

Abstract
The assembly and budding of a new virus is a fundamental step in retroviral replication. Yet, despite substantial progress in the structural and biochemical characterization of retroviral budding, the underlying physical mechanism remains poorly understood, particularly with respect to the mechanism by which the virus overcomes the energy barrier associated with the formation of high membrane curvature during viral budding. Using atomic force, fluorescence, and transmission electron microscopy, we find that both human immunodeficiency virus and Moloney murine leukemia virus remodel the actin cytoskeleton of their host. These actin-filamentous structures assemble simultaneously with or immediately after the beginning of budding, and disappear as soon as the nascent virus is released from the cell membrane. Analysis of sections of cryopreserved virus-infected cells by transmission electron microscopy reveals similar actin filament structures emerging from every nascent virus. Substitution of the nucleocapsid domain implicated in actin binding by a leucine-zipper domain results in the budding of virus-like particles without remodeling of the cell's cytoskeleton. Notably, viruses carrying the modified nucleocapsid domains bud more slowly by an order of magnitude compared to the wild-type. The results of this study show that retroviruses utilize the cell cytoskeleton to expedite their assembly and budding.
Introduction
The cytoskeleton is a dynamic network of tubulin, actin, and intermediate filaments that play important roles in cells by providing mechanical stability, generating forces, and acting as tracks for intracellular transport. Although the actin network is primarily associated with mechanical stability, cell motility, and cell contraction, it is also important for internal transport, particularly near the plasma membrane. Cargos can be transported both by riding on myosin motors along actin filaments and by the pushing forces exerted by actin as it undergoes polymerization (1,2). The force generated by the polymerization of actin filaments can be appreciated by studying filopodia formation (3). The growth of actin bundles induces these protrusions, which are formed at the edge of many cells and are important for locomotion.
Similarly, many parasites have developed intricate mechanisms to take control of the cytoskeleton so that they may harness the forces generated by actin filaments to leave the host cell (for a review, see Gouin et al. (4)). Bacteria within the genera of Listeria and Shigela induce actin-polymerization, which leads to the formation of a comet tail that pushes the bacteria within the cell and into target cells (5,6). The vaccinia virus exits the host cell but stays bound to its surface, and induces actin-polymerization from the outside, giving rise to an actin tail that pushes the virus into the target cell (7). The African swine fever virus has been shown to induce the formation of very long filopodia-like protrusions that are thought to enhance virus spreading (8).
The role of the host-cell's cytoskeleton in retroviral replication is less understood. Moloney murine leukemia virus (MLV) can move along filopodial bridges from an infected cell toward an uninfected target cell (9,10). In several types of retroviruses, interactions between viral factors and the host cell's cytoskeleton have been shown to exist during the early stages of infection (11). Actin has been identified inside human immunodeficiency virus (HIV-1) particles, but evidence for a direct interaction during assembly and budding has so far been inconclusive (11). The formation of a new virus is a fundamental step in the late stages of retroviral replication. In HIV, MLV, and other retroviruses, both assembly and budding are thought to occur primarily at the plasma membrane of the infected cell. According to the current model, self-assembly of viral proteins induces a protrusion in the plasma membrane, which grows until the nascent virion is tethered to the cell membrane by a stalk (12). Finally, the virus is released from the plasma membrane by membrane fission mediated by cellular proteins normally involved in the endosomal pathway (13). It is widely accepted that an active actin network is important for efficient formation of nascent virions; however, the mechanism by which the cytoskeleton influences budding is unknown (11,14,15).
To shed light on the role of the cytoskeleton in retroviral budding, we monitored the formation of single viruses using fluorescence and atomic force microscopy (AFM), as well as transmission electron microscopy (TEM) of cryopreserved MLV-infected cells. We found large-scale changes in the host-cell cytoskeleton that impact the morphology of the cell. Intriguingly, both HIV and MLV budding sites are characterized by highly dynamic actin-filamentous structures that emanate from the virion during its assembly. Based on our finding, we propose what we believe is a novel mechanism by which the cytoskeleton directly influences retroviral assembly and budding.
Materials and Methods
Cell culture and transfections
Standard NIH-3T3 murine fibroblasts and 3T3 cells stably infected with wild-type (WT) MoMLV (CL-1, a generous gift from Professor J. M. Cunningham, Harvard Medical School, Boston, MA), as well as HIV-infected HeLa cells (i.e., human epithelial cells) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Israel) supplemented with 10% fetal calf serum (Gibco) and antibiotics (complete medium). For AFM imaging, cells were plated in 35 mm cell culture dishes and grown to ∼50% confluence. CL-1 cells were typically imaged 24–48 h after plating. To maintain a high density of viral budding events, only cells that were cultured for <10 passages were used. For the production of HIV-expressing cells, NIH-3T3 or HeLa cells were cotransfected with the HIV vector pNL-4.3 ΔEnv-Int and HXB2-Env (generous gifts of Professor M. S. Kay, University of Utah, Salt-Lake City, UT). Cells producing virus-like particles (VLPs) were transfected with WT-Gag plasmid (pCMV55M1-10) or a Gag chimera plasmid (Leu-Gag (16)) in which the nucleocapsid (NC) domain of Gag is replaced by a leucine-zipper domain (both plasmids were generous gifts from Professor A. Rein, National Cancer Institute, Frederick, MD). All transfections were performed using the Lipofectamine 2000 transfection agent (Invitrogen, Carlsbad, CA). For enhanced transfection yield, the pAdvantage vector (Promega, Madison, WI) was included. Cells were cotransfected with GFP for visual detection of successfully transfected cells, and typically were imaged 30–40 h after transfection.
AFM imaging of live cells
AFM imaging was carried out using a Bioscope with a Nanoscope IV controller (Veeco, Santa Barbara, CA) that was mounted onto an inverted optical microscope (Axiovert 200M; Carl Zeiss, Heidelberg, Germany). Images of cells were acquired in AFM tapping mode in a fluid environment. Pyramidal silicon nitride triangular cantilevers (DNP) with a nominal stiffness of 0.2 N/m were used. The cantilevers had a nominal tip radius of 20 nm. To properly maintain the cells, a 5% CO2 atmosphere and temperature of 37°C were maintained for the entire duration of the measurements by means of a CO2 controller (Carl Zeiss, Heidelberg, Germany) and a temperature controller (model 3040; Newport, Irvine, CA). This was achieved in a micro-incubator perfusion chamber (model PDMI-2; Harvard Apparatus, Holliston, MA) that held a 35 mm cell culture dish and was attached to the microscope stage. Images of cells were rendered with the use of WSxM software (Nanotec Electronica, http://www.nanotec.es/progcorn.htm) (17). The kinetics of viral budding was obtained by means of a method we described previously (18). Briefly, the progress of budding was determined by measuring the viral protrusion height as a function of time. With this method, single particles were tracked frame by frame to extract their budding kinetics pattern.
Fluorescence microscopy
Cells were cultured on a microscope coverglass (EMS No. 1.5, Hatfield, PA). For costaining of actin and viral proteins, cells were fixed and permeabilized by incubation with a solution of 3% paraformaldehyde (PF) (Baker Chemical Co., Philipsburg, NJ) and 0.5% Triton X-100 in phosphate-buffered saline (PBS; Gibco), pH 7.4, for 3 min at 37°C, followed by fixation in 3% PF for 30 min at 37°C. The cells were washed in PBS with 0.1% bovine serum albumin (BSA), blocked in 1% BSA for 30 min, incubated with rabbit α-gp70 (MLV Env, a gift from Professor A. Rein) for 1 h, washed in PBS with 0.1% BSA, and then incubated in fluorescein (FITC)-conjugated donkey anti-rabbit secondary antibody for 1 h. F-actin was stained by incubation with 1 μM tetramethyl-rhodamine-isothiocyanate (TRITC) conjugated to phalloidin (Fluka, Buchs, SG, Switzerland) in PBS for 20 min. After extensive washing, VectaShield mounting medium (Vector Laboratories, Burlingame, CA) was added to the coverslides before they were mounted and sealed with transparent nail polish. To image the fine architecture of actin, cells were fixed and then permeabilized in a solution containing 3% PF, 0.05% glutaraldehyde (EM grade), and 0.25% Triton X-100 for 15 min at 37°C. They were then washed with PBS, treated with NaBH4, stained with TRITC-phalloidin, and mounted. The cells were imaged by epifluorescence (Axiovert 200M; Carl Zeiss, Heidelberg, Germany) and by confocal laser-scanning microscopy (Model, Olympus). Images were deconvolved and rendered using the Huygens (SVI, Hilversum, Holland) and Imaris (Bitplane AG, Zurich, Switzerland) software programs, respectively.
Electron microscopy
Cells were grown on sapphire disks (30 μm thick, 3 mm in diameter; Bruegger, Minusio, Switzerland) and subjected to high-pressure freezing in an HPM 010 high-pressure freezing machine (Bal-Tec, Balzers, Liechtenstein). Freeze substitution was performed using an AFS2 freeze substitution device (Leica Microsystems, Vienna, Austria) in anhydrous acetone containing 2% glutaraldehyde and 0.2% tannic acid for 3 days at −90°C and then warmed to 0°C over 24 h. After 1 h of incubation in anhydrous acetone containing 2% osmium tetroxide and 0.2 uranyl acetate at room temperature, the samples were washed with anhydrous acetone, infiltrated in a series of increasing concentrations of Epon (Agar Scientific, UK), and polymerized at 60°C. Sections (60–80 nm thick) were obtained using an Ultracut UCT microtome (Leica Microsystems, Vienna, Austria), double-stained with uranyl acetate and lead citrate, and examined in a Tecnai T12 electron microscope (FEI, Eindhoven, The Netherlands) operating at 120 kV. Images were recorded with an Eagle 2k × 2k charge-coupled device camera (FEI).
Results and Discussion
The overall morphology of MLV chronically infected NIH-3T3 cells differs from that of noninfected NIH-3T3 cells when viewed by transmitted light optical microscopy, with the noninfected cells appearing more spread out. Fluorescent labeling of F-actin reveals that virus infection significantly reduces actin fiber formation. This is most clearly shown by an inspection of stress fibers in the basal plane (Fig. 1), but can also be observed in the apical plane. Moreover, punctuate staining of F-actin, which is likely to represent aggregates, is often observed in the cytoplasm and in the vicinity of the plasma membrane of virus-infected cells. Similar large-scale cytoskeleton alterations are observed in other cell lines acutely infected by MLV (19). These observations suggest that the cytoskeletal changes induced by viral infection are not cell line-specific.
Figure 1.

Microscale changes in the actin cytoskeleton induced by MLV infection. (A) A fluorescence image and (B) a high-resolution 3D deconvolved image of control NIH 3T3 cells. (C) A fluorescence image and (D) a high-resolution 3D deconvolved image of MLV-infected NIH 3T3 cells. MLV infection significantly reduces actin fiber formation, whereas F-actin aggregates accumulate in the cell cytoplasm. The scale bars represent 20, 15, 30, and 5 μm in A–D, respectively.
We imaged the cytoskeletons of infected and uninfected live 3T3 cells using AFM to probe local ultrastructural changes in their actin networks (Fig. 2). To improve the contrast of the cytoskeletal features underneath the cell membrane, we operated the AFM in the torsional force mode. In this mode, the AFM probe scans perpendicularly to the long axis of the cantilever. As the tip bumps into a cytoskeletal fiber, which is stiffer than the cell membrane, the cantilever twists slightly, giving rise to an increase in the torsional signal. During imaging by AFM, budding virus particles are observed as protrusions on the surface of infected cells (Fig. 2, A and D). We demonstrated the assignment of these protrusions as budding viruses in a previous study (18) (also see Fig. S1 in the Supporting Material). In that study, we showed that the budding of MLV virions often occurs in conjunction with cytoskeletal elements (18). In the study presented here, imaging by lateral-force microscopy frequently revealed structures of the cytoskeleton, in the shape of an aster, emanating from budding MLV (Fig. 2, B and C) and HIV (Fig. 2, D and E) particles. The infected cells in these images are NIH-3T3, which in our hands allow for optimal AFM live-cell imaging. The use of this cell line to image the budding of both MLV and HIV has two possible shortcomings: 1), the structures observed to be formed may represent a cell-type-specific phenotype; and 2), more importantly, mouse fibroblasts are not a physiologically relevant cell line for HIV replication. To address these issues, we also imaged human epithelial cells from the HeLa line infected with HIV (Fig. 2F). As can be seen in Fig. 2F, similar aster-shaped structures are formed during the budding of HIV from a HeLa cell. Similar microstructures are absent from the nearly 100 uninfected control cells we observed (a typical uninfected cell is shown in Fig. 2, G and H). In virally infected cells, these aster structures are very dynamic and vary in size over time (Fig. 3). The length of the “arms” of the asters varies between ∼0.5 μm and 2.5 μm. Aster formation is detected simultaneously with or immediately after the initiation of budding, and its disappearance correlates with the release of the newly formed particle (Fig. 3).
Figure 2.

Imaging the cytoskeletons of live cells using AFM in the tapping and torsion modes. (A) A tapping-mode AFM image of an NIH-3T3 cell (CL-1) infected with MLV. Virus particles appear as a protrusion on the cell membrane. (B) An AFM torsion image of the cell shown in A reveals the fine actin architecture beneath the cell membrane (an enlargement is shown in C). (D) Tapping-mode and (E) enlarged torsion-mode AFM images of an NIH-3T3 cell infected with HIV. The nascent virus particles colocalize with actin star-like shapes. (F) A tapping-mode AFM image of a HeLa cell infected with HIV. (G) An AFM amplitude image, acquired in tapping mode, of an uninfected live NIH-3T3 cell. (H) A torsion-mode AFM image of the cell shown in G. The apparent fibril structures, which are enhanced in H and lie beneath the cell membrane, likely represent cytoskeletal actin. The scan area is 20 × 20 μm, 512 × 512 pixels.
Figure 3.

Dynamics of the aster structures. AFM height topographic time-series images of an HIV particle emerging from a live cell are shown. At t1, the emerging bud is seen at the center of an aster (arm lengths: ∼0.5–1.5 μm). After 10 min (t2), the bud virion has grown to its maximal height together with elongation of the aster arms (∼3–4 μm). Once the virus is released (t3), the aster structure gradually disassembles until it is mostly decomposed (t4). A newly viral bud is observed in image t4 (white arrow). Images are separated by 10 min intervals. Image scan area is 7 × 7 μm. Scale bar is 1.5 μm.
When AFM is used to image virally infected cells, asters are not observed in each budding site. In this imaging mode, however, only features that are in close proximity to the cell membrane can be detected. Therefore, if the remaining budding particles have inward-pointing filaments attached to them, the filaments will not be detected by AFM. To explore this possibility, we imaged sections of cryopreserved infected cells by TEM, which revealed that all of the analyzed particles (n = 32) were linked to several cytoskeletal filaments (Fig. 4). Analysis of a series of tilted TEM images confirmed the existence of physical contacts between the virus and the filaments. The early stages of budding are characterized by a dense layer of Gag proteins that form a small protrusion in the membrane, with short filaments extending from these protein clusters (Fig. 4A). As budding progresses, the particle becomes more spherical and the attached filaments elongate (Fig. 4, B–D). In a few cases (∼3%), the virus remains attached to the tip of long, filopodia-like structures (Fig. 4E). In contrast to the previously reported (9,10) filopodia bridges (and as shown in Fig. 4C, white arrow), these long “fingers” are not connected to two cells, and have an immature prerelease virus attached at their tip. Once the virus is released from the cell membrane, it is no longer attached to filaments (Fig. 4, C and D, white arrows).
Figure 4.
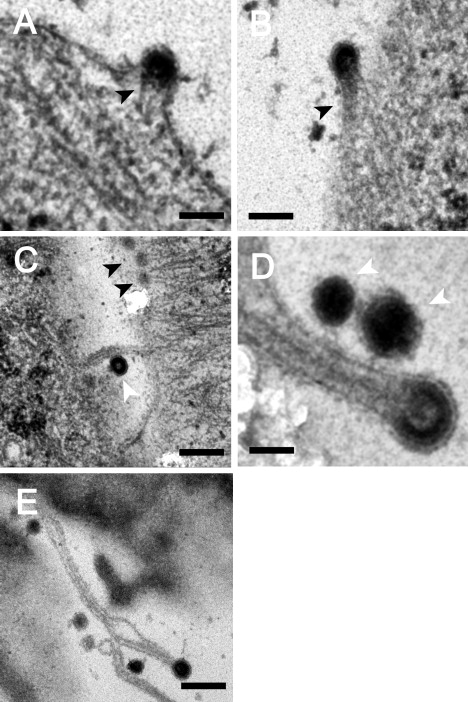
Budding of MLV and the cytoskeleton of the host cell. TEM images of cryopreserved MLV-infected cells show different stages of viral budding taking place at the cell periphery. (A) Early stages of budding, represented by the formation of a dense layer of viral proteins attached to short actin filaments (black arrowheads). (B) Semispherical viral particle attached to denser cytoskeleton filaments. (C and D) Late stages of budding, represented by nearly closed spheres attached to long actin filaments. (E) Viral particle attached to an extended cytoskeleton filament. Released particles are characterized by a complete sphere that is no longer attached to cytoskeletal filaments (C and D, white arrowheads). Scale bars: (A and B) 100 nm; (C–E) 200, 50, and 250 nm, respectively.
To properly understand our findings and their implications, it is necessary to gain insights into the molecular identity of the observed structures. Cytoskeletal images obtained by AFM mostly represent the actin fiber network, rather than other cytoskeletal elements, such as microtubules. Correlated AFM and confocal microscopy reveals that the cytoskeletal features observed in the AFM image are correlated with actin staining (20). Moreover, these features disappear from the AFM images after cells are treated with an actin-disrupting drug, whereas application of a microtubule-disrupting drug has no effect (20,21). To further support the molecular identity of these aster-shaped structures, we fixed MLV-infected cells with glutaraldehyde, and fluorescently labeled F-actin with phalloidin. Fig. 5A shows a fluorescence microscopy image of actin star-like structures that are identical in size and shape to those observed in our AFM images (Fig. 2). Cell fixation with glutaraldehyde was crucial to preserve this fine actin structure, which was not observed when milder fixation agents, such as paraformaldehyde, were used. Unfortunately, glutaraldehyde is generally known to damage immune labeling, which prevented us from colabeling the MLV particles using several different antibodies. The long filament structures (Fig. 4E) observed in the TEM images, however, are preserved by PFA. Such a long actin filament with a virus attached at its tip is shown in Fig. 5B and enlarged in Fig. 5 C. Based on the above results, we conclude that the observed star-shaped structures, as well as the filaments that are linked to budding virus particles, are made of actin fibers.
Figure 5.
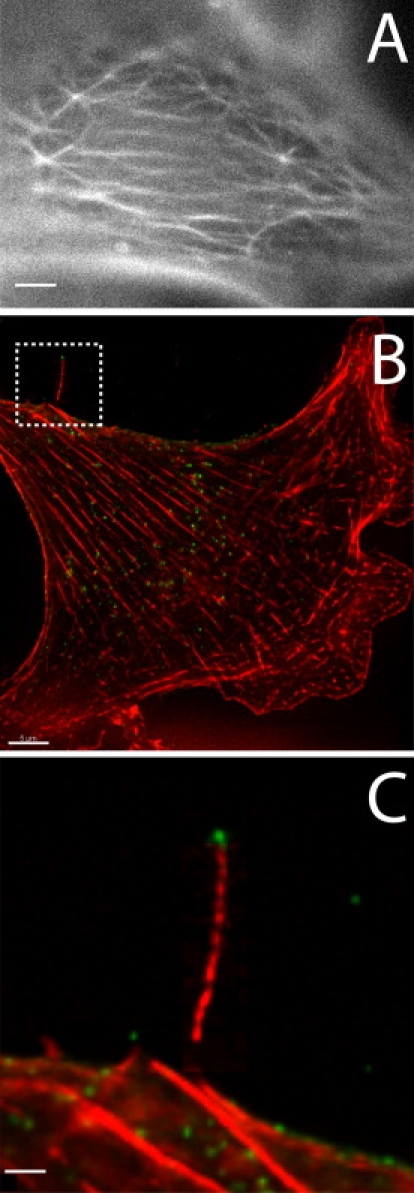
Remodeling of the actin network of retrovirus-infected cells. (A) The fine architecture of an MLV-infected, actin-labeled cell fixed with glutaraldehyde. Aster-shaped structures in the actin architecture, similar to those observed in Fig. 2, are seen at the apical surface of the cell. (B) Fluorescently labeled MLV viral particles (green) and the actin cytoskeleton (red) of an infected cell fixed with paraformaldehyde. The boxed area in B, which is magnified in C, reveals a virus attached at the tip of a long actin filament. The scale bars represent 1.5, 5, and 1.5 μm in A–C, respectively. All images were obtained by fluorescence microscopy.
To address the question of what functional role this actin remodeling plays during retroviral budding, we compared the assembly and budding of virus-like particles (VLPs) made of WT HIV-1 Gag with that of VLPs made of HIV-1 Gag chimeras in which the NC domain that is thought to bind actin (22,23) is replaced by a leucine-zipper domain (Leu-Gag) (16,24). Indeed, in contrast to WT-Gag VLPs, actin remodeling is not observed during the formation of these leucine-zipper VLPs (compare Fig. 6, A and B). We next determined the progress of the budding of these VLPs by measuring the protrusion height of individual particles as a function of time. Particle heights were determined according to a previously reported method (18). Strikingly, the budding kinetics of these VLPs (∼25–40 min, n = 20) is more than five times slower than that of WT-Gag-VLPs (<8 min, n = 30) as observed by AFM (inset of Fig. 6, A and B, respectively).
Figure 6.

Budding of virus-like particles made of leucine-zipper HIV-1 Gag chimera (Leu-Gag) and WT HIV-1 Gag from live cells. Tapping-mode AFM images of an NIH-3T3 cell infected with Leu-Gag (A) and WT-Gag (B). Remodeling of the cytoskeleton is not observed during the budding of the Leu-Gag-VLPs, in contrast to budding of WT-Gag VLPs (white arrows). Representative kinetics trajectories of three individual Leu-Gag-VLP budding events with typical lifetimes ranging between 25 and 40 min, and two budding events of WT-Gag VLP are shown in the inset of A and B, respectively. Due to the relatively low temporal resolution (which is a direct outcome of the high image acquisition time), these fast-released particles are captured in a single frame, which thus provides an upper limit for their budding kinetics.
To image budding events, cells must be imaged repeatedly for 1–3 h. In contrast to HeLa and several other cell lines that were tested, it is possible to image NIH-3T3 cells for such long durations while maintaining an image quality sufficient to enable kinetic analysis. However, the fact that HIV does not natively and efficiently replicate in these murine cells potentially reduces the relevance of the above VLP kinetics results. Several studies have shown that the targeting of Gag to the plasma membrane in murine fibroblasts is inefficient, resulting in the release of much fewer virus particles (25–27). Indeed, we detected far fewer budding events on the surface of NIH-3T3 than on the surface of HeLa cells. However, there is no evidence to suggest that budding from the surface of murine fibroblasts differs significantly from budding from the surface of human cells. In fact, the HIV particles released from NIH-3T3 cells were shown to be fully infectious (27). Moreover, the kinetics of WT HIV Gag VLPs budding from NIH-3T3 is similar to that from HeLa, as observed by fluorescent microscopy techniques (28,29). This implies that, although HIV replication in NIH-3T3 results in a lower viral titer, the kinetics of assembly and budding is not significantly changed. Taken together, these results indicate that, although remodeling of the cytoskeleton does not appear to be critical for the budding of VLPs, retroviruses take advantage of this mechanism to expedite their assembly and budding.
On the basis of our observations in this study, we conclude that this specific actin remodeling is induced by viral infection and is intimately connected to viral assembly and budding processes at the cell membrane. The formation of these structures is observed in both HIV- and MLV-infected cells, and thus their formation is likely to represent a general mechanism in retrovirus budding.
Similar actin structures were previously reproduced in vitro by means of a mixture of actin filaments and myosin-II motors (30). In those experiments, asters were produced by the contractile forces of myosin-II motors, which act to concentrate the barbed ends of actin filaments at the center of the asters. An additional in vitro study examining the pathway of filopodia formation showed that beads coated with the actin-nucleating protein Arp2/3 can give rise to an actin-star radiating from the bead (31) when introduced into a solution containing actin. Intriguingly, the bead appeared to be lifted from the surface, with the “arms” of the star pushing it upward. According to the proposed mechanism, the node of the aster (where the bead is situated) is the focal point on which the protrusive forces act. This configuration resembles the asters we detected, which had a virus particle located at the center.
Recent theoretical studies (32,33) have described the dynamics of membrane proteins (or clusters of proteins) that possess the following properties: 1), they spontaneously adopt a convex curvature; 2), they serve as nucleation centers for actin polymerization; and 3), they can diffuse on the fluid cell membrane. Membrane proteins that possess these three properties go through a dynamic instability transition in which they spontaneously aggregate and form membrane protrusions (buds). This robust instability occurs through a positive-feedback mechanism whereby a small initial membrane protrusion attracts more such proteins due to their spontaneous convex curvature. This in turn increases the local pushing force of the actin filaments, which further increases the local protrusion height and curvature.
The viral Gag structural protein forms a closed spherical shell and therefore naturally possesses a spontaneous convex curvature. Additionally, actin has been shown to associate with Gag in budded virions (23). Based on our observations from this study, as well as those from the previous in vitro work (30,31) and the above-mentioned theoretical studies (32,33), we propose an actin-driven mechanism for retroviral assembly and simultaneous budding at the cell membrane (see Fig. 7). According to this model, Gag (in the form of dimers or small clusters) aggregates at the cell membrane due to an instability mechanism that relies on a combination of its convex curvature and the protrusive force of actin. This protrusive force increases the local Gag concentration and simultaneously drives the outward growth of the nascent virus. According to our model, active actin treadmilling is required to drive assembly at the cell membrane and budding. This requirement resolves previous contradictory observations that blocking either actin polymerization or depolymerization inhibits virus budding (reviewed by Fackler and Krausslich (11)).
Figure 7.
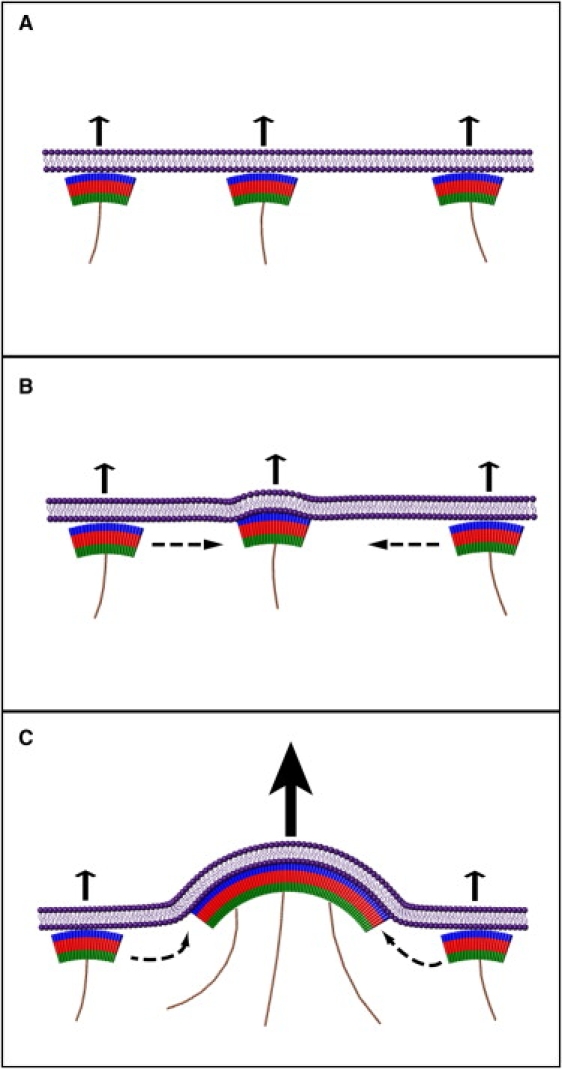
Schematic drawing of the actin-driven mechanism for retroviral budding. (A) Viral Gag proteins (represented by arches) adsorb to the cell membrane, and induce a local outward protrusive force (arrows) due to the recruitment and nucleation of actin polymerization (thin filaments). These membrane proteins, in the form of dimers or larger units, possess a spontaneous convex curvature that enables them to link to form the spherical viral shell. This system is unstable. A small perturbation (B) bends the membrane outward and draws more viral proteins to the unit, due to their spontaneous curvature. This protein aggregation leads to a larger protrusive force (C), which further bends the membrane outward and enhances aggregation. This positive feedback (dynamic instability) simultaneously results in the aggregation of the viral proteins and the budding of the virus from the cell membrane. This model describes the initiation stages of the viral aggregate and bud formation. During later stages, the virion develops a fully spherical shape and detaches from the cell.
Within the framework of the above theoretical model, the observed relations between the virus-induced actin activity and viral budding kinetics and density can be explained (for a detailed description of the theoretical model, see the Supporting Material). As shown in Fig. 8, the model predicts an inverse relationship between the critical actin protrusive force needed to initiate assembly and budding, and the average density of the viral structural protein (Gag) at the cell membrane. Specifically, in the absence of actin forces, budding and assembly require a higher Gag surface density (ϕ0; Fig. 8) than is required when actin is involved (ϕi).
Figure 8.

Typical calculated phase diagram for a membrane containing convex proteins that recruit actin polymerization (protrusive forces) (32). The thick black line denotes the critical density of membrane proteins as a function of the actin force. To the left of this line, the uniform state of the membrane is stable, whereas to the right of this line, the system spontaneously forms aggregates and buds. The contours denote increasing rates of growth of the most unstable mode (from left to right), with each contour representing a twofold increase in the growth rate. The density is given as the averaged area of the membrane covered by the viral proteins, normalized by the specific area of a Gag unit. Dashed horizontal arrows denote the trajectories of the system as the Gag density increases after infection. The critical density differs depending on the strength of the actin force. Thus, in the absence of actin forces, budding and assembly require a higher surface density of Gag (ϕ0) than is required when actin is involved (ϕi).
After viral infection occurs, viral proteins are synthesized and transported to the cell membrane, where they begin to accumulate. In our model, this process is described by moving across the density contour lines of Fig. 8 (i.e., as per the dashed horizontal trajectories superimposed on the contour map). As the system reaches the critical density (Fig. 8, thick line), the uniform membrane composition becomes unstable and, as a consequence, aggregation and budding take place. However, along the critical density line, the rate of spontaneous viral assembly and budding is zero. To initiate budding, the Gag membrane concentration must increase beyond the critical density line. Gag density will further increase until the rate of proteins released through viral budding matches the (assumed constant) rate of viral protein production inside the cell. At this stage, the Gag membrane level remains constant (steady state). Assuming that each virus contains approximately a constant number of Gag proteins (Nv), the area from which Gag proteins are gathered to form a virus is given by sv = Nv/ϕ, where ϕ is the density of Gag proteins at the cell membrane. The number of budding sites over the whole cell surface is therefore given by S/sv, so the total amount of viral proteins leaving the cell surface per unit time is ωNvS/sv, where ω is the rate of viral budding at each budding site. At steady state, this rate of viral protein release from the cell is equal to the accumulation rate of Gag at the membrane (α), so that we finally get the relation ω = α/(Sϕ). As mention above, higher Gag densities are required in the absence of actin forces, and thus our model implies a slower steady-state release rate (ω) of viruses from the individual budding sites. The predictions of this theoretical model can therefore explain our experimental results. Alternatively, the observed change in budding kinetics could be the outcome of large differences between the surface accumulation rates of WT- and Leu-Gag. The matrix domain of Gag has a key role in targeting Gag to the plasma membrane (34–36). Since this domain is identical in WT- and Leu-Gag proteins, their surface accumulation rates are expected to be similar. Thus, while we cannot completely rule out the effect of assembly rates, we think it is less likely.
In this study, the assembly and budding of individual virus particles in living cells were observed simultaneously with the ultrastructure of the local actin cytoskeleton. Although it has been speculated that the actin cytoskeleton plays a role in retrovirus assembly and budding, the specific mechanism by which it facilitates these processes has remained vague. Our results demonstrate that retroviruses utilize the forces produced by the cellular cytoskeletal machinery to drive their assembly and budding. Elaboration of these single-particle measurements to study mutated viruses and cells will allow us to address fundamental questions about viral assembly that might otherwise remain unanswered.
Acknowledgments
We thank Benny Geiger for helpful comments on this work, and Alan Rein for providing several of the reagents required for this study.
This work was supported in part by a grant from the Clore Center for Biological Physics, the Jean-Jacques Brunschwig Fund for the Molecular Genetics of Cancer, and the Kimmelman Center for Macromolecular Assemblies. I.R. holds the Robert Edwards and Roselyn Rich Manson Career Development Chair. N.S.G. holds the Alvin and Gertrude Levine Career Development Chair and is supported by the Israel Science Foundation (grant No. 337/05). The electron microscopy studies were conducted at the Irving and Cherna Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science.
Supporting Material
References
- 1.Kaksonen M., Peng H.B., Rauvala H. Association of cortactin with dynamic actin in lamellipodia and on endosomal vesicles. J. Cell Sci. 2000;113:4421–4426. doi: 10.1242/jcs.113.24.4421. [DOI] [PubMed] [Google Scholar]
- 2.Welch M.D., Mullins R.D. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 2002;18:247–288. doi: 10.1146/annurev.cellbio.18.040202.112133. [DOI] [PubMed] [Google Scholar]
- 3.Cojoc D., Difato F., Ferrari E., Shahapure R.B., Laishram J. Properties of the force exerted by filopodia and lamellipodia and the involvement of cytoskeletal components. PLoS ONE. 2007;2:e1072. doi: 10.1371/journal.pone.0001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gouin E., Welch M.D., Cossart P. Actin-based motility of intracellular pathogens. Curr. Opin. Microbiol. 2005;8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Gouin E., Gantelet H., Egile C., Lasa I., Ohayon H. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J. Cell Sci. 1999;112:1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- 6.Tilney L.G., Derosier D.J., Weber A., Tilney M.S. How Listeria exploits host-cell actin to form its own cytoskeleton. 2. Nucleation, actin filament polarity, filament assembly, and evidence for a pointed end capper. J. Cell Biol. 1992;118:83–93. doi: 10.1083/jcb.118.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cudmore S., Cossart P., Griffiths G., Way M. Actin-based motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- 8.Jouvenet N., Windsor M., Rietdorf J., Hawes P., Monaghan P. African swine fever virus induces filopodia-like projections at the plasma membrane. Cell Microbiol. 2006;8:1803–1811. doi: 10.1111/j.1462-5822.2006.00750.x. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann M.J., Sherer N.M., Marks C.B., Pypaert M., Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005;170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherer N.M., Lehmann M.J., Jimenez-Soto L.F., Horensavitz C., Pypaert M. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007;9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fackler O., Krausslich H. Interactions of human retroviruses with the host cell cytoskeleton. Curr. Opin. Microbiol. 2006;9:409–415. doi: 10.1016/j.mib.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Gelderblom H.R., Hausmann E.H., Ozel M., Pauli G., Koch M.A. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology. 1987;156:171–176. doi: 10.1016/0042-6822(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 13.Morita E., Sundquist W.I. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 14.Naghavi M.H., Goff S.P. Retroviral proteins that interact with the host cell cytoskeleton. Curr. Opin. Immunol. 2007;19:402–407. doi: 10.1016/j.coi.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C., Weisz O.A., Stolz D.B., Watkins S.C., Montelaro R.C. Differential effects of actin cytoskeleton dynamics on equine infectious anemia virus particle production. J. Virol. 2004;78:882–891. doi: 10.1128/JVI.78.2.882-891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crist R.M., Datta S.A.K., Stephen A.G., Soheilian F., Mirro J. Assembly properties of human immunodeficiency virus type 1 gag-leucine zipper chimeras: implications for retrovirus assembly. J. Virol. 2009;83:2216–2225. doi: 10.1128/JVI.02031-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horcas I., Fernandez R., Gomez-Rodriguez J.M., Colchero J., Gomez-Herrero J. WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007;78:013705. doi: 10.1063/1.2432410. [DOI] [PubMed] [Google Scholar]
- 18.Gladnikoff M., Rousso I. Directly monitoring individual retrovirus budding events using atomic force microscopy. Biophys. J. 2008;94:320–326. doi: 10.1529/biophysj.107.114579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luftig R.B., Lupo L.D. Viral interactions with the host-cell cytoskeleton: the role of retroviral proteases. Trends Microbiol. 1994;2:178–182. doi: 10.1016/0966-842x(94)90669-6. [DOI] [PubMed] [Google Scholar]
- 20.Pesen D., Hoh J.H. Micromechanical architecture of the endothelial cell cortex. Biophys. J. 2005;88:670–679. doi: 10.1529/biophysj.104.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rotsch C., Radmacher M. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys. J. 2000;78:520–535. doi: 10.1016/S0006-3495(00)76614-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B.D., Dai R.K., Tian C.J., Dawson L., Gorelick R. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 1999;73:2901–2908. doi: 10.1128/jvi.73.4.2901-2908.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilk T., Gowen B., Fuller S.D. Actin associates with the nucleocapsid domain of the human immunodeficiency virus Gag polyprotein. J. Virol. 1999;73:1931–1940. doi: 10.1128/jvi.73.3.1931-1940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y.Q., Qian H.Y., Love Z., Barklis E. Analysis of the assembly function of the human immunodeficiency virus type 1 gag protein nucleocapsid domain. J. Virol. 1998;72:1782–1789. doi: 10.1128/jvi.72.3.1782-1789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bieniasz P.D., Cullen B.R. Multiple blocks to human immunodeficiency virus type 1 replication in rodent cells. J. Virol. 2000;74:9868–9877. doi: 10.1128/jvi.74.21.9868-9877.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garber M.E., Wei P., KewalRamani V.N., Mayall T.P., Herrmann C.H. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mariani R., Rutter G., Harris M.E., Hope T.J., Krausslich H.G. A block to human immunodeficiency virus type 1 assembly in murine cells. J. Virol. 2000;74:3859–3870. doi: 10.1128/jvi.74.8.3859-3870.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jouvenet N., Bieniasz P.D., Simon S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature. 2008;454:236–240. doi: 10.1038/nature06998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanchenko S., Godinez W.J., Lampe M., Kräusslich H.G., Eils R. HIV budding and release. Euro. Biophys. J. 2009;38:S49. [Google Scholar]
- 30.Backouche F., Haviv L., Groswasser D., Bernheim-Groswasser A. Active gels: dynamics of patterning and self-organization. Phys. Biol. 2006;3:264–273. doi: 10.1088/1478-3975/3/4/004. [DOI] [PubMed] [Google Scholar]
- 31.Vignjevic D., Yarar D., Welch M.D., Peloquin J., Svitkina T. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 2003;160:951–962. doi: 10.1083/jcb.200208059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gov N.S., Gopinathan A. Dynamics of membranes driven by actin polymerization. Biophys. J. 2006;90:454–469. doi: 10.1529/biophysj.105.062224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veksler A., Gov N.S. Phase transitions of the coupled membrane-cytoskeleton modify cellular shape. Biophys. J. 2007;93:3798–3810. doi: 10.1529/biophysj.107.113282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermida-Matsumoto L., Resh M.D. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 2000;74:8670–8679. doi: 10.1128/jvi.74.18.8670-8679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ono A., Orenstein J.M., Freed E.O. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 2000;74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saad J.S., Miller J., Tai J., Kim A., Ghanam R.H. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.