSA analysis for NF-κB DNA binding activity in FACS-sorted T1, T2, and mature Fo B (M) cells following stimulation with anti-IgM F(ab′)2 or 5 mg/ml LPS (as a positive control) or left nonstimulated. Equal amounts (2.0 μg) of nuclear extracts per lane were used in each DNA binding reaction. The NF-Y band was used as loading control. These experiments are representative of three independent experiments using five to ten mice as a source of B cells for FACS sorting. E, Fold change for total NF-κB activity from D normalized to NF-Y and calibrated to the nonstimulated sample. F, Cytoplasmic extracts were prepared from equivalent number of cells (1 × 106) and analyzed by immunoblotting for A1, Bcl-2, Bcl-xL, and Mcl-1. Blot was stripped and probed for A1, Bcl-2, Bcl-xL, Mcl-1, and anti-p38 sequentially as a loading control. Data are representative of at least three experiments.
FIGURE 3.

BCR signaling induces sustained c-Rel expression in T2 and mature, but not in T1 B cells. A, Purification of T1, T2, and mature CD23+ B cell populations by FACS. Splenocytes from six to eight spleens from 7- to 12-wk-old Bcl-2 Tg mice were depleted of RBCs. T1 and T2 B cells were obtained by MACS enrichment of AA4.1+ transitional B cells, followed by FACS-sorting as described in Fig. 2A. B, Analysis of BCR-induced steady state levels of c-Rel mRNA in FACS-purified T1, T2, and mature B cells from Bcl-2 Tg mice. Cells were stimulated with 10 μg/ml anti-IgM F(ab′)2 for 16 h and analyzed for c-Rel mRNA by qRT-PCR. Relative fold levels of c-Rel was normalized to GAPDH and calibrated to the nonstimulated sample for each population. *, p = 0.0279; **, p = 0.0043. Data are mean with SD calculated from five experiments using five to ten mice as a source of B cells for cell sorting. C, BCR signaling induces accumulation c-Rel in T2 and follicular mature B cells (CD23+) but not in T1 B cells. Immunoblot analysis of total cellular extracts from FACS-sorted T1, T2, and mature CD23+ B cells from Bcl-2 transgenic mice following stimulation with anti-IgM F(ab′)2 for 16 h. Blot was probed sequentially with anti-cRel and –p38 Abs and fluorescently labeled secondary Abs. Data were collected and quantified by Odyssey. Data are representative of three experiments. D, FCM analysis of c-Rel expression in MACS enriched splenic B cells after 16 h culture with or without anti-IgM F(ab′)2 (10 μg/ml) Abs. Cells were stained for B cell subset identification (T1, T2, and mature Fo B cells) as in A, fixed, permeabilized, and intracellularly labeled with Abs against c-Rel as described in Materials and Methods. c-Rel levels in nonstimulated and stimulated cells (open solid line histograms) and competition with unlabeled anti-c-Rel Abs (filled gray histograms) are shown for each splenic B cell subset. Data are displayed as mean fluorescence intensity (MFI). E, Fold change for c-Rel protein levels over nonstimulated samples from D and two additional experiments. *, p = 0.05.
In the data presented in Fig. 4D, bcl-2 Tg T1 B cells were purified using a series of MACS depletions and positive selections, which included CD62L depletion followed by AA4.1 positive selection. The T2/M cells were obtained by MACS enrichment of CD23+ cells. The purity of T1 (CD62−AA4.1+) and T2 (CD23+) B cell populations was 65–90 and 90%, respectively, as determined by FCM.
FIGURE 4.

T1 B cells display reduced ability to up-regulate BAFF-R and p100 than T2 and mature B cells in response to BCR signaling. A, BCR signaling induces BAFF-R expression in T2 and mature but not in T1 B cells. FACS-sorted B cell subsets (as in Fig. 2A) were stimulated with 10 μg/ml anti-IgM F(ab′)2 for the 4 h. BAFF-R RNA levels were determined by qRT-PCR analysis. Relative fold induction was normalized to 18S and calibrated to the nonstimulated samples. B, FACS-sorted splenic B cell subsets (as in Fig. 2A) from Bcl-2 transgenic (Bcl-2-Tg) mice were stimulated with 10 μg/ml anti-IgM F(ab′)2 for the indicated time. RNA levels of p100 were determined by qRT-PCR analysis. Relative fold induction was normalized to GAPDH and calibrated to the nonstimulated sample for each population. Data are mean with SD calculated based on five independent experiments using more than five mice in each experiment. *, p = 0.0016; **, p = 0.0188. C, FACS-sorted splenic B cell subsets from Bcl-2 Tg mice (as in Fig. 3A) were stimulated with 10 μg/ml anti-IgM for 16 h and analyzed as in B. *, p = 0.0234; **, p = 0.0086. Mean data with SD calculated from five independent experiments using more than five mice in each experiment. D, BCR signaling results in accumulation of p100 in CD23+ (T2 + M) B cells but not in T1 B cells. Immunoblot analysis of total cellular extracts from MACS-enriched T1 and T2 + M B cells from Bcl-2 transgenic mice, as described in Materials and Methods following stimulation with anti-IgM F(ab′)2 for 36 h. Blots were probed sequentially with anti-p100 and anti-β-tubulin Abs. Data are representative of three experiments.
Purified B cells were cultured in RPMI 1640 (HyClone Laboratories) supplemented with 10% FCS, 55 nM 2-ME, 2 mM L-glutamine, and 100 IU penicillin/streptomycin in a 37°C humidified incubator.
Immunoblotting
For kinetics studies of cRel and RelA, whole-cell extracts were prepared from total splenic B cells following lysis with RIPA buffer, and nuclear extracts were prepared as described previously (34). Ten micrograms of whole-cell extracts or 3 μg of nuclear extracts were separated on SDS-PAGE gel and subjected to Western blot analysis. Abs for RelA, cRel, PKCμ (Santa Cruz Biotechnology), and TATA box binding protein (Ab-cam) were used according to the manufacturer’s instructions.
For studies performed with btk−/−, c-rel−/− or Bcl-2 Tg mice, cells were stimulated with anti-IgM (Jackson ImmunoResearch Laboratories) or anti-CD40 from (BD Pharmigen). After culture, btk−/− live cells were obtained using Lympholite M (Cedarlane Laboratories). Whole cell extracts for these experiments were resolved as described in Ref. 35 and immunoblotted with anti-cRel (sc-70; see Fig. 6B) or (sc-71; see Figs. 1F and 3C), anti-p100/p52 (sc-7386) p38 (sc-535) from Santa Cruz Biotechnology and anti-β-tubulin (T0198) was purchased from Sigma-Aldrich. For quantitative analysis of c-Rel protein by immunoblotting, c-Rel and p38 (loading control) were visualized by infrared imaging (Odyssey, Licor) using anti-Ig Alexa 670 (Invitrogen).
FIGURE 6.

BCR-induced c-Rel synthesis is mediated via a Btk dependent pathway. A, Analysis of BCR-induced c-Rel mRNA expression in WT and Btk-deficient (btk−/−) B cells. Cells stimulated with 10 μg/ml anti-IgM F(ab′)2 for the indicated times and analyzed for c-Rel mRNA expression by qRT-PCR. Relative fold induction of c-Rel was normalized to GAPDH and calibrated to the nonstimulated samples. Mean data and SD from three experiments. B, Immunoblot analysis of cellular extracts from AA4.1+ live immature B cells from WT and btk−/− mice following stimulation with 10 μg/ml anti-IgM F(ab′)2 or 5 μg/ml anti-CD40 for 14 h. Blot was probed sequentially with anti-c-Rel and β-Actin. Represents three independent experiments. C, Btk-deficient B cells display reduced c-Rel DNA binding in response to BCR stimulation. EMSA analysis for NF-κB binding activity in WT and btk−/− splenic B cells stimulated with 10 μg/ml anti-IgM F(ab′)2 for 14 h. Equal amounts (1.0 μg) of nuclear extracts per lane were used in each DNA binding reaction. The c-Rel and p50 subunits in NF-κB complexes were identified by addition of subunit-specific Abs (supershift) as indicated. D, Btk is required for BCR-induced p100 expression. Immunoblot analysis of cellular extracts from WT and btk−/− splenic B cells following stimulation with anti-IgM F(ab′)2 for 18 h. Blot was probed sequentially with p100 and p38. p38 was used as loading control. Data represent three or more experiments using multiple mice in all the above panels.
FIGURE 1.

BCR-Induced long-term induction of c-Rel coincides with expression of A1 and Bcl-xL. A and B, Splenic mature B cells were stimulated with anti-IgM F(ab′)2 (10 μg/ml) for indicated times; nuclear (NE) and whole cell extracts (WCE) were prepared as described in methods. Three micrograms NE or 10 μg WCE were fractionated by SDS-PAGE and transferred to PVDF membranes, which were probed with Abs against NF-κB proteins RelA and c-Rel as indicated. TATA box binding protein and protein kinase C (PKC) μ were used as loading controls for NE and WCE, respectively. C, Analysis of BCR-induced c-Rel mRNA expression in naive splenic B cells. Cells were stimulated with 10 μg/ml anti-IgM F(ab′)2 for the indicated time points and RNA levels of c-Rel were determined by qRT-PCR. Relative fold induction of each gene was normalized to GAPDH and calibrated to the corresponding nonstimulated sample. Data are mean with SD from three experiments. D and E, c-Rel regulates long-term expression of A1 and Bcl-xL in mouse splenic B cells following BCR stimulation. Naive splenic B cells from WT and c-Rel deficient (c-rel−/−) splenic B cells were stimulated for the indicated times with 10 μg/ml anti-IgM F(ab′)2 and qRT-PCR was performed to determine the fold change in the steady-state levels of A1 and Bcl-xL mRNA compared with the endogenous control GAPDH and calibrated to the nonstimulated sample. Data are mean with SD from three experiments. F, Western blot analysis was performed on total cellular extracts from WT and c-Rel deficient (c-rel−/−) splenic B cells stimulated with anti-IgM F(ab′)2 for the indicated times. Blot was probed with anti-Bcl-xL, -A1, -c-Rel, and -β-Tubulin Abs. The data in all panels are representative of three independent experiments using different mice for each experiment.
For the Bcl-2 family protein analyses, cellular extracts were immunoblotted with anti-A1 (R&D Systems) anti-Bcl-xL (BD Transduction Laboratory), anti-Bcl-2 (Santa Cruz Biotechnology), anti-MCl-1 from (Rockland Immunochemicals), anti-p38, and β-actin (sc-1616) purchased from Santa Cruz Biotechnology.
EMSA analysis
Equal amounts of nuclear extracts (1.0 μg) were prepared with the Nuclear and Cytoplasmic Extraction Reagent kit (NE-PER; Pierce), and were pre-incubated for 20 min at room temperature in the presence or absence of polyclonal Abs specific for p50(sc-1192X), c-Rel(sc-70X) from Santa Cruz Biotechnology. Subsequently, [γ-32P]ATP radio-labeled probe derived from κB enhancer sequences in the IL-2R promoter (36) was added and incubated on ice for 15 min. DNA binding reactions were performed as previously described (37). DNA protein complexes were resolved by electrophoresis on 4% native polyacrylamide gels and exposed to x-ray film. Total NF-κB nuclear activity was quantified and the composition of the NF-κB complexes identified using a phosphoimager (FujiFilm Medical Systems).
Real-time PCR
RNA was extracted from T1, T2, M, or total B cells using the RNeasy Mini Kit (Qiagen) and used to synthesize cDNA. For real-time PCR, Taq-man Universal Master Mix (Applied Biosystems) and Stratagene Max 3000p Detection Systems were used. Primers and FAM-labeled probes were obtained from Applied Biosystems (TaqMan Assay on Demand): NF-κB2 (Mm00479807), c-Rel (Mm00485657_m1), RelB (Mm00485672), Bcl-xL (Mm00437783_m1), and BAFF-R (Mm00840578_g1). The sequence of the primers and probe used for A1 were also obtained from (AB): A1 forward, CAGGAGAATGGATACGGCAGA; A1 reverse, CAGATCTGTCCTGTCATCTGCAG; (MGB): TCTTCCCAACCTCCA (38). RNA was quantified before use in RT-PCR for experiments that used Bcl-2 Tg mice and for kinetics with WT, btk−/− and crel−/− B cells. Data are expressed as the relative RNA concentrations relative to 18S or GAPDH expression and calibrated to nonstimulated cells.
Intracellular staining
MACS-enriched B cells were stimulated with 10 μg/ml goat anti-mouse IgM F(ab′)2 Abs for 18 h, stained for CD19, CD23, CD24, and CD21 for identification of T1 (CD19+CD23−CD24highCD21−), T2 (CD19+CD23+ CD24highCD21int), and mature Fo B cells (CD19+CD23+CD24low CD21+). After staining, the cells were fixed with 2% paraformaldehyde, permeabilized with 0.3% Triton X-100, and stained with AlexaFluor 647 labeled anti-c-Rel (SC-71). Cold competition was preformed by staining first with unlabeled anti-c-Rel, followed by staining with AlexaFluor 647 anti-c-Rel. Data were acquired on BD LSR II flow cytometer.
Statistical analyses
Data were compared with Student’s t test. All data are represented as mean ± SEM where indicated. Values of *, p ≤ 0.05 were considered statistically significant.
Results
Enhanced c-Rel gene activation at the T2 stage
Prior studies have shown that several anti-apoptotic genes are regulated by c-Rel in mature B cells after BCR stimulation (26, 27). Because T1 and T2 B cells display distinct survival properties in response to BCR cross-linking, we explored the possibility that c-Rel was differentially regulated in T1 and T2 B cell populations. We first examined the c-Rel response of mature splenic B cells to BCR stimulation. CD43-depleted naive splenic B cells were activated with anti-IgM for various times followed by analysis of nuclear or whole cell extracts by immunoblotting. We found that BCR stimulation resulted in long-term c-Rel, but not p65/RelA, nuclear expression (Fig. 1A). Extended c-Rel induction coincided with increased total cellular c-Rel protein levels (Fig. 1B) that was likely mediated by increased c-Rel gene transcription (Fig. 1C). Total p65/RelA levels did not change over this time course (data not shown). These results are consistent with and extend our previous observation in T cells (39). We conclude that BCR stimulation activates two distinct phases of nuclear c-Rel. To test whether anti-apoptotic gene expression coincides with c-Rel induction, we performed kinetic studies of A1 and Bcl-xL in WT and c-rel−/− B cells. The kinetics of A1 and Bcl-xL appear to largely coincide with the biphasic nature of BCR-induced c-Rel (Fig. 1, D–F). The correspondence of c-Rel induction to A1 and Bcl-xL gene expression suggested that inducible transcription of the c-Rel gene may be a key factor in its effectiveness as an anti-apoptotic transcription factor.
After determining that c-Rel was induced upon BCR stimulation, we compared c-Rel gene transcription in transitional B cell subsets. T1, T2, and mature Fo B cells were purified by flow cytometry (Fig. 2A) as previously described (3, 4), activated with anti-IgM for 4 h before c-Rel mRNA analyses. We observed increased c-Rel transcripts in T2 and mature B cells, but not in T1 cells (Fig. 2B). Consistent with this, BCR-induced expression of anti-apoptotic genes A1 and Bcl-xL was also more increased in T2 and mature B cells relative to T1 B cells (Fig. 2C). As a control, activation of T1 cells via CD40 efficiently induced these anti-apoptotic genes excluding the possibility that T1 B cells are generally nonresponsive to stimulation (data not shown). As expected, this difference between T1 and T2 cells also reflected in increased NF-κB DNA binding activity in T2 relative to T1 cells (Fig. 2, D and E). The reduced NF-κB DNA binding activity that we observe in T1 cells are consistent with the findings previously reported using immature transitional B cells (25). Our results extend these findings by analyzing distinct populations within transitional B cells. Based on our findings in the mature splenic B cells (Fig. 1) and an increase at early time point (Fig. 2, B and C) we asked whether T1, T2, and mature B cells display differences in the anti-apoptotic gene expression after long-term BCR stimulation. Levels of several anti-apoptotic proteins were analyzed in FACS-sorted T1, T2, and mature Fo B cells after 16 h of culture with or without anti-IgM. Like mature splenic B cells a BCR signaling-dependent increase was observed in T2 and mature B cells (Fig. 2F). In contrast, T1 B cells which did not show an increase in the transcripts for c-Rel or A1 and Bcl-xL also did not display an increase in the protein levels encoded by these anti-apoptotic genes. These results are consistent with a higher caspase activity and sensitivity to apoptosis of T1 cells after long-term BCR stimulation and may reflect a relatively higher rate of cell death in T1 B cells. Together, these data suggest that unlike T1 and like naive splenic mature B cells, T2 B cells are capable of inducing anti-apoptotic genes and their protein products.
Given the close relationship between the long-term induction of c-Rel and anti-apoptotic genes (Fig. 1), we analyzed the effects of long-term BCR stimulation on c-Rel expression in transitional B cell subsets. For these studies, we FACS-sorted transitional B cell subsets from apoptosis resistant Bcl-2 transgenic mice (as in Fig. 2A) to avoid potential effects of differential sensitivity of T1 and T2 cells to BCR-induced cell death (Fig. 3A) (31). c-Rel transcripts were not induced in T1 cells even after 16 h of BCR activation, whereas this response was evident in T2 B cells (Fig. 3B). Accumulation of c-Rel protein also significantly increased in T2 and mature B cells following BCR stimulation when compared with T1 B cells (Fig. 3, C–E). These results suggested that one key feature that distinguishes T2 from T1 cells was their ability to up-regulate c-Rel expression following BCR stimulation. Taken together, these results demonstrate that T2 cells induce the c-Rel gene transcription and protein production that is required for sustained nuclear c-Rel activity and anti-apoptotic gene expression, whereas T1 cells fail to do so rendering them sensitive to apoptosis.
c-Rel contributes to the expression of BAFF-R and p100 in T2 and mature B cells
BAFF-R plays a critical role in B cell survival beyond the T1 stage (17). The findings that B cells become further resistant to apoptosis during the T1 to T2 transition by gaining responsiveness to BAFF further support a unique role for BAFF beginning at the T2 stage (4). This is, in part, due to increased expression of BAFF-R, perhaps as a consequence of BCR signaling (15). Consistent with an enhanced responsiveness of T2 cells to BAFF (4), we found that BCR-induced BAFF-R expression increased in T2 and mature but not in T1 B cells (Fig. 4A). Because BAFF-dependent survival is mediated by both canonical and the noncanonical NF-κB pathways (20, 21, 35), effective anti-apoptotic signaling by BAFF requires not only increased BAFF-R expression, but also sustained availability of the noncanonical pathway substrate p100. We therefore examined whether substrate availability was also differentially regulated in transitional B cell subsets.
BCR stimulation increased p100 mRNA in T2, but not in T1 cells after 4 h of BCR stimulation (Fig. 4B). To establish a link between long-term c-Rel induction, we activated FACS-sorted T1, T2, and mature B cells from Bcl-2 Tg mice and analyzed p100 mRNA after 16 h of culture with or without anti-IgM. We found elevated levels of p100 in T2 and further enhanced in mature B cells, however, this increase was minor in T1 B cells (Fig. 4C), which reflects severely reduced levels of BCR-induced p100 protein in T1 cells in even after long-term stimulation via the BCR (Fig. 4D). The levels of p100 protein in T2 + M B cells were increased significantly, conceivably by p100 production by both T2 and mature B cells (Fig. 4D). These results extend previous findings with BAFF-R to the regulation of its downstream substrates p100 (15). Together, these data suggest that increased expression of BAFF-R and noncanonical NF-κB pathway mediator p100 contribute to the higher survival response of T2 and mature Fo B cells compared with T1 cells in response to BAFF. A recent report showing a critical role for the noncanonical NF-κB pathway in B cell survival further supports this hypothesis (20).
NF-κB2 has been shown to be an NF-κB target gene (35, 40). The coordinate gain of BAFF-R, p100, and c-Rel inducibility in T2 cells prompted us to examine the c-Rel dependence of genes involved in BAFF-mediated signaling. WT and c-Rel-deficient B cells were activated with anti-IgM for varying times followed by RNA and protein analyses. We observed a time-dependent increase of BAFF-R, and p100 mRNA in WT but not c-Rel-deficient B cells (Fig. 5, A and B). In concordance with the RNA data, p100 protein was robustly up-regulated in normal B cells activated via the BCR, but not in c-Rel-deficient B cells (Fig. 5C). Further, the increase in BAFF-R and p100 mRNA and p100 protein occurred more robustly at later time points, which is consistent with their induction by long-term c-Rel induction by BCR signaling. These results indicate that c-Rel plays a key role in the inducible activation of BAFF-R and p100 genes. This role is in addition to the well-established function of c-Rel in the regulation of anti-apoptotic genes under canonical NF-κB control, including Bcl-xL and A1 (Fig. 2). We note, however, that basal expression of all genes analyzed was comparable in WT and c-Rel-deficient B cells. This may be due to compensatory NF-κB activity in c-Rel-deficient B cells or it may be mediated by other constitutively active transcription factors. Together, these data demonstrate that c-Rel pathway is particularly important not only for expression of anti-apoptotic genes of the Bcl-2 family but also for long-term expression of BAFF-R, and p100 in response to BCR stimulation.
FIGURE 5.
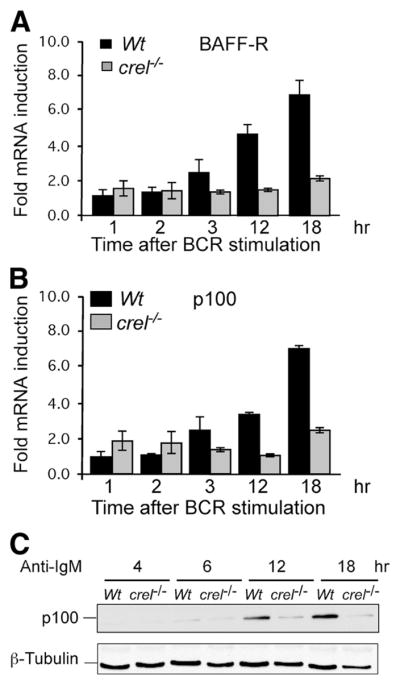
BCR Induction of BAFF-R and its substrate p100 requires c-Rel. A–C, Kinetic analysis of BAFF-R and p100 in WT and crel−/− B cells. Naive splenic B cells from WT and c-Rel deficient mice were stimulated with anti-IgM F(ab′)2 10 μg/ml for the indicated times. RNA levels of BAFF-R, p100 were determined by qRT-PCR analysis. Relative fold induction was normalized to GAPDH and calibrated to the nonstimulated sample for each population. Mean data and SD from three experiments. C, Western blot analysis of total cellular extracts from WT and c-Rel deficient splenic B cells stimulated as in A for the indicated times. Blot was probed with anti-p100 and β-tubulin Abs. Data are representative of three experiments.
BCR-induced c-Rel expression requires Btk
We and others have previously shown that like T1 cells, Btk-deficient B cells fail to induce anti-apoptotic genes and undergo apoptosis in response to BCR engagement (6, 7). If the failure to induce c-Rel contributed causally to the enhanced death of T1 cells, we expect impaired c-Rel expression in Btk-deficient B cells after BCR stimulation. We tested this prediction by activating btk−/− B cells with anti-IgM and assaying for c-Rel mRNA and protein. BCR stimulation resulted in c-Rel transcriptional induction and protein accumulation in WT, but not btk−/−, B cells (Fig. 6, A and B). CD40 signaling, which is known to promote survival of WT and btk−/− B cells resulted in comparable c-Rel induction (Fig. 6B). Lack of BCR-induced c-Rel expression also affected its nuclear DNA binding in btk−/− B cells (Fig. 6C). Consistent with a role for Btk in the regulation of c-Rel, Btk-deficient B cells also failed to increase p100 protein in response to BCR signals (Fig. 6D). Taken together, results in Figs. 5 and 6 show that btk−/− and c-rel−/− B cells are defective in mechanisms that promote survival in a manner similar to T1 B cells and suggest that Btk/c-Rel signaling axis is critical in the regulation of B cell survival.
Discussion
Establishing tolerance is an essential aspect of B cell development, whereby cells that recognize self-Ags are functionally discarded. Positive and negative selection of clonotypes in the periphery is modulated by the differential responses of transitional B cells to BCR engagement. In this study, we provide evidence that reduced c-Rel response in T1 cells controls their hypersensitivity to BCR cross-linking. As a consequence, anti-apoptotic genes targeted by c-Rel, such as Bcl-xL and A1, are poorly induced in T1 B cells. In contrast, T2 B cells resist BCR-induced apoptosis due their ability to activate and sustain signal-induced expression of c-Rel, which leads to increased anti-apoptotic gene expression relative to T1 cells. Our observations also explain the enhanced sensitivity of Btk-deficient B cells to BCR-induced apoptosis by placing c-Rel transcriptional induction downstream of Btk as an essential mediator of survival signals.
We previously showed that T1 cells fail to produce diacylglycerol and activate PKC in response to BCR engagement (3). This suggested that T1 cells would not activate NF-κB leading to reduced expression of NF-κB-dependent anti-apoptotic genes. We and others tested this hypothesis and were surprised to find that IKK was phosphorylated, IκBα was degraded, NF-κB was translocated to the nucleus and bound DNA comparably in T1, T2, and mature B cell populations (Ref. 41 and data not shown). Yet, NF-κB-dependent anti-apoptotic and c-Rel genes are differentially regulated in T1, T2, and mature B cells (Figs. 1, D and E and 2, B and C). One potential explanation for this unexpected finding may be that unlike T2 and mature, T1 B cells are in a state of nuclear nonresponsiveness as shown by BCR-specific defect in the assembly of active transcriptional machinery in T1 cells (41). This view is supported by our data showing that the first phase of A1 and Bcl-xL transcription is also defective in T1 B cells, whereas these genes are clearly up-regulated within the first 1– 4 h following BCR stimulation in T2 and mature B cells (Figs. 1, D and E and 2C).
Our data show that in addition to the early phase, T2 and mature B cells produce a second and long-term phase of c-Rel induction. Importantly, T1 B cells are defective in this response as well. The second and late phase of c-Rel induction and consequent A1 and Bcl-xL gene transcription in T2 and mature Fo B cells may be critical for an effective resistance to apoptosis in response to BCR engagement because the later phase coincides with a substantial increase in the corresponding anti-apoptotic proteins. It is unclear at present whether a reduced long-term c-Rel induction in T1 B cells in response to BCR stimulation is also associated with their nuclear nonresponsiveness. However, two distinct mechanisms operate in T2 and mature Fo B cells that allow early as well as sustained expression of c-Rel and its downstream target genes. In contrast, neither of the two phases of c-Rel induction operates in T1 B cells, resulting in an inability to produce anti-apoptotic factors and rendering them susceptible to BCR-induced cell death. Although the mechanism of long-term c-Rel activation remains unknown, it may be largely independent of the regulation by classical NF-κB pathway as newly synthesized c-Rel may not be sequestered in the cytoplasm by IκB proteins, as previously suggested (42). Regardless of the mechanism, our data suggest de novo synthesis of c-Rel in T2 cells as a mechanism to limit the expression of the anti-apoptotic genes to this apoptosis-resistant transitional B cell population. Together, our results suggest that an ability to produce a c-Rel activation response following BCR engagement is associated with resistance to apoptosis in T2 and mature Fo B cells.
In addition to BCR-initiated signals, B lymphocyte survival and selection requires activation of the BAFF/BAFF-R pathway (17, 20). We show in this study that the Btk/c-Rel pathway also up-regulates expression of BAFF-R and its substrates p100 in T2 cells. Thus, proapoptotic BCR-mediated signaling in T1 cells is converted to an anti-apoptotic response during the T1 to T2 transition by two c-Rel-regulated mechanisms: First, c-Rel directly activates anti-apoptotic gene expression and second, c-Rel enhances the levels of both BAFF-R and its substrate, p100, which likely increases BAFF responsiveness of T2 relative to T1 cells. This hypothesis is consistent with the recent findings showing that T2 cells express higher levels of cell surface BAFF-R and respond more robustly to BAFF than T1 B cells (4). In addition, prior studies are consistent with a critical role for c-Rel, anti-apoptotic genes, and BAFF-R in B cell survival. Although mature B cells develop in c-Rel-deficient mice, these B cells respond similarly to normal immature B cells in their sensitivity to BCR-induced apoptosis, whereas induced expression of Bcl-xL and cyclin E can rescue immature B cells from this form of cell death (25, 28, 29, 43, 44). Likewise, B cells deficient for Btk show similar but more pronounced defects in the survival pathways discussed above in the context of c-Rel deficiency. A vital function for BAFF-R in B cell survival beyond the T2 stage has also been well documented. Thus, loss of either BAFF or BAFF-R results in a severe deficiency of T2 and mature B cells (17, 18, 45, 46). In addition, reduced sensitivity of T2 B cells to BCR-induced apoptosis is further accentuated by developmental alterations in the BAFF/BAFF-R pathway of survival signaling (4, 13, 15, 18, 47, 48).
BAFF/BAFF-R interaction activates the alternative NF-κB pathway by IKKα-mediated phosphorylation of p100 (NF-κB2) that results in its proteolytic processing to p52 (19, 40, 49). The active transcription factor consists of the p52/RelB heterodimer, which activates anti-apoptotic genes such as Pim-2 (20). These and earlier studies show that constitutive, as well as BCR-inducible, expression of BAFF-R is higher in T2 B cells compared with T1 B cells (4, 15). For increased surface expression of BAFF-R to be effective, however, components of the alternative NF-κB pathway must be readily available. We find that p100 is induced preferentially in T2 B cells after BCR cross-linking; like c-Rel, this induction requires the presence of Btk. Re-enforcement of the BAFF-R signaling pathway by the BCR thus confines survival advantage among transitional B cells to the T2 population potentially facilitating their positive selection into mature B cells.
Taken together with earlier studies of signaling differences between T1 and T2 B cells, our observations lead us to the following model for the gain of apoptosis resistance to BCR signaling during the T1 to T2 transition; in contrast to T2, T1 B cells do not induce sustained c-Rel function that is mediated by Btk. This long-term c-Rel activation in T2 B cells results in stable induction of anti-apoptotic genes, which in turn rescues T2 B cells from BCR-induced cell death. Our observations account for the enhanced sensitivity of Btk-deficient B cells to apoptosis in response to the BCR by placing c-Rel induction downstream of Btk (6, 7, 22). Moreover, we demonstrate a unique role for Btk and c-Rel in the production of p100 in response to BCR, thus providing additional support for the re-enforcement of BAFF-R induced activation of the alternate NF-κB pathway by the BCR. The requirement for Btk in the up-regulation of p100 strongly implicates the long-term c-Rel activation in cross-talk between the canonical and alternate NF-κB pathways in productive B cell activation. Thus, the ability of transitional B cells to maintain anti-apoptotic c-Rel and BAFF-R activity following BCR engagement will determine whether these cells will survive and undergo positive selection or be negatively selected by apoptosis. Thus, our findings suggest that the ability to produce sustained c-Rel responses contributes to the gain of survival potential in T2 B cells, thus facilitating their survival and differentiation in vivo.
Acknowledgments
We thank M. Pia Arrate for expert technical assistance. We thank Dr. Emily Clark for assistance in establishing the intracellular staining protocol for c-Rel in primary splenic B cells. We acknowledge the skilled assistance of Kevin P. Weller, David K. Flaherty, and Jim Higginbotham of Vanderbilt University Medical Center Flow Cytometry and Dr. Oliver Umland of University of Miami Diabetes Research Institute Flow Cytometry Core. We thank Dr. Hsiou-Chi Liou for permission to use and Dr. Mark Boothby for providing c-Rel-deficient mice.
Footnotes
Abbreviations used in this paper: Fo B, follicular B cell; MZ, marginal zone; BAFF, B cell activating factor; Btk, Bruton’s tyrosine kinase; WT, wild type; FCM, flow cytometric; Tg, transgenic; qRT-PCR, quantitative RT-PCR.
Disclosures
The authors have no financial conflict of interest.
This study is supported in part by National Institutes of Health AI060729 (to W.N.K.), AI041054 and AI057463 (to R.T.W), AI043534 (to R.M.G), P30DK32520 (to University of Massachusetts Medical Flow Cytometry Core) and DK32520 (to the University of Miami Miller School of Medicine Diabetes and Endocrinology Center). K.L.H and N.P.S. are supported by NIH T32 HL69715–0, I.C. by NIH T32 CA09385–20 and J.A.W. by NIH 5 T32 HL069765. B.D. and R.S. are supported by intramural research program of the National Institute of Health on Aging, Baltimore, MD.
References
- 1.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 2.Cancro MP, Kearney JF. B cell positive selection: road map to the primary repertoire? J Immunol. 2004;173:15–19. doi: 10.4049/jimmunol.173.1.15. [DOI] [PubMed] [Google Scholar]
- 3.Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, Wang D, Gerstein RM, Khan WN. Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. J Immunol. 2006;177:5405–5413. doi: 10.4049/jimmunol.177.8.5405. [DOI] [PubMed] [Google Scholar]
- 4.Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J Exp Med. 2008;205:155–168. doi: 10.1084/jem.20071088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su TT, Guo B, Wei B, Braun J, Rawlings DJ. Signaling in transitional type 2 B cells is critical for peripheral B-cell development. Immunol Rev. 2004;197:161–178. doi: 10.1111/j.0105-2896.2004.0102.x. [DOI] [PubMed] [Google Scholar]
- 6.Petro JB, Gerstein RM, Lowe J, Carter RS, Shinners N, Khan WN. Transitional type 1 and 2 B lymphocyte subsets are differentially responsive to antigen receptor signaling. J Biol Chem. 2002;277:48009–48019. doi: 10.1074/jbc.M200305200. [DOI] [PubMed] [Google Scholar]
- 7.Su TT, Rawlings DJ. Transitional B lymphocyte subsets operate as distinct checkpoints in murine splenic B cell development. J Immunol. 2002;168:2101–2110. doi: 10.4049/jimmunol.168.5.2101. [DOI] [PubMed] [Google Scholar]
- 8.Russell DM, Dembic Z, Morahan G, Miller JF, Burki K, Nemazee D. Peripheral deletion of self-reactive B cells. Nature. 1991;354:308–311. doi: 10.1038/354308a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 10.Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine MH, Haberman AM, Sant’Angelo DB, Hannum LG, Cancro MP, Janeway CA, Jr, Shlomchik MJ. A B-cell receptor-specific selection step governs immature to mature B cell differentiation. Proc Natl Acad Sci USA. 2000;97:2743–2748. doi: 10.1073/pnas.050552997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Clarke SH. Evidence for a ligand-mediated positive selection signal in differentiation to a mature B cell. J Immunol. 2003;171:6381–6388. doi: 10.4049/jimmunol.171.12.6381. [DOI] [PubMed] [Google Scholar]
- 13.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sen R. Control of B lymphocyte apoptosis by the transcription factor NF-κ B. Immunity. 2006;25:871–883. doi: 10.1016/j.immuni.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Smith SH, Cancro MP. Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J Immunol. 2003;170:5820–5823. doi: 10.4049/jimmunol.170.12.5820. [DOI] [PubMed] [Google Scholar]
- 16.Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 18.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 19.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-κ B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 20.Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, Anzelon-Mills A, Rickert RC, Karin M. Alternative and classical NF-κ B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity. 2006;25:403–415. doi: 10.1016/j.immuni.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 21.Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-κ B activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Anderson JS, Teutsch M, Dong Z, Wortis HH. An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci USA. 1996;93:10966–10971. doi: 10.1073/pnas.93.20.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J Exp Med. 2005;202:1363–1374. doi: 10.1084/jem.20051283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solvason N, Wu WW, Kabra N, Lund-Johansen F, Roncarolo MG, Behrens TW, Grillot DA, Nunez G, Lees E, Howard M. Trans-gene expression of bcl-xL permits anti-immunoglobulin (Ig)-induced proliferation in xid B cells. J Exp Med. 1998;187:1081–1091. doi: 10.1084/jem.187.7.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng B, Cheng S, Hsia CY, King LB, Monroe JG, Liou HC. NF-κ B inducible genes BCL-X and cyclin E promote immature B-cell proliferation and survival. Cell Immunol. 2004;232:9–20. doi: 10.1016/j.cellimm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Grossmann M, O’Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S. The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J. 2000;19:6351–6360. doi: 10.1093/emboj/19.23.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gugasyan R, Grumont R, Grossmann M, Nakamura Y, Pohl T, Nesic D, Gerondakis S. Rel/NF-κ B transcription factors: key mediators of B-cell activation. Immunol Rev. 2000;176:134–140. doi: 10.1034/j.1600-065x.2000.00615.x. [DOI] [PubMed] [Google Scholar]
- 28.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 29.Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. 1999;11:361–371. doi: 10.1093/intimm/11.3.361. [DOI] [PubMed] [Google Scholar]
- 30.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Muller S, Kantor AB, Herzenberg LA, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 31.Woodland RT, Schmidt MR, Korsmeyer SJ, Gravel KA. Regulation of B cell survival in xid mice by the proto-oncogene bcl-2. J Immunol. 1996;156:2143–2154. [PubMed] [Google Scholar]
- 32.Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava B, Quinn WJ, 3rd, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. J Exp Med. 2005;202:1225–1234. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerji L, Glassford J, Lea NC, Thomas NS, Klaus GG, Lam EW. BCR signals target p27(Kip1) and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene. 2001;20:7352–7367. doi: 10.1038/sj.onc.1204951. [DOI] [PubMed] [Google Scholar]
- 35.Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RL, Scott ML, Wang D, Khan WN. Bruton’s tyrosine kinase mediates NF-κ B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179:3872–3880. doi: 10.4049/jimmunol.179.6.3872. [DOI] [PubMed] [Google Scholar]
- 36.Ballard DW, Walker WH, Doerre S, Sista P, Molitor JA, Dixon EP, Peffer NJ, Hannink M, Greene WC. The v-rel oncogene encodes a κ B enhancer binding protein that inhibits NF-κB function. Cell. 1990;63:803–814. doi: 10.1016/0092-8674(90)90146-6. [DOI] [PubMed] [Google Scholar]
- 37.Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)- κ B. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vigorito E, Gambardella L, Colucci F, McAdam S, Turner M. Vav proteins regulate peripheral B-cell survival. Blood. 2005;106:2391–2398. doi: 10.1182/blood-2004-12-4894. [DOI] [PubMed] [Google Scholar]
- 39.Venkataraman L, Wang W, Sen R. Differential regulation of c-Rel translocation in activated B and T cells. J Immunol. 1996;157:1149–1155. [PubMed] [Google Scholar]
- 40.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 41.Andrews SF, Rawlings DJ. Transitional B cells exhibit a B cell receptor-specific nuclear defect in gene transcription. J Immunol. 2009;182:2868–2878. doi: 10.4049/jimmunol.0802368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mittal A, Papa S, Franzoso G, Sen R. NF-κ B-dependent regulation of the timing of activation-induced cell death of T lymphocytes. J Immunol. 2006;176:2183–2189. doi: 10.4049/jimmunol.176.4.2183. [DOI] [PubMed] [Google Scholar]
- 43.Cariappa A, Liou HC, Horwitz BH, Pillai S. Nuclear factor κ B is required for the development of marginal zone B lymphocytes. J Exp Med. 2000;192:1175–1182. doi: 10.1084/jem.192.8.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tumang JR, Owyang A, Andjelic S, Jin Z, Hardy RR, Liou ML, Liou HC. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur J Immunol. 1998;28:4299–4312. doi: 10.1002/(SICI)1521-4141(199812)28:12<4299::AID-IMMU4299>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 45.Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, Rixon M, Schou O, Foley KP, Haugen H, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: impaired B cell maturation in mice lacking BLyS. Immunity. 2001;15:289–302. doi: 10.1016/s1074-7613(01)00183-2. [DOI] [PubMed] [Google Scholar]
- 46.Shulga-Morskaya S, Dobles M, Walsh ME, Ng LG, MacKay F, Rao SP, Kalled SL, Scott ML. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004;173:2331–2341. doi: 10.4049/jimmunol.173.4.2331. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, Di Liberto M, Cunningham AF, Kang L, Cheng S, Ely S, Liou HC, Maclennan IC, Chen-Kiang S. Homeostatic cell-cycle control by BLyS: Induction of cell-cycle entry but not G1/S transition in opposition to p18INK4c and p27Kip1. Proc Natl Acad Sci USA. 2004;101:17789–17794. doi: 10.1073/pnas.0406111101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brink R. Regulation of B cell self-tolerance by BAFF. Semin Immunol. 2006;18:276–283. doi: 10.1016/j.smim.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]