

Abstract

We report here the development of a corona discharge (CD) initiated electrochemical (EC) electrospray ionization (ESI) technique using a standard electrospray ion source. This is a new ionization technique distinct from ESI, electrochemistry inherent to ESI, APCI, and techniques using hydroxyl radicals produced under atmospheric pressure conditions. By maximizing the observable CD at the tip of a stainless steel ESI capillary, efficient electrochemical oxidation of electrochemically active compounds is observed. For electrochemical oxidation to be observed, the ionization potential of the analyte must be lower than Fe. Ferrocene labeled compounds were chosen as the electrochemically active moiety. The electrochemical cell in the ESI source was robust and generated ions with selectivity according to the ionization potential of the analytes and up to zeptomolar sensitivity. Our results indicate that CD initiated electrochemical ionization has the potential to become a powerful technique to increase the dynamic range, sensitivity and selectivity of ESI experiments.
Synopsis
Using a standard ESI source a corona discharge initiated electrochemical ionization technique was established resulting from the electrochemistry occurring at the CD electrode surface.
Introduction
Corona Discharge (CD) is a widely studied and applied electrical phenomenon where a gas surrounding a high voltage electrode forms an ionized gaseous plasma [1, 2]. Most mass spectrometrists are familiar with corona discharge in the context of APCI, where it is used to create an ionizing plasma [3].
When observed under ES conditions at the tip of the metal ES capillary, CD is mostly regarded as an unwanted side effect. In two of the reported cases the observed discharge resulted in atmospheric pressure chemical ionization (APCI) conditions where the discharge plasma formed product ions resulting from ion/molecule reactions [4, 5]. When operating at high potential energies under ES conditions, Van Berkel [4] noted a current surge measured using an ammeter (A). The observed current surge was attributed to CD. Van Berkel noted CD conditions were characterized by both currents in excess of 10-6 A and the presence of protonated cluster ions. Under these discharge conditions a degradation of the [M+H]+ ion intensity was observed. An increase in electrochemically generated radical cations ([M]·+) from neutral analyte molecules was not reported, however. CD was also considered unfavorable to good ES conditions by Hail and Mylchreest [6]. They designed a concentric gas desolvation tube surrounding the ES capillary to increase gas desolvation capabilities and to eliminate unwanted CD.
However, one aspect of CD ignored so far, as it relates to ES, is that CD could be an effective mechanism for electron removal from the metal tip of the ES capillary into the surrounding gas [7-9 and references cited within]. Earlier reports only consider the electrons, plasma and subsequent ion/molecule reactions produced by the discharge and do not consider the electrochemistry occurring at the metal discharge needle.
Maximizing the CD at the tip of the ES capillary might significantly enhance the amount of electrochemically generated radical cations observed in the ES spectrum by providing an efficient pathway for electron removal from the tip of the ES capillary. Optimizing CD likely will degrade the protonated species in the ES spectrum but in this paper we are interested in attempting to establish conditions where EC ionization becomes the dominant origin of ions in the ES spectrum. The observation that CD could create EC conditions directly in the ES ion source has so far not been addressed. Therefore, we sought to develop a sensitive and selective electrochemical ionization (ECI) method using the standard features of ES design and hardware. By optimizing rather than suppressing the observable CD, plasma physics theory suggested that an increase of [M]·+ intensity could be observed.
Previous approaches to enhancing EC capabilities to ES have been to exploit EC inherent to ESI as reported by van Berkel [4, 10-16] and Young [17-18] and to add an EC cell inline, upstream of the ES ion source as reported by Karst [19-26]. In the later case, the electrochemical cell is placed inline with the LC pump. Electrochemically generated ions can be detected by UV and/or mass spectrometry, but short-lived ions may undergo multiple reactions until they arrive at the detector. The additional limitation of this setup to isocratic elution has prevented its widespread use [26]. Nevertheless, these studies show the usefulness of EC in ES.
In this paper we describe our initial efforts in converting a normal ES stainless steel capillary into an efficient, robust electrochemical cell capable of high sensitivity gradient reversed phase high pressure liquid chromatography-mass spectrometry (RP HPLC-MS) analyses. After selecting the appropriate standard ES design and hardware, we explored conditions to create and optimize any observable CD. Then, we determined whether we were able to observe electrochemistry as a result of the CD processes. Then, we carried out proof-of-principle RP HPLC-MS EC experiments under standard gradient conditions and determined achievable sensitivity.
Experimental
Reagents and Materials
HPLC grade water and acetonitrile, ammonium bicarbonate 99.0%, formic acid 96%, dibasic potassium phosphate 99.9%, ethynylferrocene (E-Fc) 97%, oxaliplatin, carboplatin and reserpine 99.0% were purchased from Sigma Aldrich (St. Louis, MO). Aminoferrocene was purchased from TCI America (Portland, OR). The Ni capillary was from VICI Valco, Houston, TX. 50 mM Potassium phosphate buffer pH 7.5 in water was prepared as a stock solution.
Synthesis and labeling
N-(Ferrocenyl)iodoacetamide (Fc-IAA) was synthesized as described by Lo et. al. [27].
High Performance Liquid Chromatography (HPLC)
A Waters 1525u Binary HPLC was operated at a flow rate of 0.200 mL/min and 2 μL of Fc-IAA was injected using the Waters 2777 Sample Manager. The HPLC column was an Agilent (Santa Clara, CA) Zorbax-SB C3 2.1 × 150 mm, 5 μm particle size. The HPLC solvents were 0.2% formic acid in water (mobile phase A) and acetonitrile containing 0.2% formic acid (mobile phase B). A gradient was run starting from 0% mobile phase B to 99% mobile phase B within 10 min. The column was heated to 55 °C with a Model TL-105 column heater from Tomberline Instruments (Boulder, CO). The eluent was directed to the MS.
Mass Spectrometry
Mass spectrometry experiments were conducted using a Waters (Milford, MA) LCT Premier Time-of-Flight mass spectrometer. With the exception of Fc-IAA, samples were injected via the injection loop of the LCT and pumped with a 1:1 mixture of mobile phases A and B. Injection volume was 2 μL. Samples were dissolved in acetonitrile:water 1:1. All CD experiments were conducted using the standard Z-Spray™ orthogonal atmospheric pressure/electrospray ion source [28] with the exception that we removed the integrated LockSpray™ device. The following features of the Z-Spray source were determined to be critical to achieving a stable, intense CD capable of inducing electrochemical oxidations at the tip of the standard stainless steel electrospray capillary: (a) The knurled nut at the top of the Z-Spray source allows for adjustment of the stainless steel capillary and needed to extend 3 mm beyond the desolvation tube. (b) The capillary voltage needed to be maximized (5 KV). (c) The desolvation temperature needed to be maximized (500 C). (d) The Z-Spray source mounts to an adjustable base where the angle and distance of the electrospray capillary relative to the ion entrance cone needed to be adjusted to maximize the observable CD. (e) Upstream of the source was a PEEK insulator at the injector. (f) A 33 MOhm current limiting resistor between the ES capillary and the HV supply. All changes to ion source parameters were achieved using the “MS Tune” page of the MassLynx 4.0 software. All parameters except capillary voltage and desolvation temperature were set to normal operating values. The desolvation and cone gases were nitrogen and CO2 where noted. The cone gas flow was 30 L/hr and the desolvation gas flow was 300 L/hr.
APCI experiments were conducted using the Waters Ion Sabre probe with nitrogen as the desolvation gas at a flow rate of 300 L/hr. The desolvation probe temperature was 450°C and the discharge current was regulated at 8 μA.
Suppression of interaction with stainless steel surface experiments
Control injections contained 500 fmol of E-Fc except the 10x injection, which contained 5 pmol. Solvent was 1:1 water: acetonitrile (no formic acid). Sample injection contained 500 fmol of E-Fc. Solvent was a 10 mM solution of ammonium carbonate in 1:1 water: acetonitrile.
Current measurements
Current was measured with a Simpson model 923 microammeter (Simpson, Palatine, IL) and a Keithley model 480 picoammeter (Keithley Instruments, Cleveland, OH.)
Sensitivity (LOD) measurements
Fc-IAA was diluted in 100% HPLC water prior to injection to a final concentration of 50, 125, 500, 5000 zmol/μL. For the blank 2 μL 100% HPLC water was injected. The LCT was operated in full scan mode from 100-1000 Da with a cycle time of 1.0 seconds. The ToF was operated in positive ion V-mode with a resolution of 5000. The mass chromatogram of 368.9 Da in profile mode was plotted with a mass width of 0.2 Da. No data smoothing was used but the baseline was corrected using the MassLynx baseline subtract routine.
Theory
As depicted in Fig. 1A [29] the flow of electrons in a standard ES ion source is from the tip of the metal ES capillary to the HV power supply. Positively charged [M+H]+ ions are formed after solvent evaporation from highly charged droplets and subsequent droplet fission [30-33]. Leakage current can either flow to an upstream ground (in those instruments that have an upstream ground) or to the ground of the HV power supply (dependent on the source configuration).
Figure 1.
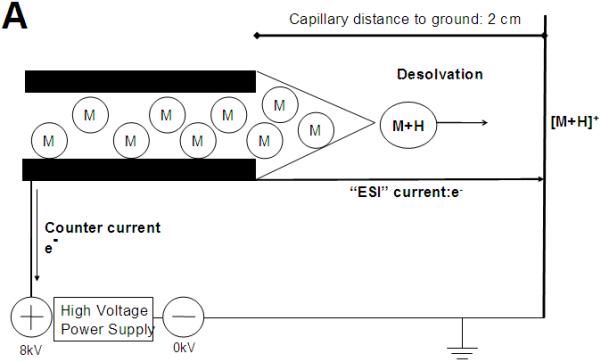
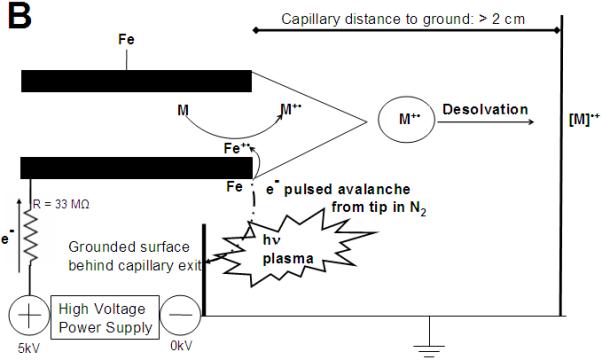
Comparison of (A) Electrospray Ionization and (B) Electrochemical Electrospray Ionization. (A). Electrons flow from the tip of the metal ES capillary to the HV power supply forming positively charged [M+H]+ after desolvation and droplet fission. Adapted from Kebarle [29] (B). Electrons with a high positive potential energy were removed from Fe (stainless steel) located at the capillary’s tip edge, oxidizing Fe to Fe·+, which subsequently ionizes all analytes with a lower ionization potential, generating a positively charged odd electron species [M]·+. Under CD conditions, high energy photons (hν) are emitted to the ground plate behind the capillary exit.
As shown in Fig. 1B and 2, our approach was fundamentally different: We have at least 1 GΩ resistance to the upstream ground, so the upstream ground is not a contributing factor in our setup. We also have a 33 MOhm current limiting resistor between the ES capillary and the HV supply eliminating any possible reverse current shown in Fig. 1A. Instead, we have established for the first time that CD at the tip of a stainless steel electrospray capillary is an effective means of electrochemically oxidizing redox-labeled compounds. For CD to occur, a sharp point surrounded by a gas in a high electric field is needed. The stainless steel capillary tube in the source has an unlimited number of sharp points at its tip. As shown in Suppl. Fig. S1, CD emission of electrons is characterized by high voltage and low currents (10-7 - 10-5 A), whereas the emission of electrons in a thermal arc occurs under just the opposite conditions, namely low voltage and high currents (100-10000 A) [1, 2]. CD seemed a better choice as the emitted electrons follow a non-Maxwellian energy distribution: the electrons with the highest energy are closest to the electrode and can thus easily remove an electron from the Fe in the electrode tip to induce an electrochemical reaction [34].
Figure 2.


(A). Overview of the commercial ion source. (B). Observed CD under ECI conditions. To achieve a stable CD, the stainless steel capillary needed to extend at least 3 mm beyond the desolvation tube. For all other settings see text and Table 1.
Our initial hypothesis was that we produce sufficient CD to ‘saturate’ the current output of the high voltage supply creating a condition at the capillary tip where CD would consume all of the available electrons from the power supply and scavenge additional electrons from the stainless steel tip subsequently creating an oxidizing environment, where one electron transfers could occur. We measured the current being consumed by the CD using a microammeter and found the maximum current to be less than 50 μA, far below the saturation value of the HV power supply (150 μA). Based on this current reading the HV power supply would have been able to provide a sufficient number of electrons to meet the demands of the CD. At a current reading of 50 μA per second saturation of the high voltage supply could not be responsible for the observed electrochemical oxidation.
We observed the blue light and the electron emission at the tip of the stainless steel ES capillary characteristic of CD, but these two processes do not explain how the observed electrochemistry can occur. However, fundamental studies by Trichel [35] and Morrow [36] provide details of the CD mechanism that can help explain the observed electrochemistry. These studies clearly demonstrate the oscillatory nature of CD. Briefly, the CD process can be described by the following series of events. As positive ions move away from the anode, an increasing electric field creates conditions at the tip of the electrode for a new electric surge, thus initiating a new current pulse [1, 2, 36]. At each current peak high-energy photons (hv) are being emitted and shortly thereafter, seed electrons are needed to trigger the next pulse. At one sharp point, the pulses occur at a frequency of approximately 1 MHz; and the width at half-height is approximately 10 ns. The ESI capillary tube has potentially thousands of points, where overlapping electron avalanches can and do occur. This is evident from the emission of blue light from the tip of the capillary as shown in Fig. 2B. In our configuration the power supply is limited to a current of 150 μA per second, which translates into 1.5 nA in any 10 μs interval. Under CD conditions, a series of narrow intense pulses of electrons are emitted and it is quite conceivable that the high voltage power supply is indeed reaching saturation but only for 10 ns intervals. It is, in fact, quite likely that the power supply is too slow to provide the seed electrons that are needed during these very short pulses. We propose that electrochemical oxidation is occurring at the apex of the pulses of the CD, where electrons were temporarily no longer available from the power supply and the necessary seed electrons were scavenged from the Fe atoms present in the stainless steel tip. This created a radical cation (Fe·+) at the tip and as analyte molecules came in contact with the oxidized Fe a subsequent oxidation reaction could occur in a collision with a molecule having an ionization potential equal to or less than the Fe/Fe·+. Once the analyte was oxidized by the Fe·+, the electrospray transported the analyte ion to the ion entrance orifice of the mass spectrometer.
As summarized in Table 1, the following parameters had to be adjusted to convert a standard ESI source to obtain ECI conditions, thereby essentially creating a dual mode use of the standard ESI source: The stainless steel capillary needed to extend at least 3 mm beyond the desolvation tube. The capillary voltage was set to 5 kV and the desolvation temperature was set to 500 °C. The LCT upstream injector comes standard with an insulated PEEK body. In addition, PEEK tubing (1 m, 127 μm) was used for the injections, so there was at least 1 GΩ resistance to ground preventing any counter current. The current was measured to flow exclusively from the HV power supply to the source and not vice versa (Suppl. Table 1). When these parameters were used, a thermal discharge (arcing) between the ESI capillary and the ion entrance was observed. Only when the ESI capillary was moved away (in z direction; see Fig. 2A) from the ion source entrance, a stable CD to the ground plate was observed as shown in Fig. 2B. It is important to note that the CD plasma was moved away from the ESI ion path, eliminating possible ion/molecule reactions as they are regularly observed in APCI or electric discharge setups [3, 37-39]. We also found the current limiting (33 MOhm resistor) standard HV power supply to be essential for this experimental setup. If the current is not limited, the current will increase beyond CD until it reaches the arcing regime of the current-voltage diagrams (see Suppl. Fig. S1). [2]
Table 1.
Parameters used for ESI and ECI/ESI conditions
| Parameter | ESI | ECI/ESI |
|---|---|---|
| Distance of stainless steel capillary to desolvation tube | max 0.5 mm | > 3 mm |
| Capillary Voltage | 2 kV | ≥ 5 kV |
| Desolvation temperature | 250°C | 500°C |
| Discharge | arcing | coronaa |
To achieve a stable CD and eliminate electrical discharges (arcing), the electrospray capillary needed to be moved away from the ion entrance cone
The following reactions take place in CD initiated ECI: as shown in equation (1) Fe from the SS metal tip is oxidized to Fe·+ and the free electron is needed as seed electron for electron avalanches observed in CD.
| (eq 1) |
Analytes (M) with lower ionization potential than Fe/Fe·+ will get ionized according to equation (2), while analytes with higher ionization potential than Fe/Fe·+ will not get ionized.
| (eq 2) |
While water clusters as shown in equation (3) and (4) likely occur, they are not part of the ionization process in CD-ECI, since the CD plasma is directed away from the ion path as shown in Fig. 1B and 2B. This is in contrast to APCI, where water clusters are the major source for ionization.
| (eq 3) |
| (eq 4) |
Taken together, we have designed an ECI/ESI source from a standard source without any hardware changes by adjusting capillary voltage, desolvation temperature, and the position of the stainless steel capillary to create a stable CD. Capillary position, the presence of a rear ground plate, and a current limiting resistor were essential to prevent the non-thermal plasma created by electrical discharge (arcing) from the capillary tip to the mass spectrometer’s ion entrance orifice from interacting with the liquid jet of the electrosprayed solution. In our model, an electron is removed from the Fe in the stainless steel capillary tip when new seed electrons are needed to maintain the characteristic oscillatory pulses during the CD. The Fe·+ then selectively removes an electron from those analytes that have a lower ionization potential than Fe/Fe·+, generating an M·+ species that is transported to the ion entrance orifice of the mass spectrometer. As shown in Fig. 1B and 2B, ion-molecule reactions that are typically observed in APCI setups are eliminated by directing the generated plasma away from the ion path to a ground plate behind the capillary.
Results and Discussion
According to the theoretical considerations organometallics with an ionization potential lower than that of Fe/Fe·+ should be oxidized. To test this hypothesis, a compound with an ionization potential lower than Fe was analyzed by ESI and ECI. As shown in Table 2, Fe has a first ionization potential of 7.83 eV, a second ionization potential of 16.5 eV, and a third ionization potential of 30.0 eV and ferrocene has an ionization potential of 6.8-7.6 eV (with variations according to the substituents). Thus, ferrocene is more likely to have an electron removed when compared to Fe/Fe·+, and Fe/Fe·+ is more likely to have an electron removed when compared to Fe·+/Fe++. Indeed, when E-Fc (I) was analyzed by ECI and ESI, it gave an intense peak at m/z 210.0 under ECI conditions (Fig. 3A) representing the ethynylferrocene cation, whereas no molecular ion was observed under ESI conditions (Fig. 3B).
Table 2.
| Ionization Potential (eV) | Compound |
|---|---|
| 6.80-7.6a | Ferrocene |
| 7.63 (1st) | Nickel |
| 7.72 (1st) | Copper |
| 7.83 (1st) | Iron |
| 7.88 | Reserpine |
| 8.9 | Platinum |
| 12.6 | Water |
| 16.5 (2nd) | Iron |
| 30.0 (3rd) | Iron |
exact ionization potential depends on the substituents on the cylcopentadienyl ring.
Figure 3.
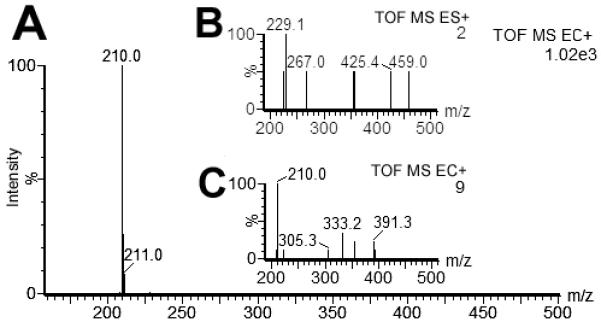
Comparison of the spectra for E-Fc (I; (C12H10Fe1; MR = 210.05 Da) under ECI (A, C) and ESI (B) conditions. Using 500 fmol (250 fmol/μL) of I under ECI conditions (A), the signal of 210 Da was intense; ESI did not show any [M+H]+ ion using 5 pmol (2.5 pmol/μL) of I. ECI (C) of E-Fc (500 fmol; 250 fmol/μL) showed a great reduction of signal intensity after the surface of the stainless steel capillary tip has been saturated with carbonate.
When corona discharge (with N2 CD showing an intense blue color) was observed at the tip of the stainless steel electrospray capillary, the pulsed electron avalanches occurring concomitant with light generation created an electron deficit resulting in the electrochemical oxidation of the Fe present in the stainless steel. The so-formed radical cation Fe·+ then reacted with an analyte that had a lower ionization potential than Fe/Fe·+. This ultimately resulted in the removal of an electron of the analyte (see above), thus ionizing it.
Organometallics that have an ionization potential higher than the first ionization potential of Fe should not be ionized by CD initiated ECI. Consistent with our proposed mechanism, platinum compounds such as oxaliplatin and/or carboplatin were not ionized under ECI conditions as they have a higher ionization potential than the first ionization potential of iron (Table 1, Suppl. Fig. S2). Only an [M+H]+ ion was observed under ESI conditions. This finding strongly supported the hypothesis that the first ionization potential of Fe was involved in the ionization mechanism. If the second or third ionization potential of Fe were involved in ECI, Pt compounds should also be ionized as the second and third ionization potential of Fe is higher than that of Pt. Similarly, if water had been oxidized in our setup, all compounds with an ionization potential lower than 12.6 eV would have been oxidized. This was not the case as the removal of one electron was selective to compounds that have a lower ionization potential than Fe/Fe·+ supporting the hypothesis that Fe/Fe·+ plays an important role in the ionization mechanism.
To further test the impact of Fe/Fe·+, a stainless steel capillary with a corroded (passivated) tip was used. Consistent with our hypothesis, no radical cations of E-Fc (I) were observed under ECI conditions (data not shown). This clearly supported the hypothesis that the nature of the capillary material effected ECI under CD conditions.
To further evaluate the impact of the capillary material, the following interaction suppression experiment was designed. It has been recently reported by Asakawa [40] that the interaction of analytes with the stainless steel surface can be suppressed by carbonate addition. The role of the carbonate is to complex the Fe in the stainless steel capillary that would otherwise form complexes with phosphate compounds. In an elegant experiment, Asakawa [40] used this suppression of interactions with the stainless steel surface to increase the signal intensity of phosphate compounds under ESI. We reasoned that carbonate should also be able to suppress the interaction of our analytes with the Fe on the tip surface, complexing the Fe+, which should lead to a significant decrease in signal intensity. Indeed, the peak intensity of (I) was drastically reduced under ECI when the interactions of the analytes with the stainless steel surface were suppressed by carbonate addition as shown in Fig. 3C. For E-Fc (I) to be oxidized to the radical cation, it must have contact to stainless steel surface tip (Fig. 1B) and a suppression of this interaction led to a significant decrease in signal intensity. Next, we tested whether the ionization was dependent on the CD by using CO2 as desolvation gas, which is commonly used to prevent CD because it has a higher electrical breakdown threshold [41]. When we used CO2 as desolvation gas no CD and no ECI of analytes were observed. ESI ions were observed as would be expected. This confirmed the importance of CD in the ionization process. To further challenge our model, we also used a Ni capillary instead of stainless steel capillary. Although we did observe CD, we did not observe ECI of ferrocene compounds (data not shown). Ni has a lower ionization potential than Fe, it thus cannot ionize ferrocene, supporting our current model.
According to our hypothesis in CD initiated ECI experiments, Fe/Fe·+ cannot oxidize reserpine (II; 1 pmol, C33H40N2O9; Mr = 608.68 Da) as reserpine has a higher ionization potential (7.88 eV) than Fe/Fe·+ (7.83 eV). When reserpine (II) was analyzed under ESI and ECI conditions, we did indeed not detect any oxidation products of reserpine (II), only the protonated species (m/z = 609) was observed under both, ESI and ECI conditions (Suppl. Fig. S3A and B). This underlines the fundamental differences between CD directed ECI and EC inherent to ESI, as the later technique can electrolyze water, which in turn is able to oxidize every analyte that has an ionization potential energy lower than water, including most organic compounds, such as reserpine (II) [42, 43]. Organic compounds usually have an ionization potential higher than 8 eV and will readily be oxidized by water according to equation (5)
| (eq 5), |
but cannot be oxidized by CD-ECI, which is selectively dependent on the ionization potential of the capillary material, in our case Fe/Fe+· at 7.83 eV.
CD initiated ECI is also not dependent on ion/molecule reactions. In fact, we eliminate ion/molecule reactions by directing CD plasma away from the ion path (Fig. 1B), and not into the ion path as is the case in electrical discharge sources shown in Suppl. Fig. S4 [37-39]. We have not observed any ions, which could be attributed to being the result of ion-molecule reactions such as the addition of oxygen or hydroxyl radicals.
The observed oxidations follow the relative ionization potential energies. In CD initated ECI, the electron transfer occurred directly from the electrode into the gas phase, so that no electrochemical oxidation of water was necessary for the ionization. Only those compounds that have a higher ionization potential energy are able to remove electrons from a compound with lower ionization potential energy giving this process a high selectivity towards analytes with lower ionization potential energy than the electrode material. In our case, we used a stainless steel capillary and Ni as electrodes. It should be noted that stainless steel is not a homogeneous material as it does contain other components than Fe, most prominently C, Cr, Ni, Mn, Mo, Si, P and S, which may also contribute to the ionization processes.
Now, that we established a new electrochemical ionization technique, we were interested to test its applicability for peptides and how ECI compares to standard ESI and APCI. Ferrocene labels are commonly used in electrochemical applications including those with in-line MS [44]. In fact, ferrocene labels are widely available for the ultrasensitive electrochemical detection of many compounds beyond the focus of our studies, including but not limited to peptides, proteins, carbohydrates, and drugs [26]. They are well characterized, making them an interesting tool to use in our studies [44-46]. To test whether peptides can also be measured under ECI conditions, peptide (III) was formed by reaction of a sulfhydryl bearing tripeptide, glutathione, with ferrocene iodoacetamide (IV). The resulting S-ferrocenyl labeled peptide (III) was investigated by ECI, ESI and APCI. Using our standard ECI conditions (III) (1 pmol; C22H28N4O7S1Fe1; MR = 548.39 Da) was analyzed by ECI-MS and gave an intense [M]·+ ion at m/z = 548 (Fig. 4A). Under ESI conditions, the signal intensity of the [M]·+ ion at m/z = 548 decreased at least ca. 75-fold and virtually no corresponding protonated species ([M+H]+) was observed when compared to ECI conditions (Fig. 4B). The remaining [M]·+ ion was attributed to low levels of dark discharge (Townsend regime) that occurs prior to CD (Suppl. Fig. S1) [1, 2]. Organometallics generally ionize poorly even under optimized ESI conditions highlighting the fundamental differences between ESI and CD initated ECI conditions. These experiments also show that the dual mode use of a standard ESI source while operating it in CD initiated ECI conditions enables the analysis of compounds that would otherwise not be amenable to standard ESI analysis.
Figure 4.
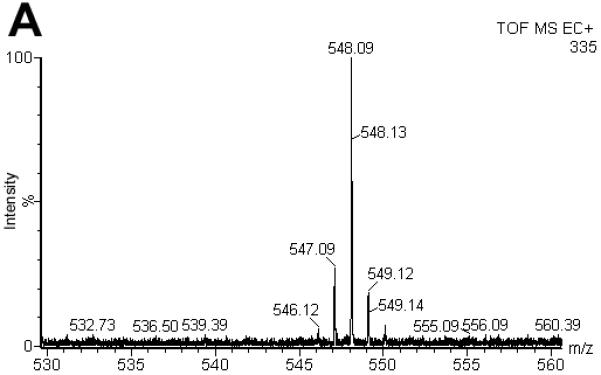
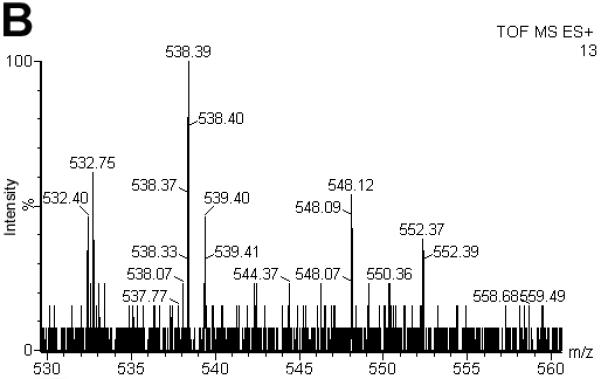
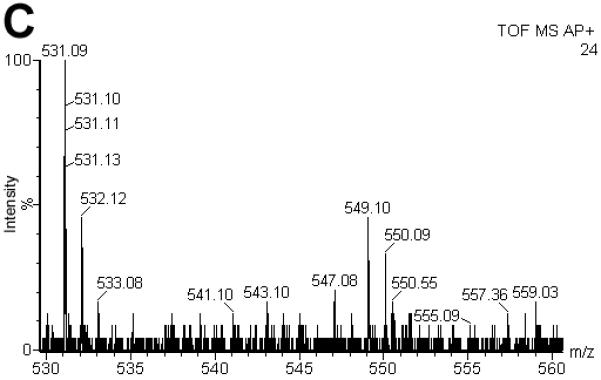
Comparison of the spectra for S-ferrocene labeled glutathione (II C22H29N4O7S1Fe1; MR = 548.39 Da) under ESI (A), ECI (B) and APCI (C) conditions. Under ECI conditions, the signal intensity of the [M]·+ ion at m/z = 548 increased at least 47-fold when compared to ESI conditions (B). It was also 2000 times more intense when compared to APCI. (C). Note, that for a signal to be obtained under APCI conditions, a 10 times more concentrated sample needed to be used. For the ESI and ECI experiments, 1 pmol (500 fmol/μL) of II was injected. For the APCI experiment, 100 pmol (50 pmol/μL) of II was injected.
Finally, (III) was investigated using the APCI Ion Sabre probe. As expected an [M+H]+ was observed. Interestingly, a 100-fold more concentrated sample was required to observe any signal. Thus, APCI had approximately a 2000-fold decreased sensitivity when compared to our ECI setup where [M]·+ was detected. Under APCI conditions, the CD plasma is generating large amounts of secondary reactant species (from water) like H3O+ and (H2O)nH+ resulting in the chemical ionization of the analyte. As mentioned earlier, to avoid this chemical ionization, we diverted the plasma away from the ion path under ECI conditions. The absence of any [M+H]+ and other ion/molecule adducts that are characteristic for APCI indicated this strategy to be successful in CD initiated ECI. The selective one-electron oxidation only produced radical cations that were directly transferred to the ion entrance orifice, thus avoiding multiple electron/ion and ion/ion reactions that could neutralize a significant number of freshly formed ions leading to tremendous increase in sensitivity in CD-ECI when compared to APCI. The increased sensitivity observed could also be attributed to a concentration effect where the analyte is accumulated at the tip of the capillary. From the carbonate suppression experiments we know that the analyte must interact with the stainless steel surface so it is plausible that analyte accumulates at the tip and is only released by the CD ionization process.
When electrochemically labeled peptides were investigated, CD-ECI was more sensitive than both, ESI and APCI. The ferrocene-based iodoacetamide label was chosen since iodoacetamide treatment is most commonly used to protect free cysteine groups in proteomics experiments prior to tryptic digestion, indicating the potential of this technique for very sensitive proteomics experiments. Particularly, the selective and ultrasensitive detection of ferrocene-labeled cysteine containing peptides that would result in a reduction of the complexity of the proteome as proposed by Aebersold [47] could benefit proteomics research.
To further explore whether ferrocene-based labels would be stable under gradient RP HPLC conditions ferrocene iodoacetamide (IV) (C12H12I1N1O1Fe1; MR = 368.98 Da) was analyzed using gradient RP HPLC. Gradient RP HPLC would be particularly advantageous as, in general, due to the slope of the background inline HPLC approaches coupled to electrochemical detection are limited to isocratic elution [26]. This is also true for a setup that uses a PFT cell as an upstream ground [14] since the upstream ground resistance is a function of the eluting solvent composition. In the ECI chromatogram obtained under CD conditions, the peak eluting at 8.7 minutes showed intense singly charged ions at m/z = 368.9 Da. This clearly indicated that the ferrocene-based labels were stable under standard gradient RP HPLC conditions. In contrast to previously established electrochemical detection, CD initiated ECI was independent of the slope of the background and thus gradient elutions can be readily used. Thus, this technique has overcome one of the major hurdles in LC-coupled electrochemical detection assays.
Finally, we investigated the sensitivity of CD initiated ECI. As shown in Fig. S5A, an injection of 250 zmol of (IV) on column gave a signal with 148 counts at 5000 resolution under full scan conditions. The comparison to a blank injection showed no interference (Fig. S5B). A linear relationship of the peak heights as a function of the injected amount was observed from 100 zmol to 10 amol (Fig S5C). To our knowledge, this is the highest sensitivity that has been so far reported using a regular, non-nanospray ES ion source highlighting the potential of this technique for ultra-sensitive measurements. It should be noted here that the achievable sensitivity in any electrochemical detection depends on the quality of the electrode and solvents.
Conclusion
We have established stable CD-initiated ECI conditions using a standard ESI source by adjusting capillary voltage, desolvation temperature, and the position of the stainless steel capillary, thus enabling a dual mode use for a standard ESI source. Using ferrocene-labeled compounds, the selective removal of one electron created an [M]·+ ion species. The CD-ECI conditions shown here represent a novel ionization technique with up to zeptomolar sensitivity for electrochemically labeled analytes not readily amenable to ESI.
Our observation of CD initiated electrochemical oxidations at the surface of a stainless steel electrospray capillary also provides a novel platform for further studies of surface electrochemistry where the oxidation products are desorbed from the capillary surface by the ESI process similar to the DESI ionization process [52, 53].
In future work, achievable CD-ECI sensitivity may be further improved by increasing capillary voltage and optimizing the ionizing surface area present at the ESI capillary tip, and by transferring this setup to a nanospray device. Successful ionization was dependent on the ionization potential of capillary material and analyte, which enables the design of different experiments by using an appropriate electrochemically active label or choosing an appropriate capillary material with an ionization potential that is higher or lower than that of the analyte(s) of interest to achieve selective ionization of compounds of interest. This novel, robust, yet flexible technique could be used as a supplementary technique to ESI to enhance the detectable dynamic range, e.g; by selectively analyzing cysteine containing peptides in a complex proteome [47].
Supplementary Material
Table 3. Figures of merit.
| Ionization mode | Sensitivity | Selectivity | Applicability | Multimode use |
|---|---|---|---|---|
| ESI | fmol | n/aa | peptides, proteins | yesc |
| APCI | pmol | n/ab | small molecules | yesd |
| CD-ECI | zmol | yes | ferrocene labeled biomolecules, small molecules | yesc |
CD-ECI achieves selectivity according to the ionization potential of analytes vs. capillary material, ESI and APCI do not have this option
under standard HPLC-MS conditions; it should be noted though that APCI can be made selective by changing the nature of the ion-molecule chemistry
ESI and CD-ECI are interchangeable without hardware change
Hardware change (APCI probe) necessary
Acknowledgements
This work was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, the Betty and Gordon Moore Foundation and the Beckman Institute. We thank Daniel Eikel for taking the photograph, Jenny TC Ho for discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chang JS, Lawless PA, Yamamoto T. Corona Discharge Processes. IEEE Transactions on Plasma Science. 1991;19:1152–1166. [Google Scholar]
- 2.Druyvesteyn MJ, Penning FM. The Mechanism of Electrical Discharges in Gases of Low Pressure. Rev. Mod. Phys. 1940;12:87–174. [Google Scholar]
- 3.Carroll DI, Dzidic I, Stillwell RN, Haegle KD, Horning EC. Atmospheric Pressure Ionization Mass Spectrometry: Corona Dicharge Ion Sources for Use in Liquid Chromatograph-Mass Spectrometer-Compyter-Analytical System. Anal. Chem. 1975;47:2369–2373. [Google Scholar]
- 4.Van Berkel GJ. The Electrolytic Nature of Electrospray. In: Cole RB, editor. Electrospray Ionization Mass Spectrometry. Wiley; New York: 1997. pp. 65–105. [Google Scholar]
- 5.Bruins AP. ESI Source Design and Dynamic Range Considerations. In: Cole RB, editor. Electrospray Ionization Mass Spectrometry. Wiley; New York: 1997. pp. 114–115. [Google Scholar]
- 6.Hail M, Mylchreest I. 5393975 U.S. Patent. 1995
- 7.Uhm HS, Lee WM. An Analytical Theory of Discharge Plasmas. Phys. Plasmas. 1997;4:3117–3128. [Google Scholar]
- 8.Gao L, Song Q, Noll RJ, Duncan J, Cooks RG, Ouyang Z. Glow Discharge Electron Impact Ionization Source for Miniature Mass Spectrometers. J. Mass Spectrom. 2007;42:675–680. doi: 10.1002/jms.1201. [DOI] [PubMed] [Google Scholar]
- 9.Higashi T, Takayama N, Nishio T, Taniguchi E, Shimada K. Procedure for increasing the detection responses of estrogens in LC-MS based on introduction of a nitrobenzene moiety followed by electron capture atmospheric pressure chemical ionization. Anal. Bioanal. Chem. 2006;386:658–665. doi: 10.1007/s00216-006-0371-z. [DOI] [PubMed] [Google Scholar]
- 10.Van Berkel GJ, Kertesz V. Using the Electrochemistry of the Electrospray Ion Source. Anal. Chem. 2007;79:5510–5520. doi: 10.1021/ac071944a. [DOI] [PubMed] [Google Scholar]
- 11.De la Mora JF, Van Berkel GJ, Enke CG, Cole RB, Martinez-Sanchez M, Fenn JB. Electrochemical processes in electrospray ionization mass spectrometry. J. Mass Spectrom. 2000;35:939–952. doi: 10.1002/1096-9888(200008)35:8<939::AID-JMS36>3.0.CO;2-V. J. Mass Spectrom. [DOI] [PubMed] [Google Scholar]
- 12.Van Berkel GJ, Quirke JME, Adams CL. Derivatization for electrospray ionization-mass spectrometry. 4. Alkenes and alkynes. Rapid Commun. Mass Spectrom. 2000;14:849–858. doi: 10.1002/(SICI)1097-0231(20000530)14:10<849::AID-RCM954>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 13.Kertesz V, Van Berkel GJ. Expanded use of a Battery-Powered Two-Electrode Emitter Cell for Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2006;17:953–961. doi: 10.1016/j.jasms.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Van Berkel GJ, Kertesz V, Ford MJ, Granger MC. Efficient analyte oxidation in an electrospray ion source using a porous flow-through electrode emitter. J Am Soc Mass Spectrom. 2004;15(12):1755–66. doi: 10.1016/j.jasms.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Van Berkel GJ, Kertesz V. Expanded Electrochemical Capabilities of the Electrospray Ion Source Using Porous Flow-Through Electrodes as the Upstream Ground and Emitter High-Voltage Contact. Anal. Chem. 2005;77:8041–8049. doi: 10.1021/ac051555l. [DOI] [PubMed] [Google Scholar]
- 16.Van Berkel GJ. Insights into analyte electrolysis in an electrospray emitter from chronopotentiometry experiments and mass transport calculations. J. Am. Soc. Mass Spectrom. 2000;11:951–960. doi: 10.1016/s1044-0305(00)00175-6. [DOI] [PubMed] [Google Scholar]
- 17.Williams D, Young MK. Analysis of neutral isomeric low molecular weight carbohydrates using ferrocenyl boronate derivatization and tandem electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2000;14:2083–2091. doi: 10.1002/1097-0231(20001130)14:22<2083::AID-RCM136>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 18.Williams D, Chen S, Young MK. Ratiometric analysis of the ferrocene boronate esters of 2- and 4-hydroxyestradiol by tandem electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2000;15:182–186. doi: 10.1002/1097-0231(20010215)15:3<182::AID-RCM208>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 19.Diehl G, Karst U. On-line electrochemistry--MS and related techniques. Anal Bioanal. Chem. 2002;373(6):390–8. doi: 10.1007/s00216-002-1281-3. [DOI] [PubMed] [Google Scholar]
- 20.Meyer J, Liesener A, Götz S, Hayen H, Karst U. Liquid chromatography with on-line electrochemical derivatization and fluorescence detection for the determination of phenols. Anal. Chem. 2003;75(4):922–6. doi: 10.1021/ac0204808. [DOI] [PubMed] [Google Scholar]
- 21.Seiwert B, Henneken H, Karst U. Ferrocenoyl piperazide as derivatizing agent for the analysis of isocyanates and related compounds using liquid chromatography/electrochemistry/mass spectrometry (LC/EC/MS) J. Am. Soc. Mass. Spectrom. 2004;15(12):1727–36. doi: 10.1016/j.jasms.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Seiwert B, Karst U. Analysis of cysteine-containing proteins using precolumn derivatization with N-(2-ferroceneethyl)maleimide and liquid chromatography/electrochemistry/mass spectrometry. Anal. Bioanal. Chem. 2007;388(8):1633–42. doi: 10.1007/s00216-007-1260-9. [DOI] [PubMed] [Google Scholar]
- 23.Seiwert B, Karst U. Simultaneous LC/MS/MS determination of thiols and disulfides in urine samples based on differential labeling with ferrocene-based maleimides. Anal Chem. 2007;79(18):7131–8. doi: 10.1021/ac071016b. [DOI] [PubMed] [Google Scholar]
- 24.Karst U. Electrochemistry/Mass Spectrometry (EC/MS) - A new tool to study drug metabolism and reaction mechanisms. Angew. Chem. Int. Ed. 2004;43:2476–2478. doi: 10.1002/anie.200301763. [DOI] [PubMed] [Google Scholar]
- 25.Seiwert B, Karst U. Analysis of cysteine-containing proteins using precolumn derivatization with N -(2-ferroceneethyl)maleimide and liquid chromatography/electrochemistry/mass spectrometry. Anal. Bioanal. Chem. 2007;388:1633–1642. doi: 10.1007/s00216-007-1260-9. [DOI] [PubMed] [Google Scholar]
- 26.Seiwert B, Karst U. Ferrocene-based derivatization in analytical chemistry. Anal. Bioanal. Chem. 2008;390:181–200. doi: 10.1007/s00216-007-1639-7. [DOI] [PubMed] [Google Scholar]
- 27.Lo KK-W, Lau JS-Y, Ng DC-M, Zhu N. Specific labelling of sulfhydryl-containing biomolecules with redox-active N-(ferrocenyl)iodoacetamide. J. Chem. Soc., Dalton Trans. 2002:1753–1756. [Google Scholar]
- 28.Bajic S. 5756994 U.S. Patent. 1996
- 29.Kebarle P, Tang L. From ions in solution to ions in the gas phase - the mechanism of electrospray mass spectrometry. Anal. Chem. 1993;65:972A–986A. [Google Scholar]
- 30.Kebarle P, Ho Y. On the Mechanism of Electrospray Mass Spectrometry. In: Cole RB, editor. Electrospray Ionization Mass Spectrometry. Wiley; New York: 1997. pp. 3–63. [Google Scholar]
- 31.Kebarle P, Peschke M. On the Mechanisms by which the Charged Droplets Produced by Electrospray Lead to Gas Phase Ions. Anal. Chim. Acta. 2000;406:11–35. [Google Scholar]
- 32.Kebarle P. A brief overview of the present status of the mechanisms involved in electrospray mass spectrometry. J. Mass. Spectrom. 2000;35:804–17. doi: 10.1002/1096-9888(200007)35:7<804::AID-JMS22>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 33.Fenn J. Electrospray wings for molecular elephants (Nobel lecture) Angew. Chem. Int. Ed. Engl. 2003;42:3871–94. doi: 10.1002/anie.200300605. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Davidson JH. Model of the Negative DC Corona Plasma: Comparison to the Positive DC Corona Plasma. Plasma Chemistry. and Plasma Processing. 2002;22:199–224. [Google Scholar]
- 35.Trichel GW. The Mechanism of the Positive Point-to Plain Corona in Air at Atmospheric Pressure. Phys Rev. 1939;55:382–390. [Google Scholar]
- 36.Morrow R. The theory of positive glow corona. J. Phys. D: Appl. Phys. 1997;30:3099–3114. [Google Scholar]
- 37.Maleknia SD, Downard K. Radical approaches to probe protein structure, folding, and interactions by mass spectrometry. Mass Spectrom Rev. 2001;20:388–401. doi: 10.1002/mas.10013. [DOI] [PubMed] [Google Scholar]
- 38.Maleknia SD, Chance MR, Downard KM. Electrospray-assisted modification of proteins: a radical probe of protein structure. Rapid Commun Mass Spectrom. 1999;13:2352–8. doi: 10.1002/(SICI)1097-0231(19991215)13:23<2352::AID-RCM798>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 39.Maleknia SD, Chance MR, Downard KM. Electrospray Generated Oxygenated Radicals to Probe Protein Structure. In: Gelpi E, editor. Advances in Mass Spectrometry. Wiley; 2001. pp. 543–544. [Google Scholar]
- 40.Asakawa Y, Tokida N, Ozawa C, Ishiba M, Tagaya O, Asakawa N. Suppression effects of carbonate on the interaction between stainless steel and phosphate groups of phosphate compounds in high-performance liquid chromatography and electrospray ionization mass spectrometry. J. Chromatogr. A. 2008;1198-1199:80–86. doi: 10.1016/j.chroma.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Tang K, Gomez A. Generation of monodisperse water droplets from electrospray in a corona-assisted cone-jet mode. J Colloid Interface Sci. 1995;175:326–332. [Google Scholar]
- 42.Kertesz V, Van Berkel GJ, Granger MC. Efficient analyte oxidation in an electrospray ion source using a porous flow-through electrode emitter. Anal Chem. 2005;77:4366–73. doi: 10.1016/j.jasms.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 43.Van Berkel GJ, Asano KG, Granger MC. Controlling Analyte Electrochemistry in Electrospray Using a Three-Electrode Emitter Cell. Anal. Chem. 2004;76:1493–9. doi: 10.1021/ac035240m. [DOI] [PubMed] [Google Scholar]
- 44.Seiwert B, Hayen H, Karst U. Differential Labeling of Free and Disulfide-Bound Thiol Functions in Proteins. J. Am. Soc. Mass Spectrom. 2008;19(1):1–7. doi: 10.1016/j.jasms.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17(10):994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 46.Kraatz H-B, Galka M. Ferrocenoyl Amino Acids and Peptides: Probing Peptide. Met Ions Biol Syst. 2001;38:385–409. [PubMed] [Google Scholar]
- 47.Van Staveren DR, Metzler-Nolte N. Bioorganometallic Chemistry of Ferrocene. Chem. Rev. 2004;104:5931–5985. doi: 10.1021/cr0101510. [DOI] [PubMed] [Google Scholar]
- 48.Parsons R. Handbook of Electrochemical Constants. Academic Press; London: 1959. [Google Scholar]
- 49.Levin RD, Lias SG. Ionization Potential and Appearance Potential Measurements. US, Department of Commerce, National Bureau of Standards; Washington, DC: 1982. 1971±1981, NSRDS-NBS 71. [Google Scholar]
- 50.Grishina AD, Vannikov AV, Khazova GO, Teodoradze MG, Koltsov YI. Photochemical processes in photoresists containing electron donor molecules. Photochem Photobiol. 1998;114:159–162. [Google Scholar]
- 51.Hsieh Y. APPI: A New Ionization Source for LC and MS/MS Assays. In: Korfmacher WA, editor. Using Mass Spectrometry for Drug Metabolism Studies. CRC Press; 2004. pp. 253–276. [Google Scholar]
- 52.Takáts Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306(5695):471–3. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 53.Venter A, Nefliu M, Cooks RG. Ambient desorption ionization mass spectrometry. Trends Anal. Chem. 2008;27(4):284–290. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.