

SUMMARY
The emergence of multidrug-resistant pathogens necessitates the search for new antibiotics acting on previously unexplored targets. Nicotinate mononucleotide adenylyltransferase of the NadD family, an essential enzyme of NAD biosynthesis in most bacteria, was selected as a target for structure-based inhibitor development. Using iterative in silico and in vitro screens we identified small molecule compounds that efficiently inhibited target enzymes from Escherichia coli (ecNadD) and Bacillus anthracis (baNadD) but had no effect on functionally equivalent human enzymes. On-target antibacterial activity was demonstrated for some of the selected inhibitors. A 3D structure of baNadD was solved in complex with one of these inhibitors (3_02) providing mechanistic insights and guidelines for further improvement. Most importantly, the results of this study help validate NadD as a target for the development of antibacterial agents with potential broad-spectrum activity.
INTRODUCTION
The versatility and resourcefulness of microbes in developing resistance to various therapies are widely recognized. Although chemical modifications of existing drugs and the development of novel inhibitors against a handful of previously established targets have proven to be successful in the short term, it is also apparent that new drug targets need to be explored to maintain and extend efficacious antibacterial therapy in the long run (McDevitt and Rosenberg, 2001). The need for new targets is further exacerbated by the emergence of bacterial pathogens with natural resistance to existing antibiotics and by a potential threat of pathogens with engineered antibiotic resistance.
Previous studies implicated NAD(P) biosynthesis as a promising, albeit relatively unexplored target pathway for the development of novel antimicrobial agents (Gerdes, et al., 2002; Osterman and Begley, 2007; Sassetti, et al., 2003). Cofactors of the NAD pool are indispensable as they are involved in hundreds of redox reactions in the cell. Additionally, NAD is utilized as a cosubstrate by a number of nonredox enzymes, e.g. by bacterial DNA ligases and protein deacetylases of the CobB/Sir2 family. This dictates the need to maintain NAD homeostasis via its active resynthesis and recycling of NAD degradation products. Recently, a number of insightful reviews have emphasized the potential of NAD(P) biosynthetic enzymes as drug targets for the treatment of cancer, autoimmune diseases, and neurodegenerative disorders (Chen, et al., 2008; Khan, et al., 2007; Lau, et al., 2009; Magni, et al., 2009). Although the early steps in NAD biogenesis and recycling vary substantially between species, the enzymes driving the downstream conversion of nicotinic acid mononucleotide (NaMN) to NAD and NADP are present in nearly all analyzed bacterial genomes (Osterman and Begley, 2007; Sorci, et al., 2009) (Fig. 1). Therefore, all three enzymes of this pathway—NaMN adenylyltransferase (EC 2.7.7.18), NAD synthetase (EC 6.3.1.5) and NAD kinase (EC 2.7.1.23)—encoded by the conserved genes nadD, nadE and nadF, represent promising broad-spectrum antibacterial targets. The observed essentiality of the respective genes is due to bacteria being unable to uptake phosphorylated pyridine nucleotides (Gerdes, et al., 2002; Osterman and Begley, 2007). Recent progress in the development of inhibitors targeting the last two enzymes, NadE (Velu, et al., 2003; Velu, et al., 2005; Velu, et al., 2007) and NadF (Bonnac, et al., 2007; Poncet-Montange, et al., 2007), provides additional validation of NAD biosynthesis as a target pathway. We have selected the NadD enzyme as a target for the development of specific inhibitors based on a number of criteria such as essentiality, broad conservation and structure–function distinction from its human counterpart (Gerdes, et al., 2002; Zhou, et al., 2002).
Figure 1. NAD(P)-biosynthesis pathways in bacteria.

NadD target enzyme is conserved in most bacteria. NaMN, a common biosynthetic intermediate in the salvage and de novo NAD biosynthetic pathways, is the substrate for NadD. The ATP substrate acts as the adenylyl moiety donor for NaMN, yielding PPi and NaAD. The latter product is then amidated (and phosphorylated) to give NAD and NADP.
NadD converts NaMN, the first intermediate shared by the most common de novo and salvage/recycling routes, to nicotinic acid adenine dinucleotide (NaAD) (Fig. 1). Therefore, this enzyme should be indispensable in all bacterial species that utilize one or both of these routes for NAD biosynthesis. This is consistent with gene essentiality data for a number of bacterial species (as reviewed in (Gerdes, et al., 2006; Gerdes, et al., 2002)). For example, the nadD gene was shown to be essential for survival in Staphylococcus aureus and Streptococcus pneumoniae that are fully dependent on niacin salvage (via PncA-PncB route). It is also essential in Escherichia coli and Mycobacterium tuberculosis, organisms that harbor both the de novo (NadB-NadA-NadC) and the salvage pathways. Remarkably, it has been recently demonstrated that NAD downstream pathway holds as an attractive target in both actively growing and nonreplicating pathogens (Boshoff, et al., 2008). NadD is present in nearly all important pathogens with only a few exceptional cases, such as Haemophilus influenzae which lacks most of NAD biosynthetic machinery and is dependent on salvage of the so-called V-factors (Gerlach and Reidl, 2006).
Many representatives of the NadD family from pathogenic and model bacteria have been characterized mechanistically and structurally (Han, et al., 2006; Lu, et al., 2008; Olland, et al., 2002; Sershon, et al.; Yoon, et al., 2005; Zhang, et al., 2002). All of these enzymes have a strong substrate preference for NaMN over its amidated analog, NMN. On the other hand, all three isoforms of the functionally equivalent human enzyme (hsNMNAT-1, hsNMNAT-2 and hsNMNAT-3) have an almost equal catalytic efficiency for either substrate, NaMN or NMN (Berger, et al., 2005; Sorci, et al., 2007). The observed difference in substrate specificity reflects the dual physiological role of the human enzyme (hereafter referred to as hsNMNAT) in the adenylation of both intermediates contributing to NAD biogenesis (Lau, et al., 2009; Magni, et al., 2008). Notably, among the three bacterial enzymes of the target pathway shown in Fig.1, NadD has the lowest sequence similarity to its human counterparts (Gerdes, et al., 2002). Comparative analysis of 3D structures of bacterial NadD and hsNMNAT revealed significant differences between their active site conformations (Zhou, et al., 2002), which are likely responsible for their distinct substrate specificities, thus opening an opportunity for selective targeting.
The goals of the present study are to use a structure-based approach to identify low-molecular weight compounds that selectively inhibit representative NadD enzymes, and to test whether such inhibitors suppress the growth of model Gram-positive and Gram-negative bacteria. Among the most important results is the validation of the NadD family as a druggable target whose specific inhibition leads to the suppression of bacterial growth in culture. Detailed interactions between NadD enzyme from Bacillus anthracis (baNadD) and one of the selected inhibitors were revealed by the X-ray cocrystal structure, information that will be instrumental in lead optimization and in development of drug prototypes.
Results
An overview of the structure-based approach applied in this study for NadD inhibitor discovery is summarized in Fig. 2. In silico screening of the large virtual library of small–molecule compounds to identify potential NadD inhibitors was performed using the ecNadD structural template. Of the ∼ 500 top-ranking in silico hits, 307 commercially available compounds were subjected to in vitro primary testing for inhibition of two representative target enzymes, ecNadD and baNadD. A series of analogs of three high-ranking compounds of distinct chemotypes (1, 3, and 15) active against both target enzymes were characterized in more detail by both enzymatic and cell-based assays. A co-crystal structure of baNadD in complex with one of the inhibitors, 3_02, revealed atomic details of its interactions with the enzyme active site, providing guidelines for future structure-based inhibitor optimization.
Figure 2. Flowchart of the structure-based approach for developing bacterial NadD inhibitors.

In silico screening of the compound library
The design of the template for in silico screening was based on the 3D structure of ecNadD reported in our earlier study (Zhang, et al., 2002). The targeted binding pocket encompassed the nicotinosyl binding site (near residues Asn40, Thr85, Phe104 and Ile106 in ecNadD) as well as the catalytic site near the conserved (H/T)xGH motif (around Phe8, Gly10 and His19). The initial screen of ∼1 million compounds targeted a single conformation of ecNadD in apo-form, which was followed by two rounds of secondary screens. In the first round, 20,000 top-scoring compounds selected in the initial screen were rescreened against two additional apo-ecNadD conformations obtained from the 1k4k crystal structure (Zhang, et al., 2002). In the second round 50,000 compounds from the initial screen were docked onto 5 ecNadD conformations obtained from an MD simulation. Of the 500 top-scoring compounds selected by these two screens, a total of 307 were purchased from vendors and subjected to experimental testing of their NadD inhibitory activity (Table S1).
Experimental testing of NadD inhibitors
To evaluate the compounds obtained from virtual screening we experimentally tested their inhibitory activity against two representative NadD target enzymes, from the model gram-negative bacterium E. coli and from the Gram-positive pathogen B. anthracis. Both recombinant enzymes were overexpressed in E. coli and purified, and their steady-state kinetic parameters were obtained using a standard coupled assay (Kurnasov, et al., 2002). An extensive kinetic analysis of baNadD enzyme, which included detection and exploration of negative cooperativity, was recently published (Sershon, et al., 2009). The results of our previously reported kinetic analysis of this enzyme, albeit less detailed, yielded comparable steady state parameters that reflect strong preference for NaMN over NMN (Sorci, et al., 2009). A similar preference was observed for ecNadD (Table S3). The experimental testing of selected compounds for their ability to inhibit NaMNATase activity of NadD enzymes was performed in the 96-well microtiter plate format using a colorimetric end-point assay, which includes an enzymatic conversion of the released PPi to Pi and a chromogenic reaction with the ammonium molybdate/Malachite Green reagent (Cogan, et al., 1999).
At this stage of analysis we aimed to detect inhibitors with moderate affinity (e.g., IC50 at least 100 µM or better). Therefore, for each of the two enzymes the testing was performed in the presence of compounds at 50–100 µM. The results of primary testing of all 307 compounds against both enzymes are shown in Table S1. At the 20% inhibition threshold, this method identified 38 ecNadD inhibitors. Remarkably, the baNadD enzyme showed on average a two-fold higher susceptibility to inhibition yielding 77 compounds at the same threshold. An appreciable correlation across the entire set of 307 analyzed compounds could be observed in their inhibitory properties against both enzymes (Table S1). This trend can be best illustrated by the comparison of two sets of ∼10% top-ranking ecNadD and baNadD inhibitors revealing that nearly one-third of them are shared between both sets (the estimated probability to get at least 12 random matches is 3·10−12, see Supplemental Data). This observation indicated that the applied in silico screening strategy was indeed successful in targeting NadD active–site components conserved between quite divergent representatives of this enzyme family. Combining this strategy with the parallel experimental testing of compounds against two divergent target enzymes allowed us to identify 12 potentially broad-spectrum NadD inhibitors. Three of these inhibitors (1, 3, and 15) with distinct scaffolds were selected for further analysis, including characterization of their analogs by enzymatic and cell-based assays and co-crystallization trials.
Selection and comparative analysis of NadD inhibitor analogs
To validate and further explore the utility of the three selected chemotypes, structurally similar and commercially available analogs of compounds 1, 3, and 15 were identified using chemical fingerprint–based similarity analysis (Butina, 1999; Godden, et al., 2005). For each of the primary compounds, 15 to 40 analogs were purchased and analyzed by the same inhibitory assay. Inhibitory activity above a 20% threshold against at least one of the analyzed NadD enzymes was confirmed for 66 of the 89 analogs (Table S2). For example, of the 29 analogs of compound 3, 23 were active against ecNadD and 24 against baNadD, whereas all 18 analogs of compound 1 turned out to be inhibitors of both enzymes. Notably, among 42 analogs of compound 15, 23 compounds were confirmed as baNadD inhibitors, but only 2 compounds had an appreciable inhibitory effect on ecNadD.
Overall, an observed frequent occurrence of analogs of compounds 1 and 3 that are active against both divergent members of NadD family supports the possibility of developing broad-spectrum NadD inhibitors. Although all the analyzed analogs were selected based only on structural similarity (without any attempts of their rational improvement), many of them displayed a moderate improvement of inhibitory properties compared to the original compounds. For example 10 analogs of compounds 1 and 3 had improved activity against ecNadD and 22 against baNadD, pointing to the possibility of their further optimization. IC50 values against ecNadD and baNadD determined for a subset of 33 compounds representing all three chemotype ranged from low micromolar to >200 micromolar (Table S2). Comparative analysis of these data revealed an appreciable correlation (r = 0.79) of the inhibitory properties of these compounds against both target enzymes over the entire subset (Fig. 3). The strongest correlation was observed for the compounds from the most active class 1 (r = 0.98).
Figure 3. Correlation analysis of IC50 values for classes 1, 3 and 15 compounds.

The analysis was restricted to compounds with IC50 values < 0.2 mM and was computed on the assumption that both IC50 values for E. coli and B. anthracis NadDs follow a Gaussian distribution.
To assess potential selectivity of these inhibitors against bacterial targets, several of the most active representatives of each chemotype were tested for their ability to inhibit human countertarget enzymes (hsNMNAT-1–3). These assays were performed at 100 µM concentration of the compounds, i.e., in the conditions leading to >90% inhibition of bacterial NadD enzymes. Remarkably, none of the tested compounds displayed any appreciable inhibitory activity against the three human isozymes (<5% for hsNMNAT-1 and hsNMNAT-3, and <10% for hsNMNAT-2). These compounds displayed the same efficacy and specificity when tested at a higher concentration of BSA (1 mg/ml) in the assay, which is a common test to eliminate promiscuous inhibitors (Coan and Shoichet, 2007; Shoichet, 2006). Overall, the observed antibacterial selectivity and versatility of the analyzed inhibitors further support NadD as a promising target for the development of potential broad-spectrum antibiotics.
Effects of NadD inhibitors on bacterial cell growth
The antibacterial activity of selected NadD inhibitors was assessed by their ability to suppress the growth of model Gram-negative (E. coli) and Gram-positive (B. subtilis) bacteria in liquid culture. To establish conditions potentially maximizing the effect of NadD inhibition in an E. coli model, we used a ΔnadA mutant strain with disrupted de novo NAD synthesis. To further restrict the flux of NaMN (the committed substrate of the NadD target enzyme) we performed the growth studies on the experimentally established lowest concentration of Nam (0.4 µM) supporting the growth of this diagnostic strain on minimal media. In these conditions, many of the selected NadD inhibitors of classes 1 and 3 showed an appreciable growth suppression effect at 100 µM (Fig. 4A and Table S2). To assess the extent of “on-target” (NadD-dependent) versus “off-target” (nonspecific) antibacterial effects of these compounds, we used a derivative of the same E. coli strain containing an overexpression plasmid vector with the E. coli nadD gene. The growth of this strain in the presence of selected inhibitors was compared to an isogenic control strain containing the same plasmid vector overexpressing a housekeeping gapA gene (unrelated to NAD synthesis). As shown in Fig. 4C and 4D, overexpression of ecNadD suppressed the antibacterial activity of the tested representatives of NadD inhibitors of classes 1 and 3. On the other hand, the bactericidal effect of the compound 15_11 (Table 3) was essentially the same in both the NadD-overexpressing and control strain (Fig. S3) suggesting that this effect is largely non-specific (NadD-independent). An alternative interpretation that the on-target activity of 15_11 is too high to be suppressed by NadD overexpression appears unlikely, as the in vitro inhibitory properties of this compound are below average (IC50, ecNadD ∼ 200 µM). Based on the structure of this compound, one may expect its hydrolysis in the medium to benzoate, a compound known to have a general and non-specific antibacterial activity. Based on a combination of these observations and considerations we excluded compounds of the class 15 from further analysis. An appreciable antibacterial activity was also observed for several analogs of compounds 1 and 3 against the model gram-positive bacteria B. subtilis (Fig. 4B and Table S2). Interestingly, the antibacterial effect of tested compounds in B. subtilis was manifested by delayed growth in contrast to E. coli where it was largely a decreased final cell density (Fig. 4B). Although establishing a rationale for this difference and confirming the actual target in Gram-positive bacteria remain to be accomplished, the growth–suppression data shown in Fig. 4 and Table S2 indicate that NadD inhibitors may indeed function as broad-spectrum antibiotics.
Figure 4. Growth inhibition of model bacteria and validation of NadD target.

(A,B) Effect of inhibitors of class 3 (100 µM for E. coli and 50 µM for B. subtilis) on cell growth as reflected in changes of the optical density at 600 nm.
(C,D) Overexpression of NadD in E. coli ΔnadA, nadD+ (red) increases resistance to inhibitors 3_15 (C) and 1_03 (D) compared to a control strain overexpressing enzyme GapA, unrelated to NAD (blue). Inhibitors were tested at ≤100 µM due to solubility issues at higher concentrations. Data shown are mean ± SD and are representative of 2 or more experiments.
Table 3.
Inhibition of target enzymes and antibacterial activity of selected compounds
| Cmpd | Structure | IC50a (µM) |
MIC50b (µM) |
|||
|---|---|---|---|---|---|---|
| ecNadD | baNadDc | E. coli | B. anth | B. subt | ||
| 3_05 | 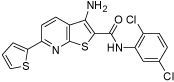 |
20 | 9 | 80 | 20 | 10 |
| 3_15 | 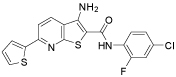 |
51 | 12 | 160 | 20 | 30 |
| 3_02 | 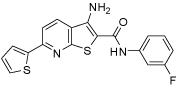 |
65 | 36 | 160 | 80 | 80 |
| 3_17 | 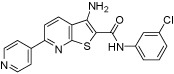 |
170 | >200 | 80 | >160 | >160 |
| 1_03 | 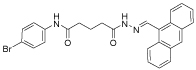 |
18 | 33 | >80 | 15 | 10 |
| 1_02 | 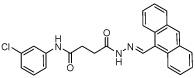 |
15 | 25 | >80 | NA | NA |
| 3_23 | 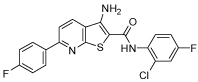 |
>200 | 63 | 40 | >80 | >80 |
| 15_11 | 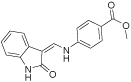 |
191 | 98 | <100c | NA | <50c |
Inhibitory efficiency of selected compounds (representative of classes 1, 3, and 15) for two target enzymes, ecNadD and baNadD is illustrated by IC50 values.
Antibacterial activity of the same compounds against Gram-negative (E. coli) and Gram-positive (B. subtilis, B. anthracis) model species is reflected by MIC50 values (the lowest concentration of compound causing more than 50% growth inhibition). Representative dose-response curves for B. subtilis and B. anthracis are provided in Fig. S4.
only single-point high estimates of MIC50 values were determined (70% growth inhibition at 100 µM for E. coli, and 96% inhibition at 50 µM for B. subtilis) for a representative of the class 15 that displayed mostly off-target antibacterial activity in E. coli model; NA, not assayed.
We further determined MIC for active compounds 3_02, 3_05, 3_15, 3_23, and 1_03 against B. anthracis sterne, B. subtilis, and E. coli. A general correlation was observed between NadD inhibition and antibacterial activity, although being less pronounced in E. coli (Table 3). Cell wall impermeability of gram-negative bacteria could be a major determinant of such weaker susceptibility. Notably, some of the less efficient ecNadD inhibitors (e.g. 3_23 and 15_11) showed a relatively strong antibacterial activity against E. coli. This observation may reflect the existence of additional targets affected by these compounds (see also Fig. S3), nonspecific or even sharing some common features with NadD (Moro, et al., 2009).
Mechanistic and structural analysis of NadD inhibition
Representatives of both classes 1 and 3 of efficient NadD inhibitors were selected for detailed kinetic characterization and co-crystallization trials. Apparent steady-state inhibitory parameters were obtained for compounds 1_02 and 3_02 against ecNadD and baNadD with respect to each substrate ATP and NaMN (Table 2 and Fig. S1). A preliminary assessment of all kinetic profiles revealed a mixed-type inhibition as indicated by α>1 values obtained by fitting initial rates to a general inhibition model (see Eq. 2 in Experimental Procedures). Despite the observed complex behavior preventing a straightforward mechanistic interpretation, the obtained data showed a substantial similarity in the inhibitory properties of both compounds with respect to both target enzymes.
Table 2.
Inhibitory parameters of representative compounds from two chemotypes
| Inhibitor: | 3_02 | 1_02 | ||||||
|---|---|---|---|---|---|---|---|---|
| Substrate: |
NaMN |
ATP |
NaMN |
ATP |
||||
| Ki | α | Ki | α | Ki | α | Ki | α | |
| (µM) | (µM) | (µM) | (µM) | |||||
| baNadD | 18±4 | 2.4 | 32±5 | 5.5 | 9±3 | 2.3 | 10±2 | 2.9 |
| ecNadD | 25±9 | 2.5 | 21±9 | 2.4 | 8±3 | 7.2 | 5±1 | 7.1 |
The apparent values of inhibitory parameters (Ki and α) of two compounds (3_02 and 1_02) were determined for both enzymes by fitting the kinetic data to the general equation for the mixed-model inhibition (Lyon and Atkins, 2002, see Experimental procedures for details). The data were collected by varying the concentration of an inhibitor and one of the two substrates (NaMN or ATP) at fixed concentration of another substrate (0.5 mM ATP or NaMN).
The 3D structure of the complex of baNadD co-crystallized with compound 3_02 and solved at 2.0-Å resolution revealed its binding in the active-site area mostly through van der Waals interactions. The planar compound stacks against two aromatic residues, Trp116 and Tyr112 (baNadD numbering), and is also in contact with Met109 and Phe103 (Fig. 5A). While there are a few water-mediated indirect interactions between 3_02 and the enzyme, there is no direct intermolecular hydrogen-bond interaction. A comparison with the 3D structure of baNadD complexed with the NaAD product solved in this study at 2.2-Å resolution and with the recently reported apo-baNadD structures (Lu, et al., 2008; Sershon, et al., 2009) provided additional insights to the structural mechanism of inhibition. This comparison revealed that the bound compound 3_02 partially overlaps with the nicotinosyl binding site and would interfere with NaMN substrate binding (Fig. 5B). In particular, inhibitor binding would potentially block the critical stacking interaction between the side-chain of the conserved Trp116 residue with the pyridine ring of the NaMN substrate (Olland, et al., 2002; Zhang, et al., 2002). This interference may contribute to a competitive aspect of the observed mixed-type inhibition.
Figure 5. Structure of baNadD in complex with inhibitor 3_02 and comparison with product-bound baNadD structure.

(A) Interactions between inhibitor (magenta) and baNadD. The two baNadD monomers are colored gold and gray, respectively. Cα traces of baNadD are shown. Protein residues that interact with 3_02 are shown as sticks. Water molecules are shown as small red spheres.
(B) Superimposition of baNadD-NaAD complex structure (cyan) with the inhibitor (3_02) bound structure (magenta). The bound product NaAD and inhibitor 3_02 are shown as blue and magenta sticks respectively.
(C) Overall structure of baNadD dimer (cyan and blue subunits) is shown with bound NaAD product. The orientation of this dimer is similar to monomers α and β in Fig. 5D.
(D) The tetrameric arrangement of Bacillus anthracis NadD-3_02 complex in the crystal. Two baNadD dimers are displayed. Only one orientation of the inhibitor 3_02 (in sticks) is shown in each binding site.
(E) The 2Fo-Fc different electron density for the bound inhibitor 3_02. The two orientations of the inhibitor are shown in magenta and yellow, respectively.
The structure comparison also revealed a substantial difference between the active-site conformations in the baNadD-3_02 and baNadD-NaAD complexes. Moreover the active-site conformation in the baNadD-3_02 complex is more similar to apo-baNadD (rmsd between Cα atoms 0.77Å) than to the baNadD-NaAD complex (rmsd of 1.32 Å). The major conformational differences occur in the regions that are involved in NaMN binding, i.e., residues 42–48 (loop connecting β2 and α2), 105–126 (helix α4), and the loop between β5 and β6 (residues 131–149) (Fig. 5B). It is tempting to speculate that, in addition to interfering with NaMN substrate binding, the interactions between the inhibitor and baNadD may partially “lock” the enzyme active site in the catalytically impaired apo-like conformation. This hypothetical mechanism may provide a rationale for the observed, largely noncompetitive mode of inhibition described above.
The baNadD enzyme has a tendency to form a homodimer as observed in the crystal structure of both, apo-form and of its complex with substrate (Fig. 5C) and confirmed by size-exclusion chromatography and analytical ultracentrifugation (AUC) (data not shown). Inspection of baNadD-3_02 complex crystal packing shows that while the native dimer interface is preserved, an additional dimer interface, related by a two-fold non-crystallographic symmetry similar to that of the “handshake” dimer observed in B. subtilis NadD (Olland, et al., 2002) is also present, resulting in a tetrameric appearance (Fig. 5D). The 3_02 inhibitor binding site is located at this hand-shake dimer interface. Because the compound binds at a site between two baNadD monomers related by a pseudo-twofold symmetry, the two symmetrical orientations of 3_02 cannot be distinguished. Therefore, we modeled 3_02 in both orientations, each with half occupancy (Fig. 5E).
Although additional interactions between the compound 3_02 and the adjacent baNadD subunit at the handshake dimer interface were observed in the crystal structure, it is unlikely that such interactions would contribute to the inhibition observed in our assay conditions. Moreover, AUC data did not reveal any changes in the oligomerization state of the protein in presence of the inhibitor. Therefore, the contribution of the handshake dimer interface to baNadD inhibition by 3_02 should be negligible under the assay conditions. This conclusion is consistent with the fact that ecNadD, being monomeric both in the crystal structure and in solution, exhibits essentially the same inhibitory properties in the presence of 3_02, including the same mixed-type mode and similar kinetic parameters.
Notably, the three most flexible regions in baNadD mentioned above also correspond to the regions that deviate the most from the hsNMNAT structure (Zhou, et al., 2002) (Fig. S2). Comparison of human NMNAT structures (as represented by hsNMNAT-1 (Zhou, et al., 2002)) with various baNadD complexes indicated that hsNMNAT active site conformation is much closer to the product-bound conformation of baNadD than to the apo form of baNadD. No significant conformational change has been observed between the apo and ligand bound human NMNAT enzymes (Garavaglia, et al., 2002; Zhang, et al., 2003; Zhou, et al., 2002). Therefore the active site of hsNMNAT, being quite dissimilar from the apo or inhibitor-bound baNadD, appears unable to accommodate or specifically interact with inhibitor 3_02 (Fig. S2). This interpretation is indirectly supported by the results of comparative virtual docking performed for the three classes of active compounds against ecNadD, baNadD, and hsNMNAT-1 (See Supplemental Data). The docking energies for the human enzyme consistently have the least favorable scores compared to the energies obtained for ecNadD and baNadD, especially in the van der Waals energy terms, suggesting that the overall shape of the binding region in hsNMNAT is sufficiently different to allow for selective inhibition of bacterial enzymes.
Discussion
In earlier studies we used a comparative-genomics approach to identify NAD cofactor biosynthesis as a target pathway for development of new anti-infective therapies (Gerdes, et al., 2002; Osterman and Begley, 2007). The NadD enzyme was chosen as one of the most attractive targets within this pathway due to its nearly universal conservation in bacterial pathogens and its essentiality directly confirmed in a number of model bacteria (Gerdes, et al., 2002). A comparative enzymatic and structural analysis revealed substantial differences between bacterial enzymes and their human counterparts, opening an opportunity for development of selective NadD inhibitors. The fact that no drugs are known to act on NadD further contributes to this choice of a target in the context of the growing challenge of multidrug-resistant bacterial pathogens.
In the current study, an integrated structure-based approach was employed to identify small-molecule compounds selectively inhibiting enzymes of the NadD family with a potential broad spectrum of antibacterial activity. Combining computational screening of a virtual compound library with experimental testing of inhibitory and antibacterial activity of selected compounds and their analogs, we have identified and characterized two classes of inhibitors with distinct chemical scaffolds (chemotypes) possessing a number of desired properties (Table 3).
Our approach to in silico screening was based on selective targeting of those active site residues that are highly conserved among bacterial NadD enzymes, yet quite distinct from the human countertarget enzymes (Zhang, et al., 2002; Zhou, et al., 2002). A focused targeting of a nicotinosyl-binding (as opposed to adenosyl-binding) site was also aimed to exploit the functional differences between the NaMN-preferring bacterial NadD and human enzymes with dual specificity for NaMN and NMN substrates (Berger, et al., 2005; Sorci, et al., 2007). We also took advantage of the large conformational differences between the apo and substrate-bound enzymes by specifically targeting the enzyme active site in the apo form so that the inhibitors would stabilize the enzyme in a catalytically impaired conformation.
The obtained results supported the efficiency of this strategy. First, we observed an appreciable correlation between inhibitory properties of compounds against two divergent members of the NadD family, from Gram-negative (ecNadD) and Gram-positive bacteria (baNadD), even at the level of the primary experimental testing of ∼300 compounds (Table S1). This trend was even more apparent in the comparison of inhibitory properties of analogs of the three compounds (1, 3 and 15) selected for detailed characterization (Fig. 3 and Table S2). These observations indicate that the level of structural conservation in the active sites of divergent representatives of the NadD family provides a potential for developing broad-spectrum inhibitors. At the same time, the three selected chemotypes showed no appreciable activity against human countertargets. This finding validated another premise of this study, that the distinction between bacterial and human enzymes is sufficient for the development of selective (bacterial-specific) NadD inhibitors.
Although the affinity of the best inhibitors identified so far (IC50 in 4–30 µM range, see Table 3 and Table S2) is not sufficient for them to be considered as candidate drugs, we were able to use them as probes to address an important question of NadD target druggability in the cell. The essentiality of the nadD gene previously established by genetic techniques, by itself, does not guarantee that inhibition of the NadD enzyme in the cell is possible and may indeed suppress the bacterial growth. Moreover, the antibacterial activity of the analyzed compounds observed in Gram-negative (E. coli) and Gram-positive (B. subtilis) model systems (Fig. 4), while being encouraging, could be due to some effects other than inhibition of the NadD enzyme. We used an E. coli model system to test whether the observed growth suppression was indeed due to the “on-target” action of representative NadD inhibitors. As illustrated in Fig. 4C–D and Fig. S3, overexpression of the target nadD gene substantially increased resistance of this strain toward compounds of classes 1 and 3 (but not 15). This finding provided a direct evidence of the on-target action of these compounds in contrast to the tested compounds of class 15 whose antibacterial activity appears to be largely due to unknown off-target effect(s). These data directly validated the NadD enzyme as a drug target amenable to inhibition in the cell, which is leading to growth suppression.
It is important to emphasize that this general conclusion holds true regardless of whether further improvement of the currently selected compounds would or would not produce sufficiently potent antibacterial agents. The major goal of the present study, to assess the druggability of NadD target, dictated the choice of a genetically tractable model system of E. coli. Whether the activity of the same compounds against B. subtilis is also due to NadD inhibition remains to be tested, especially since the observed type of growth suppression in B. subtilis culture is different from E. coli (growth delay versus decreased final cell density).
Finally, it was important to test the binding mode of NadD inhibitors. This seemed particularly important as the steady-state kinetic analysis of the representative compounds of both classes 1 and 3 revealed a mixed-type inhibition with a strong noncompetitive component. To assess inhibitor binding mode(s) and to obtain a basis for rational improvement of the inhibitors, we attempted co-crystallization of both bacterial NadD enzymes with a panel of compounds of classes 1 and 3. The structure of baNadD in complex with the compound 3_02 reported here confirmed that this inhibitor indeed binds in the active site area partially overlapping with the targeted NaMN substrate binding site (Fig. 5B). The experimentally determined location of the inhibitor is adjacent to that predicted by the in silico docking (Fig. S5). Moreover the conformation of the baNadD active site in this complex is drastically different from its product-bound conformation, which is also reported in this study. In fact, the inhibitor binding appears to stabilize the baNadD conformation in its apo form, incompatible with substrate binding and catalysis (Lu, et al., 2008; Sershon, et al., 2009). Inhibitor interference at the level of substrate binding and the stabilization of alternative enzyme conformation may provide a rationale for the observed complex (mixed-type) kinetics of inhibition. Although the actual inhibitory mechanism is not fully clear, the obtained structural information is useful for further inhibitor optimization via structure-based design and synthesis of analogs. For example, engineering additional functional groups that may form specific hydrogen–bond interactions with the enzyme should enhance the binding affinity of the compound.
Significance
In this study, we have demonstrated the feasibility of developing NadD inhibitors against divergent members of NadD family with strong selectivity for bacterial (versus human) enzymes. Some of the selected inhibitors displayed an appreciable antibacterial activity in both Gram-positive and Gram-negative model systems. The established “on-target” action of these inhibitors confirmed that NadD is a tractable drug target for the development of novel antibacterial agents. The co-crystal structure of the compound 3_02 in complex with baNadD confirmed its binding at the active site and provided guidelines for inhibitor improvement. These results, taken together, illustrate the power of combining comparative genomics with a structure-based approach for the development of new classes of antiinfective agents.
EXPERIMENTAL PROCEDURES
Materials
All common buffers and reagents were purchased from Sigma-Aldrich. Small molecules selected from in silico screening were obtained from Chembridge, Specs.net and ChemDiv Inc.
In silico Database Screening
The substrate binding site of ecNadD (Zhang, et al., 2002) was selected as the target for docking. System preparation involved analysis of the target protein structure, selection of inhibitor binding site, and generation of the sphere set used to direct the docking (See Supplemental Data for details). All database screening calculations were carried out with DOCK 4.0 (Krumrine, et al., 2003; Kuntz, et al., 1982). The primary screening was performed on a 3D database of over 1 million low–molecular–weight commercially available compounds developed in the University of Maryland Computer-Aided Drug Design (CADD) Center (Huang, et al., 2004; Zhong, et al., 2007). Ligand flexibility was incorporated during docking via the anchor-based search method (Ewing and Kuntz, 1997). Compounds from the initial primary screen were docked onto the protein based on the total ligand–protein interaction energy and scored based on the van der Waals (vdW) attractive energies normalized for molecular size (Pan, et al., 2003).
Top scoring compounds from the primary screen were subjected to more rigorous secondary docking, where additional optimization of the ligand was performed during the build up procedure. Additionally, conformational flexibility of ecNadD was taken into account via the inclusion of multiple protein conformations either from the crystallographic studies (secondary screen A) or from a molecular dynamics (MD) simulation of ecNadD (secondary screen B). In secondary screen A, the top 20,000 scoring compounds from the primary screening were individually docked to the three conformations of apo ecNadD obtained from the 1k4k crystal structure. In secondary screen B, multiple protein conformations were obtained from the MD simulation of apo ecNadD (see Supplemental Data). The top 50,000 scoring compounds from the primary screen were then docked against five MD–generated conformations and ranked using the normalized total interaction energy for each compound. The top scoring compounds from the two separate secondary screens, totaling 500 and 1000, respectively, were then separately subjected to the final compound selection based on physical properties and chemical similarity. Determination of chemical similarity and further selection of compounds were performed according to standard procedures (Supplemental Data). Finally, a total of 529 unique compounds were selected; of these, 307 were purchased from the commercial vendors for the in vitro inhibition assay. After primary testing (see below), three chemotypes (classes 1, 3 and 15) were selected for further analysis of chemical analogs. A total of 89 analogs were purchased and experimentally tested.
Kinetic analysis of NadD and primary testing of selected compounds
A discontinuous assay was utilized to determine the steady-state kinetics parameters kcat and Km for NadD and for inhibitory testing of selected compounds. This assay couples pyrophosphate (PPi) byproduct formation of NaMNATase activity to colorimetric detection of free phosphate released upon enzymatic hydrolysis.
-
NaMN Adenylyltransferase (NaMNATase) reaction
NaMN + ATP + Mg2+ ➔ NaAD+ + PPi + Mg2+
-
Inorganic pyrophosphatase (IPase) reaction
PPi + H2O ➔ 2Pi
Excess IPase is used to ensure rapid conversion of pyrophosphate to orthophosphate so that the rate-limiting step in this system is the NaMN adenylyltransferase reaction. Excess inorganic phosphate also decreases the probability that observed inhibition is due to the inhibition of IPase and not the target enzyme. We have later confirmed that the best NadD inhibitors (with IC50 values ranging from 5 to 25 µM) did not inhibit IPase.
Steady-state kinetic analysis of ecNadD and baNadD target enzymes was performed by varying substrate (NaMN or ATP) concentrations were 0, 10, 30, 60, 200, 500 µM at fixed saturating concentration of second substrate (0.5 mM). Apparent values of Km and kcat were calculated by fitting initial rates to a standard Michaelis–Menten model using the software Prism 4 (GraphPad).
The standard inhibition assay was configured in a 96-well format for automated liquid-handling and convenient readout. Each compound was prepared as a 10 mM stock solution in dimethyl sulfoxide (DMSO) and diluted ten-fold (10% DMSO) before usage. Each reaction contained 2.3 nM ecNadD (or 1.2 nM baNadD) in 100 mM Hepes, pH 7.5 buffer, 0.2 mM ATP, 0.07 or 0.2 mM NaMN,10 mM MgCl2, 0.1 mg/ml Bovine Serum Albumin, 0.2 U inorganic pyrophosphatase, and 50 or 100 µM tested compound (the complete lists of tested compounds with structure and vendor information is provided in Tables S1 and S2). Although the use of surfactants is a common practice to reduce the effects of promiscuous inhibitors, an observed interference of Triton-X100 at 0.01% with our assay prompted us to include bovine serum albumin (0.1–1 mg/ml) in the assay mixture to prevent the selection of promiscuous aggregating inhibitors (Coan and Shoichet, 2007). The choice of two-fold Km(app) concentrations of both NaMN and ATP substrates was necessary to ensure a good signal/noise ratio under the initial velocity phase of enzymatic reactions (10–20% substrate depletion), while retaining a linear signal response (0–15 µM PPi). The same assay setup was applied when testing small–molecule inhibitors against human countertargets. Concentrations of hsNMNAT-1 and hsNMNAT-2 were 3 nM, whereas hsNMNAT-3 was tested at 15 nM. After preincubation of the enzyme with the compounds for 5 min at room temperature, the reaction was initiated by addition of NaMN substrate. The reaction was allowed to progress for 20 min at room temperature prior to quenching by addition of 100 µL of Malachite Green Reagent in 1.2 M sulfuric acid prepared as described by (Cogan, et al., 1999)). After 20–30 min incubation to allow for complex/color formation, the absorbance in each well was measured at 620 nm using a microplate reader (Beckman DTX-880). To account for contribution of free Pi and/or PPi (present in the sample or released due to nonspecific hydrolysis of ATP during incubation) as well as of background absorbance (color) of the tested compounds, parallel reactions were run for each experimental point without addition of NadD enzymes, and their OD620 values were subtracted from the measurements of enzyme activity in their respective samples. Reaction in the presence of 2% DMSO but without inhibitory compound served as a positive control. Each measurement was made in triplicate. Based on the sensitivity and reproducibility of the assay, inhibition ≥20% was considered reliable. A continuous coupled assay that detected reduction of NAD+ (Kurnasov, et al., 2002) was used for preliminary assessment of NaMNTase activity and to corroborate kinetic parameters obtained with Malachite Green discontinuous assay.
IC50 measurements and Ki determination
The compounds selected based on the results of primary testing were further characterized using the malachite green end-point assay. The initial rate of enzymatic reaction was measured at fixed NaMN and ATP concentrations (equal to two-fold Km values) and various concentrations of an inhibitory compound. The IC50 value was determined by plotting the relative NaMNATase activity versus inhibitor concentration and fitting to the equation (1) using GraphPad Prism®.
| (1) |
V0 and Vi represent initial rates in the absence and presence of inhibitor concentration [I].
For Ki determination, the enzyme was preincubated with various fixed concentrations of inhibitors for 5 min. The reaction was initiated by the addition of fixed concentration of NaMN (five-fold Km) at varying concentrations of ATP (ranging from 0.2 to five-fold Km) and vice versa. The inhibition constant and inhibition pattern were evaluated by fitting the data to the Michealis-Menten rate equation (2) for general (mixed-type) inhibition (Lyon and Atkins, 2002) with the program GraphPad Prism®.
| (2) |
Vmax and Km are standard Michaelis-Menten parameters, and Ki is the equilibrium dissociation constant for the enzyme–inhibitor complex. The parameter α defines the degree to which the inhibitor binding affects the affinity of the enzyme for the substrate and is diagnostic of the inhibition mode, which may be purely competitive (α≫1), purely noncompetitive (α=1), uncompetitive (α≪1), or mixed-type (α>1 or α<1).
Suppression of bacterial growth in culture
Diagnostic E. coli strains used for growth–suppression experiments and for target verification were prepared in the background of the E. coli K-12 BW25113 (ΔnadA) knockout strain with disrupted NAD de novo synthesis pathway from the Keio collection (a gift by Dr. H. Mori, Keio University, Japan) (Baba, et al., 2006). This strain was used in combination with one of the two expression plasmids from the E. coli ASKA library (Kitagawa, et al., 2005) enabling inducible overexpression of the: (i) E. coli nadD gene (to test for the increased resistance against NadD inhibitors) or the (ii) E. coli gapA gene, a housekeeping metabolic enzyme glyceraldehyde-3-phosphate dehydrogenase (as a negative control). Starter cultures were grown overnight in LB medium. Cells were harvested, washed, and resuspended in the M9 minimal growth medium containing 1% glycerol, 0.1 mM IPTG, 50 mg/l kanamicin, 35 mg/l chloramphenicol and a limiting amount of nicotinamide (Nam, 0.4 µM). Upon reaching an optical density at 600 nm of 0.05, cells were used to initiate growth experiments in 96-well plate at various concentrations of inhibitors.
The bacterial growth at 37°C in these (and other) experiments was monitored by continuous absorbance measurement at 600 nm using an orbital shaker/microplate reader ELx808™. The area under the curve (AUGC) was used to calculate the growth inhibition (Firsov, et al., 2001) and was compared to the respective amount of DMSO. The AUGC was integrated and calculated with GraphPad Prism 4. Growth suppression studies of B. subtilis 168 (Bs168) were performed following a similar procedure in a chemically defined medium(Rodionov, et al., 2009) containing glucose (4 g/l), tryptophan (50 mg/l), glutamine (2 g/l), K2HPO4 (10 g/l), KH2PO4 (6 g/l), sodium citrate (1 g/l), MgSO4 (0.2 g/l), K2SO4 (2 g/l), FeCl3 (4 mg/l), MnSO4 (0.2 mg/l). B. anthracis was grown in the same minimal medium containing additionally 10% LB medium for robust growth.
Selected compounds causing an appreciable growth inhibition were subject of minimal inhibitory concentration (MIC) determination in a series of dilutions from 160 µM down to 2.5 µM. The high concentration limit was determined by solubility problems observed for many compounds. In this concentration range only some of the analyzed compounds displayed >90% growth inhibition. Therefore, for consistency, the value of MIC was defined as the lowest concentration of compound that caused more than 50% growth inhibition (as determined by AUGC method).
Purification and crystallography
The expression and purification of ecNadD and baNadD were performed as described previously (Zhang, et al., 2002). The human proteins hsNMNAT-1, hsNMNAT-2, and hsNMNAT-3 were also expressed in Escherichia coli and purified as reported previously (Gerdes, et al., 2002; Zhang, et al., 2003; Zhou, et al., 2002). For crystallization, baNadD was first purified with Ni Sepharose HP column (GE Healthcare) and treated with TEV protease overnight before being passed through the Ni column again to remove the 6xHis tag. The tag-free protein was further purified by a Resource Q anion exchange column and Superdex 75 16/60 gel filtration column (GE Healthcare). The purified protein was concentrated to 20 mg/ml before crystallization. The baNadD–NaAD complex crystal was obtained in a hanging drop vapor diffusion setup in the presence of 10 mM NaAD at 20°C. The reservoir contained 20% PEG 3350 and 0.2M calcium acetate. The cocrystals of baNadD with compound 3_02 were obtained in the presence of 1mM 3_02 in similar conditions as those for the NaAD complex crystals. Before data collection the baNadD–NaAD cocrystals were transferred to a cryoprotectant solution containing 40% PEG 3350 and 0.2 M calcium acetate before being frozen in liquid nitrogen. The baNadD-3_02 cocrystals were not stable in such a cryoprotectant solution; therefore a different cryprotectant containing 20% PEG 3350, 0.2 M calcium acetate and 15% DMSO was used to stabilize the inhibitor complex crystals before being freezing them liquid nitrogen. The X-ray diffraction data were collected on a RAXIS IV++ image plate detector with a FR-E Superbright X-ray generator coupled with Osmic VariMax optics. The data were processed with HKL2000 package (Otwinowski and Minor, 1997). The baNadD-NaAD complex structure was first determined by the molecular replacement methods in the CNS package (Brunger, et al., 1998) using the B. subtilis NadD structure (Olland, et al., 2002) as the initial search model (pdb code 1kam). The baNadD inhibitor complex was subsequently solved with molecular–replacement methods as implemented in the program Molrep (Vagin and Teplyakov, 2000) using the baNadD product complex structure (excluding the NaAD ligand) as the search model. Both structures were refined with REFMAC (Murshudov, et al., 1997) in the CCP4 package (Collaborative Computational Project, 1994) and deposited in the Protein Data Bank with accession codes 3E27 and 3HFJ, respectively. The data collection and refinement statistics for both structures are listed in Table 1. The models were analyzed by MolProbity program (Lovell, et al., 2003).
Table 1.
Crystal Data and refinement statistics
| Data sets | baNadD·product | baNadD·3_02 |
|---|---|---|
| Data Statistics | ||
| Space group | P212121 | P21212 |
| Unit cell (Å) | a=41.8, b=137.41, c=143.97 | a=88.79, b=97.53, c=44.30 |
| Resolution (Å) | 50-2.2 | 50-2.0 |
| Total observations | 157926 | 113171 |
| Unique Reflections | 43353 | 26467 |
| Completeness (outer shell) (%) | 98.8 (89.9) | 99.9 (100.0) |
| Rsym (outer shell) | 0.085 (0.555) | 0.037 (0.279) |
| I/δ (outer shell) | 15.9 (2.0) | 36.8 (5.4) |
| Refinement | ||
| Rworkb | 0.206 | 0.205 |
| Rfreec | 0.276 | 0.266 |
| r.m.s.d bond length (Å) | 0.011 | 0.012 |
| r.m.s.d bond angle (°) | 1.49 | 1.44 |
| Monomers/asymmetric unit | 4 | 2 |
| Protein atoms | 6196 | 3102 |
| Water molecules | 494 | 309 |
| Ligand atoms | 180 | 53 |
| Average B-factors (Å2) | ||
| Protein | 32.4 | 28.0 |
| Ligands | 24.4 | 41.1 |
| Water | 39.9 | 34.4 |
| Ramachandran Plot | ||
| Favored region (%) | 97.0 | 98.7 |
| Allowed region (%) | 99.3 | 100.0 |
Rsym = ΣhklΣj|Ij−<I>|/ΣhklΣj|Ij|.
Rwork = Σhkl|Fo − Fc|/Σhkl|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
Five percent randomly selected reflections were excluded from refinement and used in the calculation of Rfree.
Analytical ultracentrifugation (AUC)
Sedimentation equilibrium experiments were performed in a ProteomeLab XL-I (BeckmanCoulter) analytical ultracentrifuge. Protein samples at 0.22 mg/ml concentration were loaded in 6-channel equilibrium cells and spun in an An-50 Ti 8-place rotor at 15,000 and 18,000 rpm, at 20 °C for 24 hours for each speed. The sample buffer contained 20 mM Hepes (pH 7.4) and 5% DMSO. Small–molecule inhibitors where tested at 50 µM. Absorbance profiles were acquired at 280 nm and analyzed with HeteroAnalysis software (National Analytical Ultracentrifugation Facility at the University of Connecticut). Apparent molecular weight (Mw) was determined by fitting the experimental data with the Ideal equilibrium model. If the apparent Mw was higher than calculated theoretical Mw for the monomer, the data were fitted using Monomer-Nmer or Monomer-Nmer-Mmer equilibrium models.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the NIAID grant AI059146 “Targeting cofactor biosynthesis in biodefense pathogens” to AO and HZ. A. M. acknowledges the support from the University of Maryland Computer-Aided Drug Design Center. We thank Darek Martynowski for help with X-ray diffraction data collection, Andrey Bobkov at the Protein Production and Analysis Facility of BIMR for AUC analysis, Marat Kazanov and Ying Zhang for data processing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Bonnac L, Chen L, Pathak R, Gao G, Ming Q, Bennett E, Felczak K, Kullberg M, Patterson SE, Mazzola F, et al. Probing binding requirements of NAD kinase with modified substrate (NAD) analogues. Bioorg Med Chem Lett. 2007;17:1512–1515. doi: 10.1016/j.bmcl.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park TH, Yun CS, Schnappinger D, Ehrt S, et al. Biosynthesis and recycling of nicotinamide cofactors in mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J Biol Chem. 2008;283:19329–19341. doi: 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Butina D. Unsupervised Data Base Clustering Based on Daylight's Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999;39:747–750. [Google Scholar]
- Chen L, Petrelli R, Felczak K, Gao G, Bonnac L, Yu JS, Bennett EM, Pankiewicz KW. Nicotinamide adenine dinucleotide based therapeutics. Curr Med Chem. 2008;15:650–670. doi: 10.2174/092986708783885282. [DOI] [PubMed] [Google Scholar]
- Coan KE, Shoichet BK. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol Biosyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- Cogan EB, Birrell GB, Griffith OH. A Robotics-Based Automated Assay for Inorganic and Organic Phosphates. Analytical Biochemistry. 1999;271:29–35. doi: 10.1006/abio.1999.4100. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. Journal of Computational Chemistry. 1997:1175–1189. [Google Scholar]
- Firsov AA, Lubenko IY, Portnoy YA, Zinner SH, Vostrov SN. Relationships of the area under the curve/MIC ratio to different integral endpoints of the antimicrobial effect: gemifloxacin pharmacodynamics in an in vitro dynamic model. Antimicrob Agents Chemother. 2001;45:927–931. doi: 10.1128/AAC.45.3.927-931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavaglia S, D'Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, Rizzi M. Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem. 2002;277:8524–8530. doi: 10.1074/jbc.M111589200. [DOI] [PubMed] [Google Scholar]
- Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, et al. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach G, Reidl J. NAD+ utilization in Pasteurellaceae: simplification of a complex pathway. J Bacteriol. 2006;188:6719–6727. doi: 10.1128/JB.00432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godden JW, Stahura FL, Bajorath J. Anatomy of fingerprint search calculations on structurally diverse sets of active compounds. J Chem Inf Model. 2005;45:1812–1819. doi: 10.1021/ci050276w. [DOI] [PubMed] [Google Scholar]
- Han S, Forman MD, Loulakis P, Rosner MH, Xie Z, Wang H, Danley DE, Yuan W, Schafer J, Xu Z. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Staphyloccocus aureus: structural basis for NaAD interaction in functional dimer. J Mol Biol. 2006;360:814–825. doi: 10.1016/j.jmb.2006.05.055. [DOI] [PubMed] [Google Scholar]
- Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr Identification of non-phosphate-containing small molecular weight inhibitors of the tyrosine kinase p56 Lck SH2 domain via in silico screening against the pY + 3 binding site. J Med Chem. 2004;47:3502–3511. doi: 10.1021/jm030470e. [DOI] [PubMed] [Google Scholar]
- Khan JA, Forouhar F, Tao X, Tong L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin Ther Targets. 2007;11:695–705. doi: 10.1517/14728222.11.5.695. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Krumrine J, Raubacher F, Brooijmans N, Kuntz I. Principles and methods of docking and ligand design. Methods Biochem Anal. 2003;44:443–476. [PubMed] [Google Scholar]
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes SY, Osterman AL. Ribosylnicotinamide Kinase Domain of NadR Protein: Identification and Implications in NAD Biosynthesis. J Bacteriol. 2002;184:6906–6917. doi: 10.1128/JB.184.24.6906-6917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Niere M, Ziegler M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci. 2009;14:410–431. doi: 10.2741/3252. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Lu S, Smith CD, Yang Z, Pruett PS, Nagy L, McCombs D, Delucas LJ, Brouillette WJ, Brouillette CG. Structure of nicotinic acid mononucleotide adenylyltransferase from Bacillus anthracis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:893–898. doi: 10.1107/S1744309108029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon RP, Atkins WM. Kinetic characterization of native and cysteine 112-modified glutathione S-transferase A1-1: reassessment of nonsubstrate ligand binding. Biochemistry. 2002;41:10920–10927. doi: 10.1021/bi0262810. [DOI] [PubMed] [Google Scholar]
- Magni G, Di Stefano M, Orsomando G, Raffaelli N, Ruggieri S. NAD(P) Biosynthesis Enzymes as Potential Targets for Selective Drug Design. Curr Med Chem. 2009;16:1372–1390. doi: 10.2174/092986709787846505. [DOI] [PubMed] [Google Scholar]
- Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci. 2008;13:6135–6154. doi: 10.2741/3143. [DOI] [PubMed] [Google Scholar]
- McDevitt D, Rosenberg M. Exploiting genomics to discover new antibiotics. Trends Microbiol. 2001;9:611–617. doi: 10.1016/s0966-842x(01)02235-1. [DOI] [PubMed] [Google Scholar]
- Moro WB, Yang Z, Kane TA, Brouillette CG, Brouillette WJ. Virtual screening to identify lead inhibitors for bacterial NAD synthetase (NADs) Bioorg Med Chem Lett. 2009;19:2001–2005. doi: 10.1016/j.bmcl.2009.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Olland AM, Underwood KW, Czerwinski RM, Lo MC, Aulabaugh A, Bard J, Stahl ML, Somers WS, Sullivan FX, Chopra R. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J Biol Chem. 2002;277:3698–3707. doi: 10.1074/jbc.M109670200. [DOI] [PubMed] [Google Scholar]
- Osterman AL, Begley TP. A subsystems-based approach to the identification of drug targets in bacterial pathogens. Prog Drug Res 64. 2007;131:133–170. doi: 10.1007/978-3-7643-7567-6_6. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Methods in Enzymology. Academic Press; 1997. [20] Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Pan Y, Huang N, Cho S, MacKerell AD., Jr Consideration of molecular weight during compound selection in virtual target-based database screening. J Chem Inf Comput Sci. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- Poncet-Montange G, Assairi L, Arold S, Pochet S, Labesse G. NAD kinases use substrate-assisted catalysis for specific recognition of NAD. J Biol Chem. 2007;282:33925–33934. doi: 10.1074/jbc.M701394200. [DOI] [PubMed] [Google Scholar]
- Rodionov DA, Hebbeln P, Eudes A, ter Beek J, Rodionova IA, Erkens GB, Slotboom DJ, Gelfand MS, Osterman AL, Hanson AD, et al. A novel class of modular transporters for vitamins in prokaryotes. J Bacteriol. 2009;191:42–51. doi: 10.1128/JB.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Sershon VC, Santarsiero BD, Mesecar AD. Kinetic and X-ray structural evidence for negative cooperativity in substrate binding to nicotinate mononucleotide adenylyltransferase (NMAT) from Bacillus anthracis. J Mol Biol. 2009;385:867–888. doi: 10.1016/j.jmb.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK. Screening in a spirit haunted world. Drug Discov Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry. 2007;46:4912–4922. doi: 10.1021/bi6023379. [DOI] [PubMed] [Google Scholar]
- Sorci L, Martynowski D, Rodionov DA, Eyobo Y, Zogaj X, Klose KE, Nikolaev EV, Magni G, Zhang H, Osterman AL. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc Natl Acad Sci U S A. 2009;106:3083–3088. doi: 10.1073/pnas.0811718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- Velu SE, Cristofoli WA, Garcia GJ, Brouillette CG, Pierson MC, Luan CH, DeLucas LJ, Brouillette WJ. Tethered dimers as NAD synthetase inhibitors with antibacterial activity. J Med Chem. 2003;46:3371–3381. doi: 10.1021/jm030003x. [DOI] [PubMed] [Google Scholar]
- Velu SE, Luan CH, Delucas LJ, Brouillette CG, Brouillette WJ. Tethered dimer inhibitors of NAD synthetase: parallel synthesis of an aryl-substituted SAR library. J Comb Chem. 2005;7:898–904. doi: 10.1021/cc050063j. [DOI] [PubMed] [Google Scholar]
- Velu SE, Mou L, Luan CH, Yang ZW, DeLucas LJ, Brouillette CG, Brouillette WJ. Antibacterial nicotinamide adenine dinucleotide synthetase inhibitors: amide- and ether-linked tethered dimers with alpha-amino acid end groups. J Med Chem. 2007;50:2612–2621. doi: 10.1021/jm061349l. [DOI] [PubMed] [Google Scholar]
- Yoon HJ, Kim HL, Mikami B, Suh SW. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Pseudomonas aeruginosa in its Apo and substrate-complexed forms reveals a fully open conformation. J Mol Biol. 2005;351:258–265. doi: 10.1016/j.jmb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou T, Kurnasov O, Cheek S, Grishin NV, Osterman A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure. 2002;10:69–79. doi: 10.1016/s0969-2126(01)00693-1. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- Zhong S, Macias AT, MacKerell AD., Jr Computational identification of inhibitors of protein-protein interactions. Curr Top Med Chem. 2007;7:63–82. doi: 10.2174/156802607779318334. [DOI] [PubMed] [Google Scholar]
- Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem. 2002;277:13148–13154. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.