

Abstract
B-1a cells constitutively express phosphorylated, activated ERK, but the origin of pERK in B-1 cells has not been determined. To address this issue, we examined specific mediators of intracellular signaling in unmanipulated B-1a cells. We found that constitutive pERK was rapidly lost from B-1a cells following addition of metabolic inhibitors that block src kinase, Syk, PI-3K, and PLC function. We examined Syk and PLC in more detail and found rapid accumulation of phosphorylated forms of these molecules in B-1a cells, but not B-2 cells, when phosphatase activity was inhibited, and this change occurred in the majority of B-1a cells. Further, we showed that inhibition of src kinase activity eliminated “downstream” pSyk and pPLC accumulation in phosphatase-inhibited B-1a cells, indicating a pathway connection. CD86 expression is greater on B-1 than B-2 cells and plays a role in antigen presentation by B1 cells to T cells. We found that when Syk or PI-3K was inhibited, CD86 expression was diminished in a reversible fashion. All together, these results indicate that continual activation of intracellular signaling leads to constitutive activation of ERK in B-1 cells, with attendant consequences for costimulatory molecule expression.
Keywords: B cells, signal transduction, protein kinases/phosphatases, rodent
Introduction
B-1a cells constitute a unique set of B lymphocytes, initially distinguished from conventional splenic B (B-2) cells by expression of the pan-T cell surface glycoprotein, CD5. Additional well-recognized identifying phenotypic characteristics include IgMhi, IgDlo, B220lo, Mac1+, CD23-, and CD43+ (reviewed in (Hardy and Hayakawa, 2001; Kantor and Herzenberg, 1993; Rothstein, 2002)). B-1b cells represent a companion B-1 cell subset that is phenotypically similar to B-1a cells but lacks CD5 expression. B-1a cells appear first in ontogeny, after which B-1a cells decline in relative number over time as B-2 cell production proceeds (Hayakawa et al., 1983; Lalor et al., 1989). Although B-1a cells are numerically much less abundant than B-2 cells in adult animals (both in mouse and human), B-1a cells maintain themselves as a distinct population through self-renewal, in contrast to B-2 cells that are replenished from early progenitor cells (Hayakawa et al., 1986; Kantor et al., 1992; Lalor et al., 1989). The distinctions between B-1a and B-2 cells extend beyond phenotype to include structural differences in terms of differential expression of various transcription factors, transcripts and proteins, as well as metabolic and functional differences such as variations in mitogenic responsiveness, antigen presentation and skewing of T cell differentiation (Frances et al., 2006; Morris and Rothstein, 1993; Morris and Rothstein, 1994; Rothstein and Kolber, 1988b; Fischer, 2001; Zhong, 2007).
B-1a cells provide a unique element in the foundation of immune defense. B-1a cells spontaneously secrete immunoglobulin (Ig) and are responsible for the majority of non-immune serum IgM and substantial amounts of “resting” IgA (Forster and Rajewsky, 1987; Kroese et al., 1993; Sidman et al., 1986; Tumang et al., 2005). The inherent and constitutive secretion of Ig by B-1a cells, in the absence of direct stimulation, distinguishes B-1a cells from B-2 cells, and separates the native functional activities of these two B cell populations. B-1a cell-derived Ig generally adheres more closely to the germline state than B-2 cell Ig, as a result of diminished somatic mutation and reduced length of non-templated N-insertions, and is thus repertoire restricted (Forster et al., 1988; Gu et al., 1990). B-1a cell Ig is often found to recognize microbial cell wall determinants, such as phosphorylcholine derived from S. pneumoniae, against which it is protective (Boes et al., 1998; Haas et al., 2005; Klinman and Holmes, 1990). This has led to the accepted notion that B-1a cells produce “natural” antibody, representing a set of broadly reactive specificities encoded in the germline and evolutionarily retained that provides (at low affinity) serological protection against a range of microorganisms prior to the immunization that accompanies microbial pathogenesis. Evidence that natural Ig plays a key role in limiting microbial and viral dissemination and insuring the survival of infected animals has produced a new appreciation of the importance of B-1a cells to the overall scheme of immunity and a renewed emphasis on understanding the nature of B-1a cells (Baumgarth et al., 2000; Boes et al., 1998; Briles et al., 1982; Forster and Rajewsky, 1987; Haas et al., 2005).
One of the more curious distinguishing features of naïve B-1a cells lies in constitutive expression of phosphorylated and activated ERK (Wong et al., 2002). B-1a cell expression of pERK distinguishes B-1a cells from naïve B-2 cells, which do not normally express pERK, whereas ERK phosphorylation is induced in B-2 cells following B cell receptor engagement. It has been suggested that B-1a cells show signs of previous activation, which might provide some explanation for constitutive pERK. Although some findings are consistent with this idea (eg, elevated CD44 expression), many other markers of lymphocyte activation (eg, elevated CD69 expression) are lacking. Thus, B-1a cells cannot be categorized as an activated form of B-2 cells, and this has been confirmed by the recent identification of a distinct B-1 cell progenitor establishing that B-1a cells constitute a distinct B cell lineage (Montecino-Rodriguez et al., 2006), as was proposed years ago (Herzenberg, 2000). Further, the transcriptional “signature” of “resting” B-1a cells is not the same as that of anti-Ig-stimulated B-2 cells, further confirming that B-1a cells are not similar to activated B-2 cells (unpublished observations). Finally, constitutive B-1a cell expression of pERK is not accompanied by constitutive expression of activated forms of signaling mediators that would be expected if pERK were produced by B-1a cell “activation”. For these reasons, the presence of pERK in B-1a cells has been considered to reflect isolated ERK activation, possibly as a result of aberrant MAPKK activity, or as a reflection of previous activation events that have long since run their course and are no longer present.
We have now evaluated the origin of B-1a cell pERK. In B-2 cells, the pathway leading to BCR-triggered ERK phosphorylation begins with src kinase activation and propagates via Syk kinase and a collection of intermediaries termed the signalosome that includes phosphoinositide-3-kinase (PI-3K), and phospholipase Cgamma2 (PLCγ2) (Fruman et al., 2000). Inhibition of these mediators blocks BCR-induced ERK phosphorylation in B-2 cells (Jacob et al., 2002). We considered the possibility that dynamic operation of this pathway might, in fact, be responsible for the presence of phosphorylated ERK in “resting”, unstimulated B-1a cells, despite the fact that B-1a cells fail to express many criteria of “activation”. To address this issue, we examined the template of BCR-triggered intracellular signaling to query the basis for constitutive pERK in B-1a cells (Morris and Rothstein, 1993; Rothstein and Kolber, 1988a; Wong et al., 2002). Our results indicate that ERK phosphorylation represents a downstream consequence of continual activation of intracellular signaling elements.
Materials and Methods
Mice
Male BALB/cByJ mice of 6–8 weeks age were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were cared for and handled in accordance with National Institutes of Health and institutional guidelines. All studies were approved by the Institutional Animal Care and Use Committee.
B Cells
B-1a cells were purified from peritoneal washout cells by positive selection for B220loCD5+ after immunofluorescent staining and fluorescence activated cell sorting (FACS), or were purified by negative selection using a combination of anti-Thy1.2+C treatment plus plate adherence, as previously described (Frances et al., 2005). Experiments presented in Figures 1–3 were performed first with B-1 cells obtained by negative selection (3 or more times each) and then confirmed at least once with sort-purified B-1a cells. B-1a cells isolated through both purification methods yielded exactly the same results for each experiment. Experiments presented in Figures 4, 5, and 6 were performed exclusively on sort-purified B-1a cells. During FACS, doublets were stringently excluded by sequentially identifying singlets initially on the basis of forward scatter height versus width (FSC-H v FSC-W) and then side scatter height versus width (SSC-H v SSC-W). In similar fashion, B-2 cells were purified from spleen cell suspensions by positive selection for B220+CD5- via FACS, or negative selection with anti-Thy1.2+C treatment, as described (Frances et al., 2005).
Western immunoblot analysis
Proteins were extracted from B cell pellets with NP-40 lysis buffer. In each experiment, equal amounts of protein for each condition were subjected to SDS-PAGE followed by immunoblotting, as previously described (Mizuno and Rothstein, 2005).
Immunoprecipitation
Whole cell lysates were precleared with protein G Sepharose (Amersham Biosciences) and immunoprecipitated as previously described (Mizuno and Rothstein, 2005). Resultant immunoprecipitates were subjected to SDS-PAGE and Western blotting as described above.
Phosphoflow analysis
Intracellular phosphospecific flow cytometry and fluorescent cell barcoding was performed as previously described (Irish et al., 2004). In brief, to examine B-1a and B-2 cells (Figure 4), B cell populations were sorted from peritoneal washout and spleen cells, respectively, were resuspended in RPMI-1640 medium, and were left untreated or were treated with 25 μM pervanadate, as previously described (Wienands et al., 1996). Cells were then fixed, permeablized, and stained with Alexa-647-conjugated pY-Syk (clone 17a/P-ZAP70, BD Biosciences) or Alexa-647-conjugated pY-PLCγ2 clone (clone K86-689.37, BD Biosciences). To compare B-1a and B-1b cells (Figure 5), peritoneal washout cells were resuspended in RPMI-1640 medium, and were left untreated or were treated with 25 μM pervanadate, as previously described (Wienands et al., 1996). Cells were then fixed, permeabilized, and stained with PE-conjugated anti-B220, PE-Cy5-conjugated anti-CD5, and either Alexa-647-conjugated pY-Syk or Alexa-647-conjugated pY-PLCγ2. B-1a cells were identified by gating on B220loCD5+ cells and B-1b cells were identified by gating on B220loCD5- cells. Flow cytometric analysis was performed using a BD Biosciences LSR II.
Reagents
Affinity-purified F(ab’)2 fragments of polyclonal goat anti-mouse IgM were obtained from Jackson ImmunoResearch Laboratories. Anti-phospho-ERK1/2 (Thr202/Tyr204), anti-ERK1/2, anti-phospho-Syk (Tyr352), and anti-Syk were obtained from Cell Signaling Technology. Anti-PLCγ2 and secondary Abs for immunoblotting were obtained from Santa Cruz Biotechnology. Anti-phosphotyrosine antibody was obtained from Millipore. LY294002, wortmannin, U73122, PP2, PP3, and Syk inhibitor were obtained from Calbiochem. PE-labeled anti-CD86, FITC-labeled anti-B220, PE-labeled anti-B220, and PE-Cy5-labeled anti-CD5 were obtained from BD Pharmingen.
Results
Constitutive pERK is rapidly lost in B-1a cells exposed to inhibitors of src kinases, Syk, PI-3K and PLCγ2
We speculated that constitutive elevation of pERK in B-1a cells might reflect an ongoing process rather than an isolated condition or a residual outcome of previous activation events, and, more specifically, might reflect an end result of continual signal propagation. If so, then interruption of the signaling process would lead to a decrease in pERK generation, producing a decline in total pERK, because the initial baseline level must of necessity represent the balance between production and degradation. To address the role of key signaling molecules in constitutive ERK activation, we treated naïve B-1a cells with one of several metabolic inhibitors for one hour, after which cell lysates were prepared and examined for levels of pERK and ERK by Western immunoblotting. B-1a cells were compared to B-2 cells, with or without prior inhibitor treatment, in which ERK phosphorylation was then induced by stimulation with anti-Ig for 10 minutes. Results are shown in Figure 1. Naïve B-2 cells did not express detectable pERK; whereas pERK was strongly induced in B-2 cells by brief stimulation with anti-Ig. Further, as expected, the anti-Ig-induced upregulation of pERK in B-2 cells was substantially blocked by interference with src kinase activity using the inhibitor, PP2, whereas treatment with the inactive analog PP3 had no effect. The anti-Ig-induced upregulation of pERK in B-2 cells was also interrupted by inhibition of Syk (Syk chemical inhibitor), PI-3K (LY294002, wortmannin), and PLC (U73122). In direct contrast, naïve B-1a cells expressed substantial amounts of pERK in the absence of any stimulation or manipulation, as previously reported (Wong et al., 2002). However, we found that this constitutive pERK was completely lost within one hour of B-1a cell treatment with the same src kinase, Syk, PI-3K and PLC inhibitors that blocked anti-Ig-induced pERK in B-2 cells. As with anti-Ig-stimulated B-2 cells, PP3 had no effect. In further experimentation we found that B-1a cell treatment with either the PI-3K or Syk inhibitors for as little as 15 minutes led to a substantial decline in pERK (data not shown). These results strongly suggest that B-1a cell pERK represents the distal outcome of a dynamic process involving well-known components of receptor-triggered intracellular signaling.
Figure 1. Constitutive pERK is rapidly lost in B-1 cells exposed to inhibitors of key signaling elements.
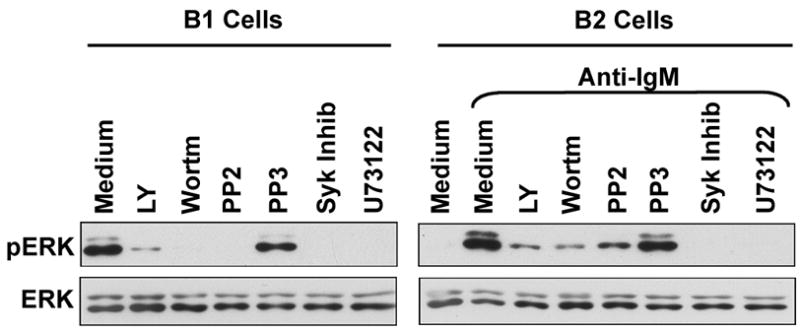
(Left hand panel) B-1 cells were cultured in medium alone (Medium); were treated with PP2 (20 μM), PP3 (20 μM), Syk inhibitor (Syk inhib, 10 μM), LY294002 (LY, 10 μM), wortmannin (wortm, 25 nM), or U73122 (0.6125 μM), for 1 hour. Whole cell extracts were prepared and Western blotted with anti-pERK antibody. Blots were stripped and reprobed with anti-ERK antibody to verify equal loading. (Right hand panel) B-2 cells were cultured in medium alone (Medium); were stimulated by anti-IgM (aIgM, 15 μg/ml) for 10 minutes; or were treated inhibitors for 1 hour as described above followed by anti-IgM for 10 minutes. Whole cell extracts were prepared and Western blotted as above. One of three comparable experiments is shown.
B-1a cells accumulate pSyk in the presence of phosphatase inhibition
The results above suggest that B-1a cells experience spontaneous signaling, which, if true, should be reflected in phosphorylation of intracellular mediators. To address this issue, we examined the phosphorylation status of Syk as an index protein. The representative pTyr352 site was evaluated in both B-1a and B-2 cells, in the presence or absence of anti-Ig stimulation. Results are shown in Figure 2A. As expected, B-2 cells expressed little pSyk at baseline, whereas BCR stimulation induced substantial Syk phosphorylation in B-2 cells. Surprisingly, we found that B-1a cells also expressed little baseline pSyk but, as in B-2 cells, Syk phosphorylation in B-1a cells was substantially induced by anti-Ig treatment. Thus, these experiments did not provide evidence for baseline activation of intracellular signaling elements in B-1a cells. However, in an actively signaling B cell, the level of pSyk at any given time represents the net balance between phosphorylation and dephosphorylation. We considered the possibility that Syk is actively phosphorylated, and dephosphorylated, continually in B-1a as opposed to B-2 cells. To test this hypothesis, we repeated the assessment of pTyr352Syk in B-1a and B-2 cells with or without treatment with the phosphatase inhibitor, sodium orthovanadate, for 30 minutes. Results are shown in Figure 2B. Phosphatase inhibition did not alter the pattern of pSyk in B-2 cells, where pSyk was not apparent at baseline but was induced by BCR stimulation. In marked contrast, we found that the addition of sodium orthovanadate dramatically increased the baseline level of pSyk in otherwise unmanipulated B-1a cells. Although it appears that the Syk content of B-1 cells is greater than that of B-2 cells, this has not been a consistent finding (see Figure 3), and, separately, examination of B-1 and B-2 cell lysates containing similar amounts of actin and calnexin showed that these populations contained similar amounts of Syk (data not shown). These results strongly suggest that Syk is being rapidly phosphorylated, and just as rapidly dephosphorylated, in B-1a cells, but not in B-2 cells, meaning that phosphorylated Syk protein is continually turning over in B-1a cells consistent with continual intracellular signaling.
Figure 2. B-1 cells accumulate pSyk in the presence of phosphatase inhibition.
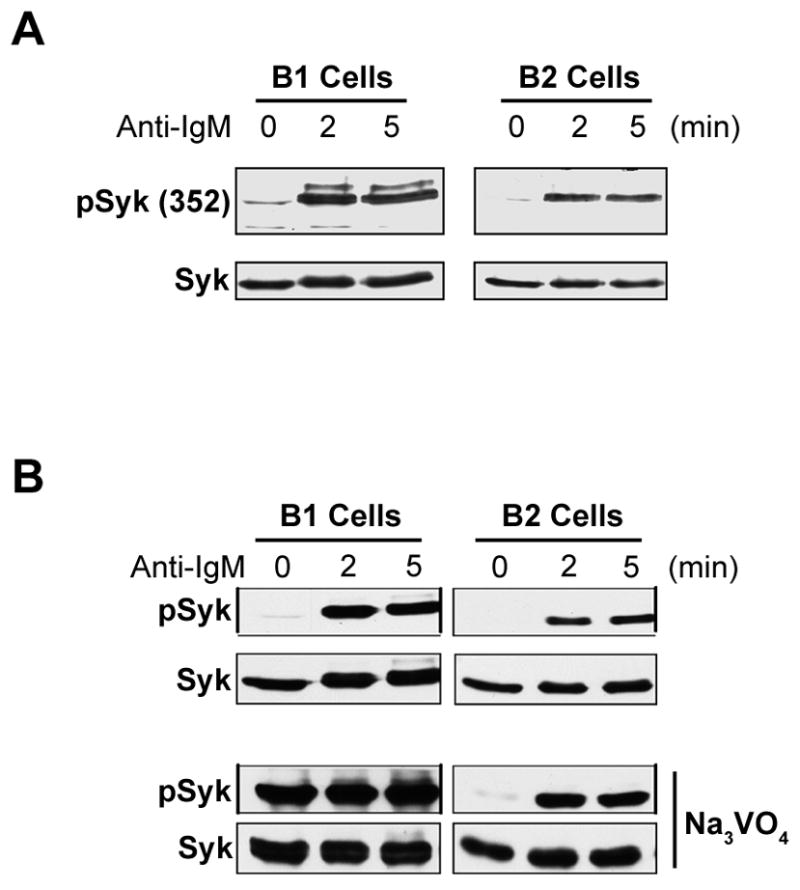
A) B-1 and B-2 cells were unstimulated (0) or were stimulated with anti-IgM (15 μg/ml) for 2 or 5 minutes. Whole cell lysates were then prepared and Western blotted sequentially with antibodies specific for tyrosine phosphorylated Syk (Tyr352) and for unphosphorylated Syk. One of five comparable experiments is shown. B) B-1 and B-2 cells were unstimulated (0) or were stimulated with anti-IgM as described above (top panels); and, B-1 and B-2 cells were treated with Na3VO4 (2 mM) for 30 minutes in the absence or presence of anti-IgM for 2 and 5 minutes. Whole cell lysates were then prepared and Western blotted with anti-pSyk352 antibody. Blots were stripped and reprobed with anti-Syk antibody to verify equal loading. One of five comparable experiments is shown.
Figure 3. Inhibition of src kinase activity blocks orthovanadate-enhanced accumulation of pSyk and pPLC.
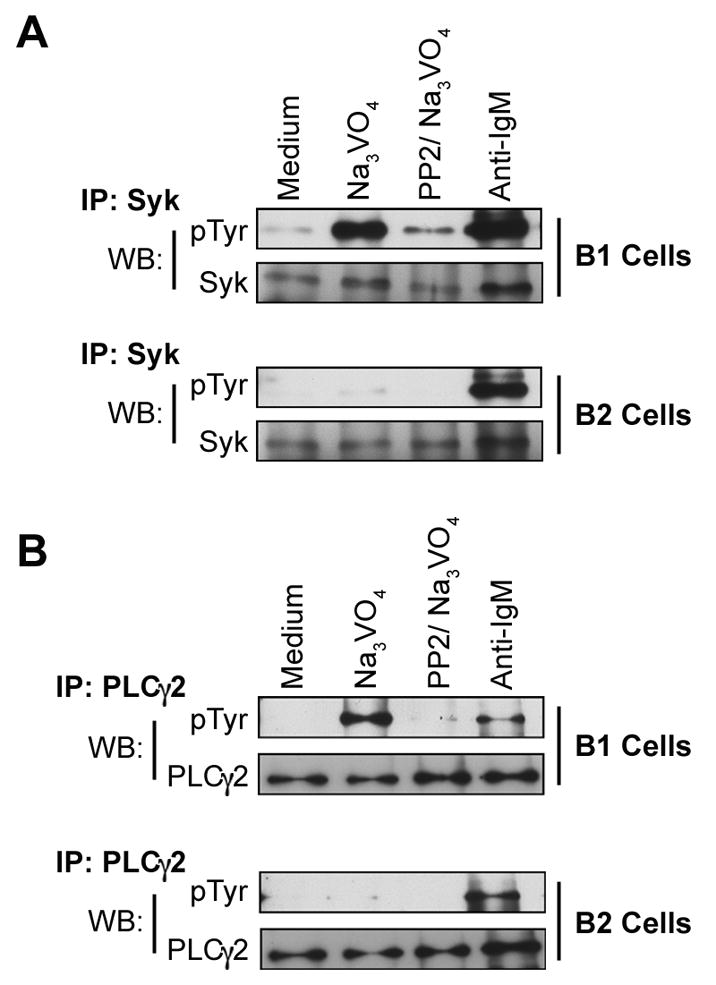
B-1 and B-2 cells were either untreated (Medium) or were treated with Na3VO4 (2 mM, 15 min); PP2 (20 μM, 1 hour) followed by Na3VO4 (2 mM, 15 min); or anti-IgM (65 μg/ml, 2 min). Whole cell lysates were prepared, pre-cleared with protein G, and then immunoprecipitated with anti-Syk (A) or anti-PLCγ2 (B) antibody and protein G agarose. Immunoprecipitates were Western blotted with anti-phosphotyrosine antibody 4G10. Blots were stripped and reprobed with anti-Syk (A) or anti-PLCγ2 (B) to verify equal loading. One of three independent experiments is shown.
Inhibition of src kinase activity blocks orthovanadate-enhanced accumulation of pSyk and pPLCγ2
Syk is a signaling intermediary that is phosphorylated by src kinase activity (Tamir and Cambier, 1998); if orthovanadate-enhanced pSyk in B-1a cells reflects ongoing signal propagation, it should be diminished by src kinase inhibition. To determine whether orthovanadate-enhanced pSyk results directly from the activation of more proximal signaling mediators, we examined the phosphorylation status of Syk in the presence of sodium orthovanadate, with or without prior PP2 treatment for one hour. In these experiments, Syk was immunoprecipitated and then immunoblotted with anti-phosphotyrosine in order to detect all tyrosine phosphorylation events. Results are shown in Figure 3A. As noted above, phosphatase inhibition with orthovanadate revealed substantial accumulation of pSyk in B-1a but not in B-2 cells. We found that initial treatment with the src kinase inhibitor, PP2, markedly reduced the level of orthovanadate-enhanced pSyk in B-1a cells, strongly suggesting that pSyk generation in B-1a cells depends on src kinase activity. Inasmuch as Syk is a relatively proximal intracellular mediator, we examined the same issues in PLCγ2, a more distal signaling component. Results are shown in Figure 3B and completely parallel the results described for Syk. Thus, as with pSyk, phosphatase inhibition with orthovanadate produced substantial accumulation of pPLCγ2 in B-1a but not in B-2 cells. And again, as with pSyk, we found that initial treatment with the src kinase inhibitor, PP2, markedly reduced the level of orthovanadate-enhanced pPLCγ2 in B-1a cells, suggesting that generation of pPLCγ2, like pSyk, depends on src kinase activity.
Phosphatase inhibition enhances pSyk and pPLCγ2 in the bulk of B-1a cells
To rule out the possibility that orthovanadate-enhanced pSyk was present in only a small fraction of B-1a cells (and thus might be attributed to an unusual subpopulation) we evaluated the number of B-1a cells in which constitutive signaling for Syk phosphorylation takes place by phosphoflow analysis (Irish et al., 2004). Baseline (uninhibited) and phosphatase-inhibited B-1a and B-2 cells were fixed and permeabilized and stained intracellularly with anti-pSyk (or with control) antibody, after which B cells were examined by flow cytometry. In these experiments phosphatase activity was blocked with pervanadate, a more rapidly acting inhibitor as compared to orthovanadate and one that is particularly suited to flow cytometric analysis (Wienands et al., 1996). Results are shown in Figure 4. In the absence of pervanadate, B-1a cells expressed a low level of pSyk; that is, staining with anti-pSyk was somewhat greater than staining with control antibody. Upon addition of pervanadate to B-1a cells there was a rapid and marked increase in the level of pSyk. These findings contrast sharply with results obtained with B-2 cells. B-2 cells showed no evidence of specific staining for pSyk at baseline; further, addition of pervanadate to B-2 cells produced only a small increase in pSyk. Thus, 2 minutes after pervanadate treatment, the difference in MFI between specific and nonspecific staining indicates that B-1a cells contained 5 times as much pSyk as B-2 cells, as shown in Figure 4B. Moreover, pSyk increased in virtually the whole population of B-1a cells, rather than in a subpopulation. We further examined the same issues in PLCγ2, a more distal signaling component. Results are shown in Figures 4C and 4D and parallel the results described for pSyk. Thus, as with pSyk, pPLCγ2 accumulated to a much greater extent in pervanadate-treated B-1a as compared to B-2 cells, with the difference in MFI for anti-pPLCγ2 and control staining amounting to 5-fold greater in B-1a versus B-2 cells within 2 minutes of pervanadate treatment. And again, as with pSyk, we found that pPLCγ2 increased in virtually the whole population of B-1a cells, rather than in a subpopulation. These findings fully support and complement the western blot results described above. Further, these results demonstrate the dynamic nature of Syk and PLCγ2 phosphorylation in B-1a cells and eliminate the possibility that pSyk and pPLCγ2 detected by western blotting represent events occurring in a minor subset of purified B-1a cells.
Figure 4. Phosphatase inhibition enhances pSyk and pPLCγ2 in the bulk of B-1a cells.

Sort-purified B-1a and B-2 cells were untreated or treated with pervanadate (25 μM) for 1 or 2 minutes. B cells were then fixed, permeabilized and stained with anti-pSyk and with control antibody (A), or with anti-pPLCγ2 and control antibody (C). The difference in MFI between anti-pSyk and control antibody is shown for B-1a and B-2 cells in (B). The difference in MFI between anti-pPLCγ2 and control antibody is shown for B-1a and B-2 cells in (D). One of 3 comparable experiments is shown.
CD5 does not regulate continual signaling in B-1a cells
CD5 has been reported to modulate BCR-mediated signaling in B-1 cells through association with SHP-1 (Bikah et al., 1996; Sen et al., 1999), although B-1b cells that lack CD5 expression are as indolent to BCR crosslinking as B-1a cells (Sen et al., 2002). To address the possibility that CD5 is involved in cycling phosphorylated Syk and PLCγ2 to the non-phosphorylated state, we compared pSyk and pPLCγ2 in B-1a and B-1b cells, at baseline and after phosphatase inhibition. Results are shown in Figure 5. B-1a and B-1b cells were similar to each other in pSyk and pPLCγ2, both before and after addition of pervanadate, and differed dramatically from B-2 cells. These results indicate that dynamic, continual signaling that finds its expression in constitutive ERK phosphorylation and becomes further apparent following phosphatase inhibtion is a property of B-1 cells per se and does not depend on, and is not regulated by, CD5.
Figure 5. CD5 does not regulate continual signaling in B-1a cells.

Peritoneal washout cells were untreated or treated with pervanadate (25 μM) for 2 and 4 minutes. B cells were then fixed, permeabilized and stained with anti-B220, anti-CD5, anti-pSyk and with control antibody, or with anti-B220, anti-CD5, anti-pPLCγ2 and control antibody. The difference in MFI between anti-pSyk and control antibody (A), and between anti-pPLCγ2 and control antibody (B), is shown for B-1a (B220loCD5+) and B-1b (B220loCD5-) cells. One of 5 comparable experiments is shown.
Constitutive CD86 surface expression in B-1a cells is blocked by inhibiting PI-3K and Syk
Like pERK, the co-stimulatory molecule CD86 (B7.2) is elevated at baseline in B-1a as compared to B-2 cells (Tumang et al., 2004). Again, as with pERK, CD86 is upregulated in B-2 cells after BCR ligation and CD86 induction is blocked in B-2 cells by inhibition of signaling mediators such as Syk and PI-3K (Richards et al., 2001). In view of these parallels, we considered that constitutive elevation of B-1a cell CD86 might result from operation of the same signaling elements responsible for constitutive pERK in B1a cells and for CD86 upregulation in BCR-triggered B2 cells. To test this, we treated B-1a cells and anti-Ig-stimulated B-2 cells with inhibitors of Syk and PI-3K and assessed surface expression of CD86. Results are shown in Figure 6. B-2 cells expressed little CD86 in the absence of stimulation, whereas BCR engagement produced upregulation of CD86 expression and this induction was blocked by inhibition of PI-3K or Syk. Naïve B-1a cells expressed more CD86 than did naïve B-2 cells (Figure 6B), as expected (Tumang et al., 2004). Most importantly, we found that B-1a cell treatment with inhibitors of either PI-3K or Syk produced a substantial decline in constitutive CD86 expression. Mean values for 5 experiments are shown in Figure 6C. Moreover, the decline of CD86 in B-1a cells was reversed after removal of the inhibitor, as shown in Figure 6D, and did not reflect cellular damage. Thus, the constitutively elevated level of CD86 expression found in B-1a cells, like the constitutively elevated level of pERK, appears to depend on the activity of intracellular signaling mediators and is reduced by inhibition thereof.
Figure 6. Constitutive CD86 surface expression in B-1a cells is blocked by inhibition of PI-3K and Syk.

A) B-2 cells were cultured in medium alone, or were stimulated with anti-IgM (15 μg/ml) alone or in the presence of LY294002 (10 μM) or Syk inhibitor (10 μM), for 18 hours. Cells were immunofluorescently stained with anti-CD86 or irrelevant control antibody and evaluated by flow cytometry, as indicated. One of five comparable experiments is shown. B) B-1a cells were cultured in medium alone, or were treated with LY294002 or Syk inhibitor for 18 hours and then stained with anti-CD86 and analyzed, as above. One of five independent experiments is shown. C) Mean proportion of CD86+ B-1a cells is shown for various conditions across 5 experiments as outlined in (B), along with lines indicating the standard errors of the means. Significance was determined using an unpaired student’s t-test; **p=0.006, ***p=0.0002. (D) B-1a cells were cultured in medium alone or with LY294002, for 18 hours and stained with anti-CD86 and with isotype control antibody, or were cultured for 18 hours with or without LY294002 and then washed, cultured in medium for an additional 24 hours, and then stained with anti-CD86 and with isotype control antibody.
Discussion
We found that inhibition of key signaling elements eliminates “constitutive” expression of phosphorylated ERK, and, in keeping with this, we identified accumulation of phosphorylated Syk and PLCγ2 that is src kinase-dependent in the bulk of B-1a cells upon phosphatase inhibition. This provides evidence for constitutive operation of a B-1a cell signaling pathway, interruption of which led to a marked decline in CD86 expression. These results suggest a new paradigm in which some unique B-1a cell characteristics that are increased at baseline in comparison to B-2 cells (eg, pERK and CD86) result from continual activity of intracellular signaling elements, whose operation is clearly apparent when phosphatase activity is blocked. It is worth noting that not all unusual B-1a characteristics can be similarly explained; for example, the constitutively elevated level of pSTAT3 that characterizes B-1a cells (Karras et al., 1997) was not altered by inhibition of Syk, PI-3K or PLC (data not shown). In previous work we reported that B-1 cells express increased tyrosine phosphorylation (of unidentified proteins) in comparison with B-2 cells (Morris and Rothstein, 1994), but we determined that any signaling derived therefrom terminated at the level of PLC because PLC activity was diminished and induction of NF-κB and stimulation of proliferation failed completely (Morris and Rothstein, 1993; Morris and Rothstein, 1994). The latter conclusion must now be revised in light of our new results that demonstrate propagation of signaling from src kinase through Syk and PLC resulting in downstream ERK phosphorylation. Thus, we have shown for the first time that src kinase activity is coupled to ERK phosphorylation/CD86 expression in a spontaneous and continual fashion in unstimulated B-1a cells at baseline. This finding was achieved by inhibiting signaling mediators and reading out pERK, and by taking steps to reveal cryptic activation of Syk and PLCγ2 through blockade of phosphatase activity. It is possible, however, that constitutive signaling and signaling attendant to anti-Ig-induced BCR crosslinking follow different paths, with the former transmitted to ERK and the latter blocked at PLC.
Our results with orthovanadate/western blotting are exceptionally clearcut: B-1a cells accumulated substantial amounts of phosphorylated Syk and PLCγ2, whereas B-2 cells did not. Our results with pervanadate/phosphoflow also showed dramatic accumulation of phosphorylated Syk and PLCγ2 in B-1a (and B-1b) cells; however, there was some, though much less, accumulation of pSyk and pPLCγ2 in B-2 cells as well. It is unclear at this time whether B-2 cells completely fail to spontaneously activate intracellular mediators, or whether this process occurs at a much reduced rate in B-2 cells as compared to B-1a cells. It may be that pervanadate inhibits more extensively the B-2 cell complement of phosphatases than does orthovanadate, as these two reagents have somewhat different mechanisms of action (Huyer et al., 1997), and that this in turn provides the means to detect the lower rate of pSyk and pPLCγ2 generation in B-2 cells. This would be consistent with previous reports that antigen receptor signaling is continually required for B-2 cell viability (Lam et al., 1997; Monroe, 2006). Regardless of the technique involved, however, B-1a cells consistently accumulated a substantially higher level of pSyk and pPLCγ2 than did B-2 cells in the presence of phosphatase inhibition, and thus B-1a cells are readily distinguished from B-2 cells as much more active in terms of spontaneous (unprovoked) phosphorylation of signalosome mediators. B-1a cells did not, however, differ substantially from B-1b cells, indicating that CD5 does not play a role in these processes and suggesting that the high level of constitutive signaling observed in B-1a cells is a feature of the B-1 cell population per se.
It has been suggested that the B-1a cell population is repertoire-skewed and antigen-selected during development (Arnold et al., 1994; Carmack et al., 1990). This positive selection may depend on a unique effect of antigen binding on early B-1 lineage cells, in contradistinction to the negative effect of antigen binding on developing B-2 cells (Casola et al., 2004; Hardy, 2006; Watanabe et al., 1999). Regardless, the work presented here is a clear indication that an intracellular signaling cascade is turned on in mature B-1 cells and, further, that it operates at a higher rate in B-1 cells as compared to B-2 cells, for which it seems to be required. In B-1 cells as with B-2 cells, cellular effects presumably produced by BCR signaling are assumed to derive from antigen binding to the BCR, although in neither case has this been proven. To the extent that this is true, however, the higher rate of signaling in B-1 cells is unexpected inasmuch as B-1 cell immunoglobulin, though broadly reactive, is generally considered to bind with low affinity.
The tolerant state of anergic B cells has been reported to depend on continual antigen binding (Gauld et al., 2005; Goodnow et al., 1991), and, as observed in B-1 cells (Wong et al., 2002), this leads to constitutive expression of phosphorylated ERK in some, but not all, models for B cell tolerance (Cambier et al., 2007). This may suggest a superficial resemblance to B-1a cells. In fact, comparisons with anergic B cells are severely hampered by the great variability that exists in the signaling and other characteristics of B cells among various antigen-specific BCR transgenic models (Cambier et al., 2007). Moreover, any superficial similarity between B-1a cells and anergic B cells fades upon careful examination. Thus, B-1a cells express elevated levels of surface IgM whereas surface immunoglobulin levels are depressed in many models of B cell anergy (Goodnow et al., 1988; Roark et al., 1997). Similarly, B-1a cells express elevated levels of CD80 and CD86 whereas these interaction molecules are displayed at normal or diminished levels in many anergic B cell models (Cambier et al., 2007; Tumang et al., 2004). And although anergic B cells in one model system express slightly more CD5 than B-2 cells, the level of CD5 expression in B-1a cells far outweighs that of anergic B cells (Hippen et al., 2000).
Functionally, B-1 cells respond early and vigorously to stimulation by LPS, whereas in at least 3 different anergic B cell systems LPS stimulation is diminished or absent (Borrero and Clarke, 2002; Cooke et al., 1994; Fischer et al., 2001; Noorchashm et al., 1999). Moreover, “resting” primary B-1 cells express “resting” B-2-like levels of intracellular Ca++, as previously reported (Cohen and Rothstein, 1991; Sen et al., 2002), whereas anergic B cells express elevated Ca++ levels (Benschop et al., 2001; Healy et al., 1997)(although it has been reported that BCR transgenic B-1a cells express elevated intracellular Ca++ much like anergic B cells (Chumley et al., 2002)). Further, B-1a cells are highly active in allogeneic stimulation of CD4+ T cells (which appears to be attributable, in large part, to elevated expression of CD86), unlike anergic B cells (Ho et al., 1994; Zhong et al., 2007). Finally, and most persuasively, B-1 cells spontaneously secrete IgM (through a mechanism not available to B-2 cells, ref (Tumang et al., 2005)), and homeostatically proliferate to self-renew (in the absence of stimulation), and thus exist in a completely different state as compared to anergic B cells (Kantor et al., 1992; Klinman and Holmes, 1990; Tumang et al., 2005).
These considerations suggest that B-1 cells and B-2 cells are wired differently, such that contact with self-antigen leads to different outcomes. Although causality remains unproven, it appears that in B-1 cells this leads to increased expression of B7 family members and enhanced antigen presentation along with differentiation to modest immunoglobulin secretion with preservation of responsiveness to certain “danger” signals such as TLR activation. In contrast, it appears that in anergic B cells this leads to a more general shutdown in terms of innate responsiveness as well as stimulatory capacity for T cells. The idea that B-1 cells are inherently different from B-2 cells and anergic B cells fits well with the recent identification of a specific B-1 cell progenitor (Montecino-Rodriguez et al., 2006), which in turn suggests that B-1 cells represent a distinct lineage, a concept originally delineated on the basis of adoptive transfer experiments (Kantor and Herzenberg, 1993; Kantor et al., 1992). Our finding that continual signaling demonstrated in the presence of phosphatase inhibition exists in B-1b cells as much as in B-1a cells indicates that this property tracks with B-1 cell status and thus fits well with the idea that B-1 cells represent a distinct lineage.
The work here shows that in B-1a cells the finding of constitutive pERK expression is not a residual effect of signaling that has long since past, but rather represents the downstream outcome of continual intracellular signaling that starts with src kinase activity and which was cryptic and inapparent in many respects until evidenced by inhibition of signaling mediators and blockade of enzymatic dephosphorylation. Operation of this signaling pathway is responsible for elevated CD86 expression in B-1a cells which in turn accounts in large measure for the extremely high antigen presentation function of this population. In sum, continual signaling in B-1 cells has physiological consequences both for B cell features and for B cell/T cell interaction, and thus for immune system function.
Acknowledgments
This work was supported by Public Health Service grants AI29690 and AI60896 awarded by the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold LW, Pennell CA, McCray SK, Clarke SH. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J Exp Med. 1994;179:1585–95. doi: 10.1084/jem.179.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med. 2000;192:271–80. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity. 2001;14:33–43. doi: 10.1016/s1074-7613(01)00087-5. [DOI] [PubMed] [Google Scholar]
- Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. 1996;274:1906–9. doi: 10.1126/science.274.5294.1906. [DOI] [PubMed] [Google Scholar]
- Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188:2381–6. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrero M, Clarke SH. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J Immunol. 2002;168:13–21. doi: 10.4049/jimmunol.168.1.13. [DOI] [PubMed] [Google Scholar]
- Briles DE, Forman C, Hudak S, Claflin JL. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J Exp Med. 1982;156:1177–85. doi: 10.1084/jem.156.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol. 2007;7:633–43. doi: 10.1038/nri2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmack CE, Shinton SA, Hayakawa K, Hardy RR. Rearrangement and selection of VH11 in the Ly-1 B cell lineage. J Exp Med. 1990;172:371–4. doi: 10.1084/jem.172.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, Rajewsky K. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317–27. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- Chumley MJ, Dal Porto JM, Cambier JC. The unique antigen receptor signaling phenotype of B-1 cells is influenced by locale but induced by antigen. J Immunol. 2002;169:1735–43. doi: 10.4049/jimmunol.169.4.1735. [DOI] [PubMed] [Google Scholar]
- Cohen DP, Rothstein TL. Elevated levels of protein kinase C activity and alpha-isoenzyme expression in murine peritoneal B cells. J Immunol. 1991;146:2921–7. [PubMed] [Google Scholar]
- Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, Howard M, Goodnow CC. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–38. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer GM, Solt LA, Hastings WD, Yang K, Gerstein RM, Nikolajczyk BS, Clarke SH, Rothstein TL. Splenic and peritoneal B-1 cells differ in terms of transcriptional and proliferative features that separate peritoneal B-1 from splenic B-2 cells. Cell Immunol. 2001;213:62–71. doi: 10.1006/cimm.2001.1860. [DOI] [PubMed] [Google Scholar]
- Forster I, Gu H, Rajewsky K. Germline antibody V regions as determinants of clonal persistence and malignant growth in the B cell compartment. EMBO J. 1988;7:3693–703. doi: 10.1002/j.1460-2075.1988.tb03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster I, Rajewsky K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur J Immunol. 1987;17:521–8. doi: 10.1002/eji.1830170414. [DOI] [PubMed] [Google Scholar]
- Frances R, Tumang JR, Kaku H, Gurdak SM, Rothstein TL. B-1 cells express transgelin 2: unexpected lymphocyte expression of a smooth muscle protein identified by proteomic analysis of peritoneal B-1 cells. Mol Immunol. 2006;43:2124–9. doi: 10.1016/j.molimm.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Frances R, Tumang JR, Rothstein TL. B-1 cells are deficient in Lck: defective B cell receptor signal transduction in B-1 cells occurs in the absence of elevated Lck expression. J Immunol. 2005;175:27–31. doi: 10.4049/jimmunol.175.1.27. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Satterthwaite AB, Witte ON. Xid-like phenotypes: a B cell signalosome takes shape. Immunity. 2000;13:1–3. doi: 10.1016/s1074-7613(00)00002-9. [DOI] [PubMed] [Google Scholar]
- Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6:1160–7. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Brink R, Adams E. Breakdown of self-tolerance in anergic B lymphocytes. Nature. 1991;352:532–6. doi: 10.1038/352532a0. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–82. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- Gu H, Forster I, Rajewsky K. Sequence homologies, N sequence insertion and JH gene utilization in VHDJH joining: implications for the joining mechanism and the ontogenetic timing of Ly1 B cell and B-CLL progenitor generation. EMBO J. 1990;9:2133–40. doi: 10.1002/j.1460-2075.1990.tb07382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Hardy RR. B-1 B cell development. J Immunol. 2006;177:2749–54. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–18. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Stall AM, Herzenberg LA. Immunoglobulin-bearing B cells reconstitute and maintain the murine Ly-1 B cell lineage. Eur J Immunol. 1986;16:1313–6. doi: 10.1002/eji.1830161021. [DOI] [PubMed] [Google Scholar]
- Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–28. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA. B-1 cells: the lineage question revisited. Immunol Rev. 2000;175:9–22. [PubMed] [Google Scholar]
- Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B cells. J Exp Med. 2000;191:883–890. doi: 10.1084/jem.191.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WY, Cooke MP, Goodnow CC, Davis MM. Resting and anergic B cells are defective in CD28-dependent costimulation of naive CD4+ T cells. J Exp Med. 1994;179:1539–49. doi: 10.1084/jem.179.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–51. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Jacob A, Cooney D, Pradhan M, Coggeshall KM. Convergence of signaling pathways on the activation of ERK in B cells. J Biol Chem. 2002;277:23420–6. doi: 10.1074/jbc.M202485200. [DOI] [PubMed] [Google Scholar]
- Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol. 1993;11:501–38. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- Kantor AB, Stall AM, Adams S, Herzenberg LA. Differential development of progenitor activity for three B-cell lineages. Proc Natl Acad Sci U S A. 1992;89:3320–4. doi: 10.1073/pnas.89.8.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras JG, Wang Z, Huo L, Howard RG, Frank DA, Rothstein TL. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J Exp Med. 1997;185:1035–42. doi: 10.1084/jem.185.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman DM, Holmes KL. Differences in the repertoire expressed by peritoneal and splenic Ly-1 (CD5)+ B cells. J Immunol. 1990;144:4520–5. [PubMed] [Google Scholar]
- Kroese FG, Ammerlaan WA, Kantor AB. Evidence that intestinal IgA plasma cells in mu, kappa transgenic mice are derived from B-1 (Ly-1 B) cells. Int Immunol. 1993;5:1317–27. doi: 10.1093/intimm/5.10.1317. [DOI] [PubMed] [Google Scholar]
- Lalor PA, Herzenberg LA, Adams S, Stall AM. Feedback regulation of murine Ly-1 B cell development. Eur J Immunol. 1989;19:507–13. doi: 10.1002/eji.1830190315. [DOI] [PubMed] [Google Scholar]
- Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–83. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Rothstein TL. B cell receptor (BCR) cross-talk: CD40 engagement creates an alternate pathway for BCR signaling that activates I kappa B kinase/I kappa B alpha/NF-kappa B without the need for PI3K and phospholipase C gamma. J Immunol. 2005;174:6062–70. doi: 10.4049/jimmunol.174.10.6062. [DOI] [PubMed] [Google Scholar]
- Monroe JG. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat Rev Immunol. 2006;6:283–94. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol. 2006;7:293–301. doi: 10.1038/ni1301. [DOI] [PubMed] [Google Scholar]
- Morris DL, Rothstein TL. Abnormal transcription factor induction through the surface immunoglobulin M receptor of B-1 lymphocytes. J Exp Med. 1993;177:857–61. doi: 10.1084/jem.177.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DL, Rothstein TL. Decreased surface IgM receptor-mediated activation of phospholipase C gamma 2 in B-1 lymphocytes. Int Immunol. 1994;6:1011–6. doi: 10.1093/intimm/6.7.1011. [DOI] [PubMed] [Google Scholar]
- Noorchashm H, Bui A, Li HL, Eaton A, Mandik-Nayak L, Sokol C, Potts KM, Pure E, Erikson J. Characterization of anergic anti-DNA B cells: B cell anergy is a T cell-independent and potentially reversible process. Int Immunol. 1999;11:765–76. doi: 10.1093/intimm/11.5.765. [DOI] [PubMed] [Google Scholar]
- Richards JD, Dave SH, Chou CH, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J Immunol. 2001;166:3855–64. doi: 10.4049/jimmunol.166.6.3855. [DOI] [PubMed] [Google Scholar]
- Roark JH, Bui A, Nguyen KA, Mandik L, Erikson J. Persistence of functionally compromised anti-double-stranded DNA B cells in the periphery of non-autoimmune mice. Int Immunol. 1997;9:1615–26. doi: 10.1093/intimm/9.11.1615. [DOI] [PubMed] [Google Scholar]
- Rothstein TL. Cutting edge commentary: two B-1 or not to be one. J Immunol. 2002;168:4257–61. doi: 10.4049/jimmunol.168.9.4257. [DOI] [PubMed] [Google Scholar]
- Rothstein TL, Kolber DL. Anti-Ig antibody inhibits the phorbol ester-induced stimulation of peritoneal B cells. J Immunol. 1988a;141:4089–93. [PubMed] [Google Scholar]
- Rothstein TL, Kolber DL. Peritoneal B cells respond to phorbol esters in the absence of co-mitogen. J Immunol. 1988b;140:2880–5. [PubMed] [Google Scholar]
- Sen G, Bikah G, Venkataraman C, Bondada S. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur J Immunol. 1999;29:3319–28. doi: 10.1002/(SICI)1521-4141(199910)29:10<3319::AID-IMMU3319>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sen G, Wu HJ, Bikah G, Venkataraman C, Robertson DA, Snow EC, Bondada S. Defective CD19-dependent signaling in B-1a and B-1b B lymphocyte subpopulations. Mol Immunol. 2002;39:57–68. doi: 10.1016/s0161-5890(02)00047-0. [DOI] [PubMed] [Google Scholar]
- Sidman CL, Shultz LD, Hardy RR, Hayakawa K, Herzenberg LA. Production of immunoglobulin isotypes by Ly-1+ B cells in viable motheaten and normal mice. Science. 1986;232:1423–5. doi: 10.1126/science.3487115. [DOI] [PubMed] [Google Scholar]
- Tamir I, Cambier JC. Antigen receptor signaling: integration of protein tyrosine kinase functions. Oncogene. 1998;17:1353–64. doi: 10.1038/sj.onc.1202187. [DOI] [PubMed] [Google Scholar]
- Tumang JR, Frances R, Yeo SG, Rothstein TL. Spontaneously Ig-secreting B-1 cells violate the accepted paradigm for expression of differentiation-associated transcription factors. J Immunol. 2005;174:3173–7. doi: 10.4049/jimmunol.174.6.3173. [DOI] [PubMed] [Google Scholar]
- Tumang JR, Hastings WD, Bai C, Rothstein TL. Peritoneal and splenic B-1 cells are separable by phenotypic, functional, and transcriptomic characteristics. Eur J Immunol. 2004;34:2158–67. doi: 10.1002/eji.200424819. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Nisitani S, Ikuta K, Suzuki M, Chiba T, Honjo T. Expression levels of B cell surface immunoglobulin regulate efficiency of allelic exclusion and size of autoreactive B-1 cell compartment. J Exp Med. 1999;190:461–69. doi: 10.1084/jem.190.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienands J, Larbolette O, Reth M. Evidence for a preformed transducer complex organized by the B cell antigen receptor. Proc Natl Acad Sci U S A. 1996;93:7865–70. doi: 10.1073/pnas.93.15.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SC, Chew WK, Tan JE, Melendez AJ, Francis F, Lam KP. Peritoneal CD5+ B-1 cells have signaling properties similar to tolerant B cells. J Biol Chem. 2002;277:30707–15. doi: 10.1074/jbc.M202460200. [DOI] [PubMed] [Google Scholar]
- Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, Oukka M, Strom TB, Rothstein TL. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007;37:2400–4. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]