

Abstract
Human serum albumin (HSA) is not only a fatty acid and drug carrier protein, it is also a potent inhibitor of Aβ self-association in plasma. However, the mechanism underlying the inhibition of Aβ fibrillization by HSA is still not fully understood. We therefore investigated the Aβ-HSA system using a combined experimental strategy based on saturation transfer difference (STD) NMR and intrinsic albumin fluorescence experiments on three Aβ peptides with different aggregation propensities (i.e., Aβ(12–28), Aβ(1–40), and Aβ(1–42)). Our data consistently show that albumin selectively binds to cross-β-structured Aβ oligomers as opposed to Aβ monomers. The HSA/Aβ oligomer complexes have KD values in the micromolar to submicromolar range and compete with the further addition of Aβ monomers to the Aβ assemblies, thus inhibiting fibril growth (“monomer competitor” model). Other putative mechanisms, according to which albumin acts as a “monomer stabilizer” or a “dissociation catalyst”, are not supported by our data, thus resolving previous discrepancies in the literature regarding Aβ-HSA interactions. In addition, the model and the experimental approaches proposed here are anticipated to have broad relevance for the characterization of other systems that involve amyloidogenic peptides and oligomerization inhibitors.
Introduction
A distinctive hallmark of Alzheimer's disease (AD) is the deposition of amyloid plaques in the brain (1). Two major components of these amyloid deposits are the amyloid-β (Aβ) peptides Aβ(1–40) and Aβ(1–42) (2). The Aβ peptides are produced through the proteolytic cleavage of the amyloid precursor protein (APP) (2–4) and are distributed in both the cerebrospinal fluid (CSF) and blood (5). The brain/blood Aβ equilibrium is shifted toward the bloodstream by agents in the peripheral serum that do not penetrate the blood–brain barrier (BBB), but bind the Aβ peptide (5). Such agents have been proposed to act like a “peripheral sink” that lowers the risk of amyloid plaque deposition in the brain and consequently the risk of AD (5). One of the most potent Aβ sequestering systems is human serum albumin (HSA), which under physiological conditions binds >90% of Aβ(1–40) and Aβ(1–42) in blood serum (6,7). Furthermore, HSA is one of the most potent endogenous inhibitors of Aβ fibrillization (6,7). Therefore, the interactions between the Aβ peptides and HSA represent a critical component of the transport and metabolism of the Aβ system, and their investigation may provide clues for possible therapeutic strategies against AD.
Despite the physiological and pharmacological relevance of the Aβ/HSA system, the mechanism underlying the inhibition of Aβ fibrillization by HSA is still not fully understood. At least three main types of models have been proposed for proteins known to prevent peptide amyloidogenesis (Fig. 1, a–c) (8–10). According to model I (Fig. 1 a), the inhibitory protein (P) selectively binds and stabilizes the monomeric form of the amyloidogenic peptide, preventing its self-association. An example of a protein that acts according to such a “monomer stabilizer” mechanism is the phage-display selected affibody ZAβ3, which binds monomeric Aβ(1–40) with nanomolar affinity, effectively preventing its fibrillization (8). Another possible mechanism to explain fibrillization inhibition (model II; Fig. 1 b) assumes that the oligomers that serve as seeds for rapid aggregation are kinetically but not thermodynamically stable (9). In this case, the inhibitory protein P, rather than selectively binding to the monomeric peptide, acts as a catalyst that accelerates the dissociation of the oligomers into monomers (model II; Fig. 1 b). An example of a system that functions according to such a dissociation catalyst mechanism is the molecular chaperone Hsp104, which dissociates the oligomeric Sup35 prion-peptide into monomeric species that are unable to interact with Hsp104 (9). An additional model that does not involve any direct peptide monomer-inhibitory protein interactions is the “monomer competitor” model (model III; Fig. 1 c) in which the inhibitory protein selectively binds to the oligomers, preventing the further addition of peptides and growth into larger oligomer assemblies. An example of such a mechanism is provided by the apolipoprotein E3 (ApoE3), which inhibits fibrillogenesis by binding soluble oligomers (10). From these examples, it is clear that different proteins with fibrillization inhibitory functions often adopt different inhibition mechanisms.
Figure 1.
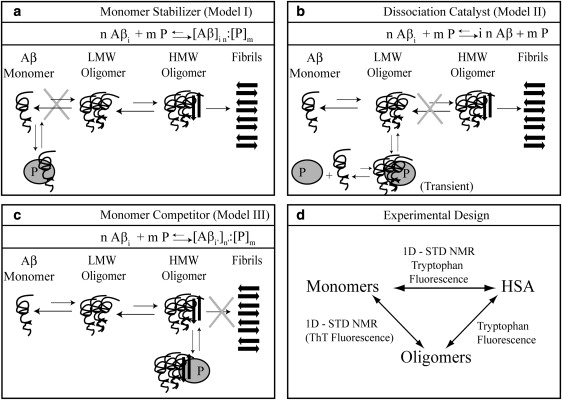
Panels a–c depict possible hypothetical models for the mechanism of oligomerization inhibition by a generic inhibitory protein P. Aβ denotes the Aβ peptide in its monomeric state, whereas Aβi and Aβi' indicate Aβ oligomers. LMW and HMW Aβ refer to low- and high-MW Aβ oligomers, respectively. The letters n, m, and n′ refer to integer numbers that define the stoichiometry of the noncovalent complexes involving the Aβ peptide and the P protein. In both models I and II, oligomers are disrupted (i.e., “cleared”) by P. Whereas an Aβ oligomer-HSA complex in model II forms only transiently, in model III it does not clear the oligomers and HSA binds stably to them, preventing their further growth into larger assemblies. To include the possibility that the inhibitory binding protein partially converts large oligomers into a higher number of smaller oligomers, the subscripts i and n were replaced by i′ and n′ for the P-bound oligomers in model III. In any case, such oligomers must remain larger than the critical size required to interact with the inhibitory protein (denoted as ics in panel c), i.e., i n = i′ n′ and ics < i′ < i. The cartoon representation of the models was used for clarity, but it does not imply a specific pathway for fibril formation or assign specific structures or stoichiometries for Aβ in different oligomerization or HSA-bound states. Panel d summarizes the experimental design to test models I–III. The STD NMR experiments mainly probe interactions with the low-MW components of the system (i.e., Aβ monomers); however, tryptophan fluorescence can probe HSA interactions with both Aβ monomers and oligomers. Due to HSA-ThT interactions, ThT fluorescence can be used to reliably probe cross-β-structured oligomers only in the absence of HSA.
In the case of HSA, based on the data published so far (6,7,11–14), it is not possible to establish consistently and conclusively which model best describes its interactions with the Aβ system and the consequent inhibition of Aβ fibrillization. The monomer stabilizer model (model I; Fig. 1 a) is apparently supported by immunoassays (12) and by circular dichroism (CD) binding studies (13) that have been interpreted in terms of an Aβ monomer:HSA complex with a 1:1 stoichiometry and a dissociation constant in the micromolar range (KD = 5 ± 1 μM) (12,13). However, these results do not agree with surface plasmon resonance (SPR) data (7) showing that albumin interferes with the incorporation of biotin-Aβ(1–40) into amyloid fibrils by selectively binding polymeric but not monomeric derivatized Aβ(1–40). The SPR evidence therefore does not support model I. Although similar conclusions were also obtained through preliminary NMR HSA-binding studies on a short Aβ fragment (11), those early SPR/NMR results were later dismissed by others (13) on the basis of the extensive use of derivatization or fragmentation of the Aβ peptides.
Considering the discrepancies currently present in the literature regarding Aβ-HSA interactions (6,7,11–14), it is important to obtain more direct and conclusive evidence to reliably establish which model (Fig. 1, a–c) best explains the inhibitory action of HSA with respect to Aβ fibrillization. In an attempt to solve these inconsistencies, dynamic light scattering (DLS) was recently employed to map the effect of bovine serum albumin (BSA) on the oligomer size distribution of Aβ(1–40), and the results revealed that albumin shifts the Aβ oligomer distribution toward low-molecular weight (MW) oligomers and possibly monomers (14). However, no conclusive model selection (Fig. 1, a–c) could be established based on this DLS investigation, because interactions of albumin with monomeric Aβ could not be ruled out (14).
Given the incongruities among previous reports (6,7,11–14), our main goal in this study was to differentiate and clarify which of the three hypothetical mechanisms (Fig. 1, a–c) applies to the inhibitory function of Aβ fibrillization by HSA. For that purpose, we investigated the HSA/Aβ system using NMR and tryptophan fluorescence, as illustrated in Fig. 1 d. These two spectroscopic techniques were chosen because they probe the multiple interactions of the HSA/Aβ system in complementary ways. NMR is ideal for sensing interactions involving low-MW species, i.e., mainly the Aβ monomer/HSA and the Aβ monomer/Aβ oligomer interactions (Fig. 1 d), whereas fluorescence takes advantage of the presence of a tryptophan residue in HSA (i.e., W214) but not in the Aβ peptide. As a result, tryptophan fluorescence probes only the interactions involving HSA, i.e., the Aβ monomer/HSA and Aβ oligomer/HSA interactions (Fig. 1 d). Thus, all possible interactions of the HSA/Aβ system are detected by this integrated experimental design, and specifically the putative binding of Aβ monomers by HSA is characterized by two independent methods, providing a solid experimental basis for the selection of the hypothetical models outlined in Fig. 1, a–c.
The experimental scheme illustrated in Fig. 1 d was applied to three different Aβ peptides with increasing length and propensity to oligomerize: Aβ(12–28), Aβ(1–40), and Aβ(1–42). The Aβ(12–28) fragment was chosen because it spans the central hydrophobic core of the Aβ peptide and contains key residues necessary for interactions with HSA (11). Furthermore, the Aβ(12–28) peptide can be easily stabilized for weeks or months in its monomeric state at high concentrations suitable for NMR (11,15). In addition, once Aβ(12–28) oligomers form, their exchange with the monomeric state is sufficiently fast to be detected by saturation transfer difference (STD) NMR experiments with high sensitivity (11,16). However, Aβ(12–28) is only a fragment of the Aβ peptide most commonly found in amyloid plaques in vivo. We therefore complemented our Aβ(12–28) studies with a parallel investigation on the more biologically relevant Aβ(1–40), for which the monomer-to-oligomer transition is sufficiently slow under conditions suitable for NMR to allow the acquisition of NMR data for the monomeric state. Finally, the Aβ(1–42) was selected for its high propensity to self-associate, thus facilitating the detection and characterization of interactions involving the Aβ oligomers. For each Aβ system, the presence of oligomers was independently monitored through STD NMR and thioflavin T (ThT) fluorescence experiments, thus providing a solid foundation for the selection of the inhibitory model (Fig. 1, a–c).
For all three tested peptides, our results consistently support a selective interaction of HSA with Aβ oligomers rather than monomers, which firmly disproves that HSA acts as a “monomer stabilizer,” i.e., model I in Fig. 1 a is excluded. Additionally, our data conclusively rule out the oligomer “dissociation catalyst” model (model II; Fig. 1 b) and confirm the “monomer competitor” mechanism (model III; Fig. 1 c). According to this model, HSA binds Aβ oligomers without causing their full dissociation into monomers, and competes with the addition of further Aβ monomers, effectively preventing the growth of the Aβ oligomers into larger assemblies.
Materials and Methods
Sample preparation
Details about the sample preparation protocols (11,16,17) are available in the Supporting Material.
NMR spectroscopy
The frequency for the saturation of HSA was optimized using a 0.5 mM HSA solution prepared in 50 mM acetic acid-d4 at pH 4.7, 10% D2O, and by setting the carrier frequency of the saturating Gaussian pulse train at -0.26, 0.57, 0.66, and 7.05 ppm while the off-resonance saturation frequency was kept constant at 30 ppm. All STD experiments were acquired using previously described pulse sequences (16,18) and a Bruker Avance 700 MHz spectrometer equipped with 5 mm TCI Cyroprobe (Karlsruhe, Germany) at 20°C, unless otherwise specified. Selective saturation was achieved using a train of 40 Gaussian-shaped pulses of 50 ms each and separated by a 1 ms interpulse delay, resulting in a total saturation time of ∼2 s, which was preceded by a 100 ms interscan delay. The strength of each saturating Gaussian pulse was 110 Hz with a 1% truncation and 1000 digitization points. The STD spectra were obtained by subtracting on-resonance and off-resonance spectra through phase cycling, and the off-resonance spectra were recorded as reference. A 0.1 ms or 30 ms spin lock (SL) was used before signal detection to respectively maximize or suppress the HSA signal. For all STR and STD spectra acquired in the presence of an Aβ peptide, the SL duration was set to 30 ms. In all experiments, the water magnetization was suppressed using the 3-9-19 Watergate gradient spin echo (19). For all STD experiments, 64 scans and 16 dummy scans were acquired, except for Aβ(1–40) and Aβ(1–42), for which 512 scans were accumulated. For the more sensitive saturation transfer reference (STR) experiments, 32 scans and 32 dummy scans were acquired. The spectral processing parameters are included in the figure captions. The methyl spectral region (0.6–1.1 ppm) was integrated and measured as a function of time to monitor the aggregation profile of the Aβ peptides. The error was estimated from the spectral noise to be ∼5%. The one-dimensional (1D) intensity loss was modeled through an offset-exponential: a × e-bt + c, where the a–c parameters were obtained through nonlinear curve-fitting.
Fluorescence spectroscopy
Tryptophan fluorescence spectra were acquired using a Varian Cary Eclipse spectrophotometer (Mulgrave, Australia) and recorded in a 0.5 × 1-cm cell (0.5 cm at the emission and 1 cm at the excitation side) with the excitation and the emission slit width set to 5 nm. An excitation wavelength of 295 nm was used to excite the single tryptophan residue of HSA, Trp-214 (20–22). When fluorescence measurements were used to investigate ligand interactions, spectra for recorded ligand solutions without HSA (blank solutions) were acquired first and then subtracted from the spectra in the presence of HSA. All spectra were recorded three times and smoothed using a 10-point average. ThT fluorescence spectra were recorded using a Tecan Safire fluorescence spectrometer and 96-well plates with 50 μL sample volumes. In this case, excitation and emission wavelengths were set at 450 and 490 nm, respectively (23). Spectra measured before the addition of 20 μM of ThT were subtracted to correct for the blank fluorescence contribution. For each sample, at least four measurements were performed and the resulting averaged values are reported. The error was calculated as the standard deviation of all measurements.
Results
Optimization and controls of STD experiments
A critical step in obtaining a reliable selection among the models outlined in Fig. 1, a–c is to test whether monomeric Aβ peptides interact with albumin. For this purpose, we used 1D STD and reference (STR) NMR spectra, which are ideal for probing interactions with KD values in the micromolar range (24), such as those previously observed between Aβ and HSA (13). In contrast to previous investigations (11), the STD experiments used here were specifically optimized to probe Aβ-HSA interactions. Details about the optimization of the STD experiments and the related controls using known HSA ligands, such as aspirin and L-tryptophan (25,26), are provided in Fig. S1, Fig. S2, and Fig. S3.
HSA does not interact with the monomeric Aβ(12–28) peptide
As the initial step in testing model I (Fig. 1 a), we characterized the interactions between HSA and monomeric Aβ(12–28) through STD NMR experiments. Specifically, we recorded STD data for the Aβ(12–28) peptide (Fig. 2, a–d) using the same acetate buffer and concentrations as in the control spectra with aspirin and L-tryptophan (Fig. S2). Under these experimental conditions, Aβ(12–28) is stable as a monomeric species due to the combination of 30 kDa-cutoff filtration and the subsequent dilution to submillimolar levels included in our sample preparation protocols (11,15–17). The absence of oligomers in the Aβ(12–28) solutions used for the STD experiments is also independently confirmed by the lack of STD signal observed in the absence of HSA (Fig. 2 a), because the 0.66 ppm saturation frequency employed for albumin causes saturation of the methyls of Aβ(12–28) as well (16). Therefore, if Aβ(12–28) oligomers were present, they would give rise to a detectable STD signal in Fig. 2 a, as the monomer/oligomer exchange in acetate buffer has been shown to be sufficiently fast to provide excellent saturation transfer (11,16).
Figure 2.
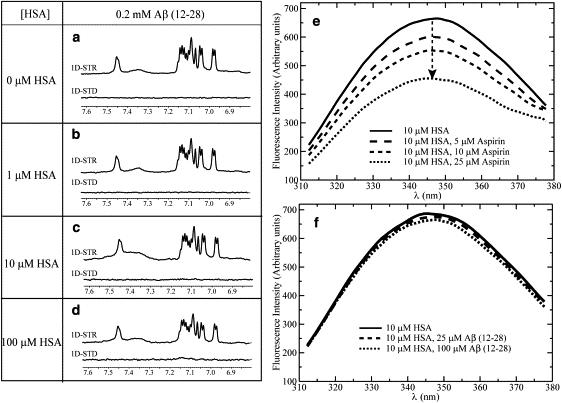
(a–d) Effects of HSA on the STR and STD spectra of 0.2 mM Aβ(12–28) in 50 mM acetic acid-d4, pH 4.7, 10% D2O. The STR and STD spectra of 10 and 100 μM HSA solutions were subtracted from the protein peptide mixture spectra to remove residual HSA signal. All spectra were acquired at 700 MHz using a TCI CryoProbe and at 20°C. A 30-ms-long SL was used to minimize the residual HSA signal. All spectra were processed using a line-broadening factor of 3 Hz. Panels e and f depict the effects of aspirin and Aβ(12–28), respectively, on the emission intrinsic fluorescence spectra of HSA.
It should also be noted that HSA is known to undergo several pH-dependent interdomain reorientations (22), which may affect its ligand interactions (25). The different HSA conformers can be probed using the maximum fluorescence emission wavelength (λmax), as shown in Fig. S3. Fig. S3 indicates that even at pH 4.7, albumin exists mainly in the physiological N-state. The STD data of Fig. 2 are therefore relevant for the N-form of albumin.
The STD and STR data of Fig. 2, b–d, show no significant STD signal or STR line-broadening for Aβ(12–28) at any HSA concentration tested. The absence of STD signal and STR line-broadening in Fig. 2, b–d clearly rules out the possibility that the N-state of HSA interacts with the monomeric Aβ(12–28) peptide with a KD in the micromolar to submillimolar range. It is also highly unlikely that monomeric Aβ(12–28) binds HSA with a lower KD value (i.e., KD ∼ nM), as these high-affinity interactions would result in significant signal losses and/or chemical shift changes in the reference spectrum of Fig. 2 d.
The NMR results for the Aβ(12–28)/HSA system were further supported by independent HSA tryptophan fluorescence experiments. Although wild-type Aβ peptides do not contain Trp residues, a single tryptophan is present in HSA (i.e., Trp-214) and is located in the proximity of Sudlow site I in subdomain IIA (26). Quenching of the intrinsic Trp-214 fluorescence was previously used to probe binding to HSA for several ligands (26). In general, ligand-dependent intrinsic tryptophan fluorescence quenching is either due to direct energy transfer from the albumin fluorophore to the bound ligand fluorophores or due to ligand-induced variations in the local Trp environment (20,21,26). For instance, Fig. 2 e indicates that a significant dose-dependent quenching of the HSA Trp-214 fluorescence occurs upon addition of aspirin in the 5–25 μM concentration range. Considering that aspirin, similarly to the Aβ peptide, does not absorb at the HSA emission wavelength region around 340 nm (27), the data of Fig. 2 e suggest that aspirin perturbs the albumin structure around Trp-214, as also expected since aspirin binds to Sudlow site I in the proximity of Trp-214. However, when similar fluorescence experiments were performed with the Aβ(12–28) peptide prepared in the monomeric state, no significant Trp fluorescence quenching was observed (Fig. 2 f), indicating that monomeric Aβ(12–28) does not affect the environment of Trp-214 in HSA, consistent with the absence of binding as supported by the previous NMR data (Fig. 2, e–d).
HSA does not interact with the monomeric Aβ(1–40) peptide
For the purpose of verifying that the absence of interactions between HSA and Aβ(12–28) is not due to the fragmentation of the Aβ peptide, a similar combined NMR and fluorescence-based experimental strategy was further extended to the biologically relevant Aβ(1–40) peptide prepared under conditions that favor the monomeric state (Figs. 3, a–c, and 4, a and b). Specifically, Aβ(1–40) samples with concentrations ≤100 μM and freshly prepared according to the protocols described in the Supporting Material are largely monomeric due to the lag time of the nucleation phase in the aggregation of Aβ(1–40) (28). This is also independently confirmed by ThT fluorescence, which does not detect any significant concentration of cross-β aggregates for 25 or 100 μM Aβ(1–40) (Fig. 4 a), and by the lack of detectable STD signal for Aβ(1–40) alone (Fig. 3 a). Considering, as discussed above, that the 0.66 ppm irradiation frequency causes saturation of the Aβ methyls as well, the observation of no significant STD signal in Fig. 3 a points to the absence of Aβ(1–40) oligomers for which the peptide exchange with the monomeric state occurs sufficiently fast to give rise to saturation transfer (ST) (16), confirming the monomeric nature of our 100 μM Aβ(1–40) sample. The lack of detectable STD signal for Aβ(1–40) in Fig. 3 a also indicates that the Aβ(1–40) monomers are sufficiently unstructured to quench possible intramonomer cross-saturation effects (16).
Figure 3.
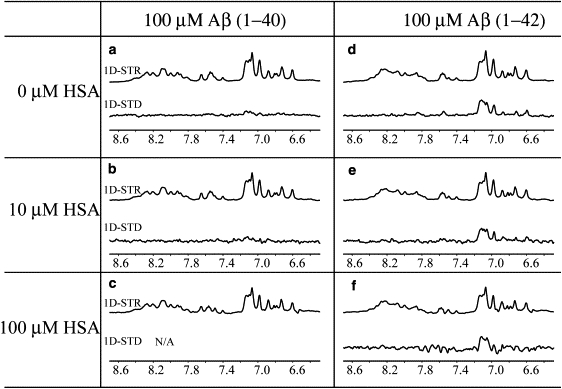
Effect of HSA on the STR and STD spectra of Aβ(1–40) and Aβ(1–42) samples. All peptide solutions were prepared at a 0.1 mM concentration in 20 mM potassium phosphate buffer, pH 7.4, 10% D2O. A 30-ms-long SL was used to minimize the HSA signal. The STR and STD spectra of the 10 and 100 μM HSA solutions were collected and then subtracted from the protein peptide mixture spectra. Although this subtraction was possible for the 100 μM Aβ(1–42) sample, it was not viable for the Aβ(1–40) sample due to the negligible STD effect arising from this peptide as compared to that originating from albumin. All spectra were acquired at 700 MHz using a TCI CryoProbe and at 20°C. The STR and STD spectra were processed using a line-broadening factor of 10 Hz. In panel f, at a 100 μM albumin concentration, the 30-ms SL becomes less effective at completely removing the protein signal, resulting in residual difference artifacts at a range of 7.4–7.8 ppm.
Figure 4.
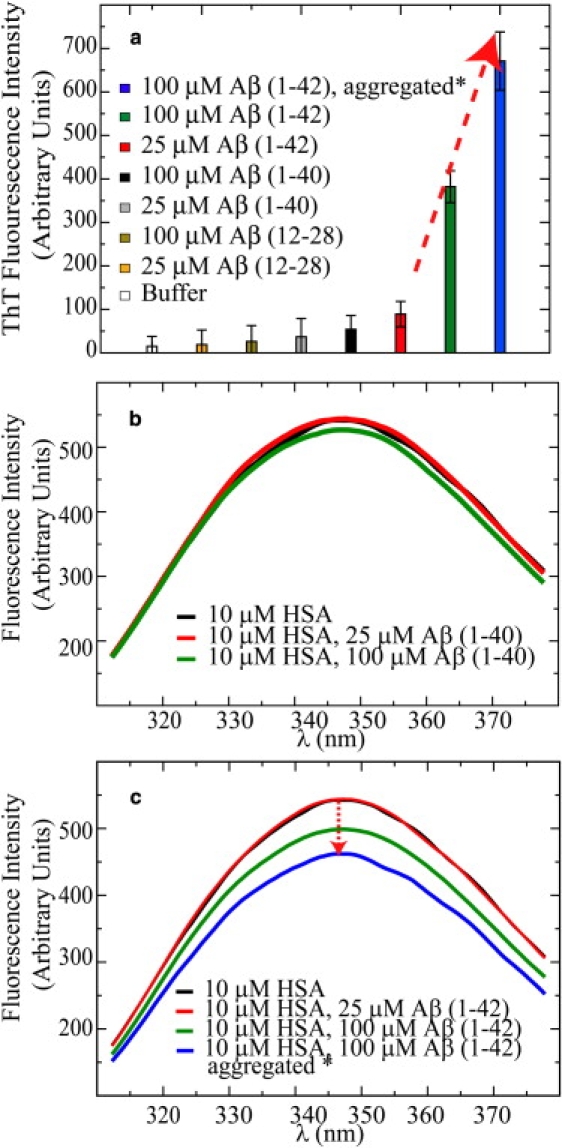
(a) Probing Aβ assemblies using ThT fluorescence. All samples were freshly prepared, with the exception of one 100 μM Aβ(1–42) sample (blue bar) that was aggregated for 3 h at 37°C before the ThT fluorescence measurements. This sample is denoted by an asterisk (∗). ThT was present in all samples at a 20 μM concentration. At least four measurements were collected for each sample, and the average values are reported. The error was calculated as the standard deviation of all measurements. (b) Interactions of HSA with Aβ(1–40) as probed by tryptophan fluorescence. (c) Interaction of HSA with Aβ(1–42) probed by HSA tryptophan fluorescence quenching at increasing Aβ(1–42) concentrations.
The STR spectra of freshly prepared 100 μM Aβ(1–40) in the absence and presence of different concentrations of HSA (Fig. 3, a–c) show no significant changes in line-broadening, chemical shift, or intensity upon addition of HSA to the peptide solution, pointing to the absence of HSA-monomeric Aβ(1–40) interactions in the micromolar KD range. This conclusion is further confirmed by the lack of signal in the STD spectrum of Fig. 3 b and by the absence of significant variations in the HSA Trp fluorescence spectra acquired even with 10-fold excess of Aβ(1−40) (Fig. 4 b), further confirming the absence of interactions in the micromolar affinity range between HSA and monomeric Aβ (1–40).
HSA selectively interacts with the oligomeric but not the monomeric Aβ(1–42)
At a peptide concentration of 25 μM, and using sample preparation protocols that minimize self-association (28), it is possible to obtain Aβ(1–42) solutions for which the ThT fluorescence is reduced close to basal levels (Fig. 4 a), suggesting the absence of significant amounts of cross-β-structured aggregates (29). Under these conditions, no appreciable quenching of the intrinsic albumin fluorescence is observed upon addition of Aβ(1–42) (Fig. 4 c), which is consistent with the absence of interactions between HSA and monomeric Aβ(1–42) with a KD in the micromolar range. However, when the self-assembly of Aβ(1–42) is promoted by increasing its concentration to 100 μM, not only is significant ThT binding observed (Fig. 4 a), pointing to the presence of assemblies with cross-β structure (29), but also the intrinsic albumin fluorescence is quenched (Fig. 4 c), suggesting that albumin selectively binds oligomeric Aβ(1–42) species. A positive control of these results is provided by a 100 μM Aβ(1–42) sample that was further aggregated for 3 h at 37°C before performing the ThT measurements and adding HSA. This sample showed increased ThT fluorescence (Fig. 4 a) and also resulted in an additional quenching of the HSA intrinsic fluorescence (Fig. 4 c), confirming that HSA selectively targets oligomers with cross-β structures as opposed to monomers.
As a further control for the absence of HSA-Aβ(1–42) interactions, the STD and STR spectra of freshly prepared 100 μM Aβ(1–42) were acquired in both the absence and presence of increasing concentrations of albumin (Fig. 3, d–f). As shown in Fig. 3, d–f, no significant change is observed in the STR spectra of Aβ(1–42) upon addition of HSA up to a 1:1 stoichiometric ratio, confirming that albumin does not bind monomeric Aβ(1–42) in the micromolar or submillimolar range. In addition, it is interesting to note that, unlike Aβ(1–40) (Fig. 3 a), Aβ(1–42) gives rise to a significant STD signal even in the absence of albumin (Fig. 3 d). This observation is consistent with intramonomer cross-saturation caused by at least partial structuring of the monomeric Aβ(1–42) peptide and/or with the presence of oligomers that are sufficiently small to give rise to a detectable NMR signal or are in fast/intermediate exchange with the monomeric state. In either case, these oligomers are likely occurring early in the fibrillization pathways. The absence of STD changes upon albumin addition (Fig. 3, d–f) is therefore consistent with a selective interaction of HSA with larger assemblies, such as those detected by ThT fluorescence but not STD, as their exchange with the monomeric state is too slow and/or their concentration is too low to be sensed by STR/STD NMR. However, when we compare ThT and STD as oligomer detection methods, we should also consider that, unlike STD, the usefulness of ThT fluorescence measurements for probing the presence of assemblies with cross-β structure is mainly limited to Aβ solutions without any albumin. This is because ThT interacts with albumin, as proven by the STD spectra in Fig. S4. In addition, recent independent studies also reported that ThT binds albumin with a KD of ∼10 μM (30). Such ThT-albumin interactions may therefore affect and bias experiments in which ThT fluorescence is employed to monitor Aβ aggregation in the presence of albumin, as previously attempted (14).
Albumin inhibits Aβ(1–42) fibril growth at substoichiometric concentrations
The analysis presented above clearly shows that HSA targets Aβ oligomers rather than monomers as entailed by models II and III (Fig. 1, a–c); however, a better differentiation between these two remaining putative mechanisms requires additional experiments. For this purpose, we measured the effect of HSA on the self-association of the Aβ(1–42) peptide at 37°C, as monitored through 1D NMR signal losses over a period of 23 h (Fig. 5 a). Fig. 5 a shows that in the absence of oligomerization inhibitors, >50% of the original 1D signal is already lost 3 h after the acquisition of the first 1D NMR spectrum. After this rapid initial NMR signal loss, a slower decay is observed that levels off at a plateau of ∼22–25% of the original 1D intensity (Fig. 5 a). No detectable line-broadening was observed in the NMR spectra of Aβ(1–42) during the course of its aggregation (Fig. 6), indicating that the exchange beteween NMR-detectable and NMR-undetectable species of Aβ(1–42) is slow on the chemical shift timescale (> ms). The absence of line-broadening over time also indicates that the observed NMR signal loss is due to the sequestration of Aβ(1–42) monomers into NMR undetectable oligomers, protofibrils, and fibrils.
Figure 5.
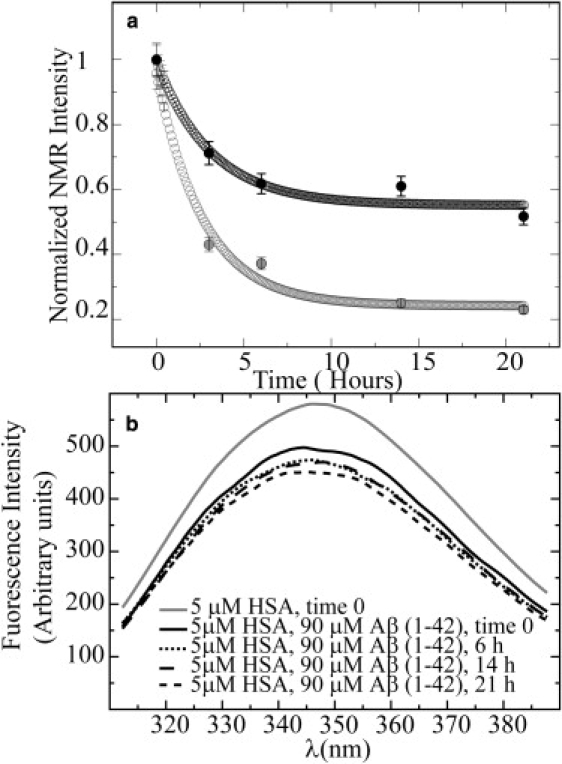
Time-dependent aggregation of 90 μM Aβ(1–42) in the absence (gray curve) and presence (black curve) of 5 μM HSA as monitored by NMR 1D NMR spectra with a 30-ms SL filter (a) and by intrinsic HSA tryptophan fluorescence (b). The experimental data were fitted using the offset decaying exponential: a × e-bt + c, where t is in hours and the a–c parameters were obtained through nonlinear curve-fitting. The actual experimental data are plotted in solid circles, and the fitted values are shown in open circles. Between the readings, samples were incubated in a water bath at 37°C. The NMR intensities reported in panel a are normalized intensities of the methyl spectral region (0.6–1.1 ppm) measured as a function of time. The error was estimated from the spectral noise to be ∼5%. NMR experiments were acquired at 700 MHz at 37°C in 20 mM potassium phosphate, pH 7.4, 10% D2O, 0.02% NaN3.
Figure 6.
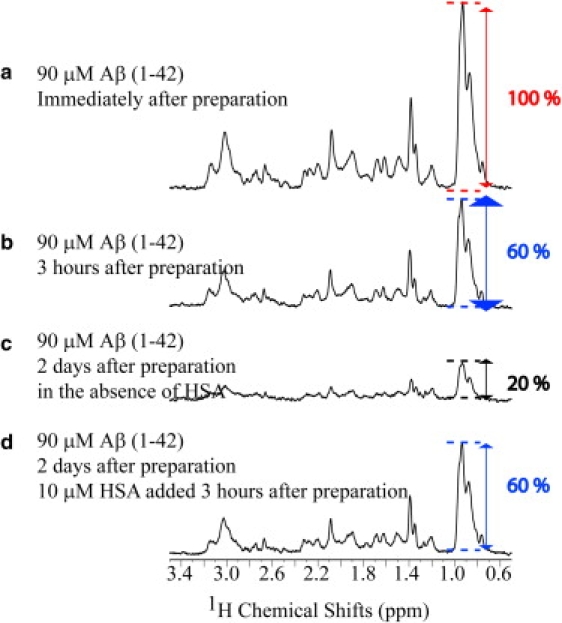
Effect of a delayed addition of HSA on the aggregation profile of Aβ(1–42). Panels a and b show the 1D NMR spectra of 90 μM Aβ(1–42) immediately after preparation and after 3 h, respectively. In the absence of HSA, 2 days after the sample was prepared, ∼80% of the initial NMR signal is lost, as shown in panel c. However, when 10 μM of HSA were added 3 h after sample preparation, no NMR signal losses were observed even after 2 days (d). These spectra were recorded at 600 MHz and 37°C. In between acquisition sessions, samples were stored in a water bath at 37°C. Note that the Aβ(1–42) samples used here and in Fig. 5 came from different stock solutions.
In the presence of HSA at substoichiometric ratios (i.e., 18/1 = [Aβ(1–42)]/[HSA]), the overall loss of the peptide NMR signal over time is significantly reduced relative to what was observed for Aβ(1–42) alone (Fig. 5 a). In the presence of albumin, the plateau value reached after the initial rapid decay stabilizes at ∼50–60% of the original NMR intensity, i.e., at approximately twice the NMR signal intensity detected in the absence of albumin. Since no direct contribution from the NMR resonances of albumin is expected in these spectra due to the low protein concentration, as well as the 30-ms SL filter employed for these 1D spectra, the NMR data in Fig. 5 a demonstrate that substoichiometric concentrations of albumin are sufficient to inhibit the transition of Aβ(1–42) from NMR-detectable monomeric species into large, NMR-undetectable assemblies. Of interest, line-fitting with an offset-exponential decay function (Fig. 5 a) shows that, unlike the plateau value, the initial rate of decay is not dramatically affected by albumin (i.e., −0.37 h−1 and −0.32 h−1 in the absence and presence of HSA, respectively). Assuming that the initial decay rate senses mostly the prenuclear early-oligomer seeded self-association, whereas the plateau height mainly probes the residual monomers or NMR-detectable oligomers in equilibrium with fibrils, protofibrils, and large oligomers, the data of Fig. 5 a support the conclusion that HSA targets mainly larger oligomers, in line with our fluorescence and STD results (Figs. 3 and 4).
The partial aggregation of Aβ(1–42) in the presence of HSA was also monitored through albumin intrinsic Trp fluorescence spectra (Fig. 5 b) acquired in parallel with the NMR data of Fig. 5 a and under experimental conditions similar to those used for the NMR experiments. The fluorescence data of Fig. 5 b show that a significant quenching occurs for all time points sampled after the addition of 90 μM Aβ(1–42). This is because at these concentrations, samples of freshly prepared Aβ(1–42) peptide already contain significant amounts of aggregates with cross-β structure, as indicated by the previous ThT fluorescence data (Fig. 4 a). Overall, Fig. 5 shows that albumin is able to inhibit the time-dependent shift of the Aβ(1–42) oligomer distribution toward high-MW assemblies (Fig. 5 a) by binding to cross-β-structured aggregates (Fig. 5 b). This figure also illustrates the complementarity of NMR and fluorescence experiments. NMR reports mainly on the low-MW Aβ species in solution, which are unlikely to interact directly with albumin, and therefore the intensity of the first point in Fig. 5 a is not affected by the addition of HSA. Conversely, the intrinsic albumin fluorescence also senses interactions with high-MW Aβ oligomers that would otherwise escape detection by NMR. This is why in Fig. 5 b, the major variation of Trp fluorescence is observed for the very first time point, unlike what is observed for the NMR time profile of Fig. 5 a.
Albumin does not cause the dissociation of Aβ oligomers into monomers
The picture emerging from the data presented above appears to be consistent with model III (Fig. 1 c) as opposed to model II (Fig. 1 b). However, model II (Fig. 1 b), according to which albumin promotes the dissociation of preexisting oligomers, is still able to explain the observed inhibition of Aβ fibrillization at substoichiometric amounts of HSA (Fig. 5 a). Therefore, before conclusively ruling out model II, we carried out additional experiments by adding albumin to solutions in which significant populations of Aβ oligomers were preformed. For instance, Fig. 6 illustrates that 3 h after preparation of the Aβ(1–42) samples, a significant loss of 1D signal intensity is already observed (Fig. 6, a and b), pointing to the initial formation of assemblies with sizes beyond the NMR detection limit. If self-association continues after 3 h in the absence of inhibitory protein, most of the NMR signal is lost after 2 days (Fig. 6 c). However, if HSA is added 3 h after preparation, the intensity of the NMR intensity remains comparable to that measured before the addition of albumin (Fig. 6, b and d), without the signal losses observed in the absence of HSA over the same time span (Fig. 6 c), but also without regaining the signal measured immediately after sample preparation (Fig. 6 a). These observations therefore support the notion that HSA does not cause the Aβ(1–42) oligomers to dissociate into monomers. Similar results were obtained for the Aβ(12–28) peptide, as indicated in Fig. S5 and Fig. S6 and explained in the Supporting Material.
Discussion
The data presented here (Figs. 2–5) consistently support the notion that albumin does not bind Aβ peptides that are predominantly in the monomeric or low-MW oligomeric states and lack a ThT-binding competent cross-β structure. This conclusion applies not only to the Aβ(12–28) fragment model system (Fig. 2), but also to the longer Aβ(1–40) and Aβ(1–42) peptides (Figs. 3–5), and is supported by both NMR data and independent HSA intrinsic Trp fluorescence experiments with an extensive series of positive and negative controls (Figs. 2–5, and Fig. S1, Fig. S2, Fig. S3, and Fig. S4). On this basis, it is therefore possible to rule out that albumin inhibits Aβ fibrillization though a “monomer stabilizer” mechanism (i.e., model I; Fig. 1 a). At this stage, the “dissociation catalyst” and “monomer competitor” models (models II and III; Fig. 1, b and c, respectively), which involve interactions with Aβ oligomers, appear more plausible. Indeed, interactions between albumin and the Aβ peptide oligomers with a KD in the micromolar to submicromolar range are fully supported by the quenching observed in the HSA intrinsic fluorescence spectra (Figs. 4 c and 5 b) whenever cross-β-structured Aβ aggregates are present as indicated by detectable levels of ThT fluorescence (Fig. 4 a). Overall, the quenching of the HSA intrinsic fluorescence observed upon addition of HSA appears strikingly well correlated with the intensity of the ThT fluorescence detected before HSA addition (Fig. 4), as one would expect if the HSA binding-competent forms of the Aβ peptides are assemblies with cross-β structure.
Our NMR data for experiments in which albumin was added to preformed Aβ assemblies (Fig. 6 and Fig. S5) show that the interaction between albumin and the Aβ oligomers does not cause the dissociation of Aβ into monomers, as predicted by the dissociation catalyst model (model II; Fig. 1 b), but it inhibits the transition of the Aβ oligomer distribution from monomeric or low-MW species into larger, NMR-undetectable, fibril-like assemblies that would otherwise occur in the absence of albumin (Fig. 5 a) (31). This HSA-dependent accumulation of monomeric and low-MW Aβ oligomers is observed even at substoichiometric (i.e., 18:1) concentrations of albumin (Fig. 5 a) and it is fully accounted for by the monomer competitor model (model III; Fig. 1 c), assuming that HSA interacts with larger Aβ oligomers at the sites where further monomers would otherwise bind. Typically, the number of such sites of monomer addition is significantly lower than the number of polypeptide chains in an oligomer or fibril, and therefore the binding of substoichiometric amounts of HSA to cross-β-structured oligomers is in principle sufficient to inhibit further addition of the monomers to the growing aggregates, thus preventing the consequent loss of NMR signal as observed in Fig. 5 a. For further details about the effect of HSA on the mechanism of homogenous nucleation growth, see the Supporting Material.
The monomer competitor model (model III; Fig. 1 c) supported by our data is consistent with previously published dynamic light scattering (DLS) (14) and surface plasmon resonance (SPR) (7) experiments. DLS profiles indicate that when Aβ(1–40) aggregates in the presence of the HSA homolog BSA, the population of large oligomers (i.e., Stokes radius ∼ 140 nm) decreases and that of smaller assemblies (i.e., Stokes radius ≤ 34 nm) increases, consistent with the accumulation of low-MW species in the presence of HSA (Fig. 5 a). However, the DLS experiments alone are not sufficient to rule out possible Aβ monomer-HSA interactions (14). In this respect, SPR is complementary to DLS. SPR does not map the oligomer redistribution of the Aβ peptide, but it can probe direct albumin-Aβ monomer interactions. Specifically, SPR did not detect any interactions with HSA or BSA when monomeric biotin-Aβ(1–40) was immobilized on the sensor chip, whereas when the SPR experiment was repeated with preformed polymers of Aβ(1–42) deposited through a monoclonal antibody, clear evidence for binding with a micromolar or higher affinity was obtained (7). These SPR based results are fully consistent with the selectivity of HSA for large cross-β-structured Aβ assemblies as opposed to Aβ monomers, indicating that the SPR data were not biased by the derivatization of the Aβ peptide with biotin and/or by the type of chip-anchoring technique used, as previously speculated (13).
Although our conclusions regarding the monomer competitor model (model III; Fig. 1 c) are consistent with and supported by previous DLS and SPR results, they do not agree with previous investigations based on CD spectra (13) and immunoassays that were interpreted in terms of binary 1:1 complexes between Aβ monomers and HSA (12). A possible explanation for this discrepancy may lie in the presence of undetected Aβ oligomers in equilibrium with Aβ monomers in the samples used for the CD and immunoassay experiments. Indeed, the Hill coefficients reported for the binding of Aβ(1–40) to HSA are in the 1.4–1.5 range (13), pointing to a higher cooperativity than that anticipated based on the formation of 1:1 complexes between Aβ monomers and HSA. Another challenge encountered when CD experiments are used to monitor Aβ-HSA interactions is the deconvolution of the contributions to the CD spectra from these two interacting components due to the poor selectivity of CD for Aβ and HSA. This problem is effectively circumvented by using either intrinsic Trp fluorescence (since Trp is present only in HSA and not in the Aβ peptides) or T1ρ-filtered NMR experiments, where the long (30 ms) SL filters out most contributions from albumin, while preserving the signal of the low-MW Aβ peptides.
Conclusions
In this work we characterized Aβ-HSA interactions using a combined experimental strategy based on NMR and intrinsic albumin fluorescence, and applied it to a family of Aβ peptides (Aβ(12–28), Aβ(1–40), and Aβ(1–42)) for which the oligomeric state was independently assessed by ThT fluorescence and STD NMR experiments. Based on our NMR and fluorescence data, and on an extensive series of positive and negative controls, we show that albumin selectively binds cross-β-structured Aβ assemblies as opposed to Aβ monomers. This binding occurs with a KD in the micromolar or submicromolar range and competes with the further growth of the HSA-bound Aβ oligomers through monomer addition. Overall, our data clearly support a monomer competitor model for the Aβ fibrillization inhibitor function of HSA, resolving previous discrepancies in the literature regarding Aβ-HSA interactions. Other putative mechanisms, according to which albumin acts as a monomer stabilizer or a dissociation catalyst, are not supported by our data.
The proposed model for the Aβ-HSA interactions not only explains how albumin is able to affect fibrillization, it will also help elucidate how inhibitory proteins affect the distribution of toxic soluble oligomers of amyloidogenic peptides in general, such as the islet amyloid polypeptide (32). Furthermore, the combined STD NMR/fluorescence approach presented here is likely to be generally suitable for the investigation of other systems involving the interactions of amyloidogenic peptides with fibrillization-inhibitory proteins.
Acknowledgments
We thank Tyler E. McNicholl, Elle Dunitz, Rajeevan Selvaratnam, Dr. J. Ortega, Dr. A. Bain, Dr. Rahul Das, and Dr. Somenath Chowdhury for helpful discussions, and Dr. J. Brennan for access to the fluorescence facilities.
G.M. received financial support from the National Sciences and Engineering Research Council and the Alzheimer Society of Canada, and a Maureen Andrew New Investigator award from the Heart and Stroke Foundation of Canada. J.M. was funded by a graduate fellowship from the National Sciences and Engineering Research Council.
Supporting Material
References
- 1.Cummings J.L. Alzheimer's disease. N. Engl. J. Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- 2.Haass C., Selkoe D.J. Cellular processing of β- amyloid precursor protein and the genesis of amyloid β -peptide. Cell. 1993;73:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- 3.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R. Naturally secreted oligomers of the Alzheimer amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 4.Dahlgren K.N., Manelli A.M., Stine W.K., Baker L.K., Jr., Krafft G.A. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 5.Zlokovic B.V. Clearing amyloid through the blood–brain barrier. J. Neurochem. 2004;89:807–811. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]
- 6.Biere A.L., Ostaszewski B., Stimson E.R., Hyman B.T., Maggio J.E. Amyloid β -peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 7.Bohrmann B., Tjernberg L., Kuner P., Poli S., Levet-Trafit B. Endogenous proteins controlling amyloid β -peptide polymerization. J. Biol. Chem. 1999;274:15990–15995. doi: 10.1074/jbc.274.23.15990. [DOI] [PubMed] [Google Scholar]
- 8.Hoyer W., Gronwall C., Jonsson A., Stahl S., Hard T. Stabilization of a β-hairpin in monomeric Alzheimer's amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narayanan S., Bosl B., Walter S., Reif B. Importance of low-oligomeric-weight species for prion propagation in the yeast prion system Sup35/Hsp104. Proc. Natl. Acad. Sci. USA. 2003;100:9286–9291. doi: 10.1073/pnas.1233535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans K.C., Berger E.P., Cho C.G., Weisgraber K.H., Lansbury P.T. Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1995;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milojevic J., Esposito V., Das R., Melacini G. Understanding the molecular basis for the inhibition of the Alzheimer's Aβ-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J. Am. Chem. Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- 12.Kuo Y.M., Kokjohn T.A., Kalback W., Luehrs D., Galasko D.R. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 2000;268:750–756. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 13.Rozga M., Koniecki M., Jabonowskaa M., Dadlez M., Bala W. The binding constant for amyloid Aβ 40 peptide interaction with human serum albumin. Biochem. Biophys. Res. Commun. 2000;364:714–718. doi: 10.1016/j.bbrc.2007.10.080. [DOI] [PubMed] [Google Scholar]
- 14.Reyes Barcelo A.A., Gonzalez-Velasquez F.J., Moss M.A. Soluble aggregates of the amyloid-β peptide are trapped by serum albumin to enhance amyloid-β activation of endothelial cells. J. Biol. Eng. 2009;3:1–8. doi: 10.1186/1754-1611-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarvet J., Damberg P., Bodell K., Erksson L.E.G., Graslund A. Reversible random coil to β-sheet transition and the early stage of aggregation of the A β (12–28) fragment from the Alzheimer peptide. J. Am. Chem. Soc. 2000;122:4261–4268. [Google Scholar]
- 16.Huang H., Milojevic J., Melacini G. Analysis and optimization of saturation transfer difference NMR experiments designed to map early self-association events in amyloidogenic peptides. J. Phys. Chem. B. 2008;112:5795–5802. doi: 10.1021/jp7118718. [DOI] [PubMed] [Google Scholar]
- 17.Esposito V., Das R., Melacini G. Mapping polypeptide self-recognition through 1H off-resonance relaxation. J. Am. Chem. Soc. 2005;127:9358–9359. doi: 10.1021/ja051714i. [DOI] [PubMed] [Google Scholar]
- 18.Mayer M., Mayer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 19.Piotta M., Saudek V., Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–666. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 20.Thumser A.E.A., Buckland A.G., Wilton D.C. Monoacylglycerol binding to human serum albumin: evidence that monooleoylglycerol binds at the dansylsarcosine site. J. Lipid Res. 1998;39:1033–1038. [PubMed] [Google Scholar]
- 21.Bojko B., Sulkowska A., Maciazek M., Rownicka J., Njau F. Changes of serum albumin affinity for aspirin induced by fatty acid. Int. J. Biol. Macromol. 2008;42:314–323. doi: 10.1016/j.ijbiomac.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Dockal M., Carter C.D., Ruker F. Conformational transitions of the three recombinant domains of human serum albumin depending on pH. J. Biol. Chem. 2000;275:3042–3050. doi: 10.1074/jbc.275.5.3042. [DOI] [PubMed] [Google Scholar]
- 23.Benseny-Cases N., Cocera M., Cladera J. Conversion of non-fibrillar β-sheet oligomers into amyloid fibrils in Alzheimer's disease amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2007;361:916–921. doi: 10.1016/j.bbrc.2007.07.082. [DOI] [PubMed] [Google Scholar]
- 24.Stockman B.J., Dalvit C. NMR screening techniques in drug discovery and drug design. Prog. Nucl. Magn. Reson. Spectrosc. 2002;41:187–231. [Google Scholar]
- 25.Yang J., Hage D.S. Role of binding capacity versus binding strength in the separation of chiral compounds on protein-based high-performance liquid chromatography columns interactions of D- and L-tryptophan with human serum albumin. J. Chromatogr. A. 1996;725:273–285. doi: 10.1016/0021-9673(95)01009-2. [DOI] [PubMed] [Google Scholar]
- 26.Bocedi A., Notaril S., Narciso P., Alessandro B., Fasano M. Binding of anti-HIV drugs to human serum albumin. IUBMB Life. 2004;56:609–614. doi: 10.1080/15216540400016286. [DOI] [PubMed] [Google Scholar]
- 27.Miles I.C., Schenk H.G. Fluorescence of acetylsalicylic acid in solution and its measurement in presence of salicylic acid. Anal. Chem. 1970;42:656–659. doi: 10.1021/ac60288a032. [DOI] [PubMed] [Google Scholar]
- 28.Hou L., Shao H., Zhang Y., Li H., Menon N.K. Solution NMR studies of the Aβ (1–40) and Aβ (1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 29.Walsh P., Yau J., Simonetti K., Sharpe S. Morphology and secondary structure of stable β-oligomers formed by amyloid peptide PrP(106–126) Biochemistry. 2009;48:5779–5781. doi: 10.1021/bi9007319. [DOI] [PubMed] [Google Scholar]
- 30.Priyankar S., Sadaf F., Basir A., Rizwan H.K. Interactions of thioflavin T with serum albumins: spectroscopic analyses. Spectrochim. Acta Part A. 2009;74:94–99. doi: 10.1016/j.saa.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Lomakin A., Chung D.S., Benedek G.B., Kirschner D.A., Teplow D.B. On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nanga R.P.R., Brender J.R., Xu J., Hartman K., Subramanian V. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J. Am. Chem. Soc. 2009;131:8252–8261. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.