

Abstract
Cyanovirin-N (CVN) is a highly potent anti-HIV carbohydrate-binding agent that establishes its microbicide activity through interaction with mannose-rich glycoprotein gp120 on the virion surface. The m4-CVN and P51G-m4-CVN mutants represent simple models for studying the high-affinity binding site, BM. A recently determined 1.35 Å high-resolution structure of P51G-m4-CVN provided details on the di-mannose binding mechanism, and suggested that the Arg-76 and Glu-41 residues are critical components of high mannose specificity and affinity. We performed molecular-dynamics simulations in solution and a crystal environment to study the role of Arg-76. Network analysis and clustering were used to characterize the dynamics of Arg-76. The results of our explicit solvent solution and crystal simulations showed a significant correlation with conformations of Arg-76 proposed from x-ray crystallographic studies. However, the crystal simulation showed that the crystal environment strongly biases conformational sampling of the Arg-76 residue. The solution simulations demonstrated no conformational preferences for Arg-76, which would support its critical role as the residue that locks the ligand in the bound state. Instead, a comparative analysis of trajectories from >50 ns of simulation for two mutants revealed the existence of a very stable eight-hydrogen-bond network between the di-mannose ligand and predominantly main-chain atoms. This network may play a key role in the specific recognition and strong binding of mannose oligomers in CVN and its homologs.
Introduction
Cyanovirin-N (CVN), a 101-residue-long lectin, belongs to the group of carbohydrate-binding agents that establish their antiviral activity through interaction with the carbohydrate shields of viruses, including HIV, Ebola, and hepatitis C (1–4). In the case of HIV, CVN binds to the mannose-rich sugar moieties of the surface envelope glycoprotein gp120 and disrupts virion attachment to the host cell receptors and virus-cell fusion mechanism (5–7). Additionally, CVN has been reported to have potential for novel antiviral therapies that result in the generation of virus modifications with depleted glycan shields (such viruses are less protected against immune response) (8). The origin of CVN's high specificity to mannose oligomers and details of the gp120 immobilization mechanism are still under intensive study and discussion. A variety of experimental and computational techniques have been used to address these questions, including site mutations of the protein and ligand, structural studies in solution and crystal, and molecular-dynamics (MD) simulations (9–13). A summary of CVN structural studies can be found in a recent review (14). An MD simulation of CVN by Fujimoto et al. (15) focused on the docking of tri-mannose to complement a corresponding experimental study (12). The results of a previous MD simulation of native CVN complexed with di-mannose (16) are of particular interest here, as discussed further below.
The CVN mutants studied in this work were initially created to determine the importance of multiple binding modes for antiviral activity (17,18). Wild-type CVN exists in solution in several oligomeric forms, including monomer and a unique domain-swapped dimer (70% and 25% at pH < 5) (13). Although at neutral pH the monomeric form is predominant in solution, only the crystal structures of the domain-swapped dimer are known to contain native CVN (14). Information on the CVN monomer geometry is based on a CVN:dimannose structure from NMR solution experiments (10). The monomer has two di-mannose specific binding sites: AM and BM (M stands for monomeric). These two binding sites are sequentially and structurally similar. However, the AM binding site has a more shallow shape that results in a lower affinity to di-mannose (Ka ∼ 6.8 × 105 M−1) compared to the BM site (Ka ∼ 7.2 × 106 M−1), which forms a deep pocket (10). Both of the mutants studied in this work (m4-CVN and P51G-m4-CVN) have four mutations (K3N, T7A, E23I, and N93A) that completely inactivate the AM binding site, whereas P51G-m4-CVN has an additional P51G mutation that stabilizes formation of the monomer (Fig. 1 a). It appears that a multiple-site binding mode is important for antiviral activity, although some ambiguity is present in the literature (17–19). Monomeric units of the mutants under investigation still bind to gp120, but as expected, and in contrast to both CVN and P51G-CVN, they do not possess antiviral activity (20,21). Therefore, the m4-CVN and P51G-m4-CVN monomeric units represent simple models for studying binding-site BM.
Figure 1.

(a) General view of the P51G-m4-CVN:di-mannose complex. Four mutations (K3N, T7A, E23I, and N93A), which completely inactivate the binding site AM (the left half of the protein), and an additional P51G mutation (binding-site BM, the right half of the protein), which stabilizes the monomer, are shown as side chains. Side chains of Glu-41 and Arg-76 are also shown. The di-mannose hydrogens are removed for clarity. (b) H-bonding of Arg-76 and Glu-41 with di-mannose residues in the binding-site BM as possible factors of CVN specificity. The picture presents one of the frames from the m4-CVN:di-mannose complex solution simulation. However, this configuration is very transient in our simulation. (c) A network created from the 500-frame sampling set (points) and six representative structures from clustering based on 5000 frames (numbered white circles). The graph is based on the RMSD pairwise matrix calculated for the structure fragment that includes nonhydrogen atoms of the Arg-76 residue and the closest residue of di-mannose. Links between nodes are attributed to <0.75 Å RMSD between the fragments of corresponding structures. The representative structures identify the most populated clusters. The size of the node is proportional to the cluster population. The most and least populated clusters, clusters 1 and 6, include 1665 and 101 frames, respectively. The first three and all six clusters respectively cover 84% and 97% of conformational space observed in the last 50 ns of simulation of the P51G-m4-CVN:di-mannose complex in solution. (d) A network created from the 950-frame sampling set from the P51G-m4-CVN:di-mannose complex simulations in solution and crystal. Nodes are coded as follows: (small circles) 500 frames from 50 ns solution simulations; (triangles and diamonds) 30 ns crystal simulations of chain a (150 frames) and chain b (300 frames), respectively; (labeled squares) experimentally observed conformations of chain a, and two alternative conformations of chain b. White circles depict the positions of the representative structures for the most populated clusters in the solution simulation. Nodes between major clusters correspond to transient conformations, whereas the peripheral nodes belong to the very low populated clusters.
The high specificity of CVN to mannose has been attributed in particular to hydrogen (H)-bonds between hydroxyl groups of sugar ligand and protein side-chain atoms (10). In the high-affinity binding site BM, H-bonding with the Glu-41 residue side chain has been proposed as an important component in the recognition and binding of the di-mannose ligand (16,22) (Fig. 1, a and b). Another interesting feature of the binding-site BM is the presence of the Arg-76 residue. An earlier MD simulation of a native CVN:di-mannose complex indicated that two side-chain nitrogen atoms of Arg-76 form H-bonds with two oxygen atoms of di-mannose (Fig. 1 b) and lock the ligand in the binding pocket (16). Arg-76 remained in that locking conformation for more than half of the entire 20 ns simulation, in contrast to the generally very high mobility of the arginine residue. A very similar “capping” conformation was observed in an NMR structural study (10). In a recent x-ray diffraction investigation of the P51G-m4-CVN:di-mannose complex, two independent polypeptide chains (“a” and “b”) were found in the asymmetric unit. Although the two chains have nearly identical conformations, in one of them the rotameric state of the Arg-76 residue is somewhat similar to previously observed ligand gating configurations (22). In the a chain, Arg-76 has one conformation and the ligand is exposed to surrounding media. In the b chain, it is disordered on two alternative positions with 80:20% occupancy, and in both conformations it partially “covers” the ligand in the binding site (22). This result was interpreted as experimental support for the previously proposed capping-and-locking function of the Arg-76 residue. However, one should be careful to avoid overinterpreting a crystal structure.
Crystal structures represent a confined medium that may introduce biophysical and biochemical artifacts due to interactions of a protein complex with neighboring molecules (23,24). In this work, we reconsider the critical importance of Arg-76 for CVN specificity to di-mannose based on the results of explicit solvent MD simulations of the P51G-m4-CVN:di-mannose complex in solution and in the crystal environment to examine whether the conformations of Arg 76 from x-ray data accurately represent the conformations in solution. We also used solution MD simulations of the P51G-m4-CVN:di-mannose and m4-CVN:di-mannose complexes to study the dynamics of the entire protein-ligand H-bond network. It appears that the interaction of hydroxyl groups of the di-mannose ligand with predominantly main-chain atoms is one of the major ligand-protein complex stabilizing factors in the binding site BM.
MD simulations in the crystal environment are still relatively rare and more methodologically challenging than MD simulations in solution (25). Applications of MD crystal simulations include studies of solute and solvent transport in protein crystals (26,27), simulations of protein dynamics in the crystal (28,29), testing of force fields (30–32), and explorations of the crystal packing influence on protein structure and dynamics. Our study falls in the last category. In a recent work on Neuroglobin (Ngb) Anselmi et al. (23) demonstrated that significant structural changes in ferrous CO-bound Ngb are caused by the crystal environment rather than peculiarities of the heme-binding mechanisms. In another investigation, Neugebauer et al. (24) showed that conformations of two important helical binding regions in the crystal structure of the human CD81 receptor are affected by crystal contacts, and the observed structure does not represent the native state of the protein. In this study, we focus on comparing the conformational sampling of a particular binding-site residue, Arg-76, in the crystal environment and in solution, and on how it relates to the mechanism of the CVN interaction with oligomannose. To describe the Arg-76 conformational sampling, we used well-established clustering techniques (33) as well as network analysis (34). Network analysis allows visualization of results of the “classical” clustering algorithms and conformational space sampling from different simulations on one plot, which can be very useful for comparative analyses and for understanding topological relationships between clusters.
Materials and Methods
General
The details of the MD simulations are summarized in Table S1 of the Supporting Material. The initial coordinates of the P51G-m4-CVN:di-mannose complex were taken from a 1.35 Å resolution crystal structure of this complex (Protein Data Bank (PDB) ID: 2RDK) (22). Residues 2–101, all of which are visible on the electron density map, were included in modeling, as was a his-tag linkage residue, Leu-102, which is also “visible” in the crystal structure. Both the C- and N-termini are far from the binding site BM. The model for m4-CVN:di-mannose was constructed with the assistance of the native CVN:di-mannose structure determined by solution NMR (PDB ID: 1IIY) (10). The di-mannose ligand used in the simulations was the Manα(1→2)Manα oligomer. All simulations were performed using the Amber 10 package (35,36). Residues were protonated to correspond to the neutral pH. The only histidine residue in the system was uncharged (proton on Nɛ), all five aspartic and five glutamic acid residues were negatively charged, and all three arginine and four lysine residues were positively charged. The histidine residue is far from the ligand-binding site, and its protonation state should not affect our results. A total negative charge of −3e on the protein was neutralized by the addition of three Na+ counterions. Input coordinates, topology, and force-field parameters files were created with the tleap program (37).
Amber FF03 and GLYCAM06 force-field parameters were used for the proteins and ligand, respectively (38–41). A discussion about the choice of force field is presented in the Supporting Material. Nonbonded and electrostatic energy scaling factors were set according to the FF03 parameterization: SCNB = 2.0 and SCEE = 1.2. Deviation of these scaling factors from unity, with which GLYCAM06 was parameterized, may degrade the accuracy of the rotamer sampling of carbohydrates; however, since di-mannose stays bound in one conformation, this should not affect our results. For simulations in both NVT and NPT ensembles, the temperature was maintained by Langevin dynamics with a collision frequency of 1 ps−1. The random number generator was reseeded at every simulation restart (every 200 ps) (42). A cutoff of 10 Å and the particle mesh Ewald method for simulation of periodic boundaries were used to calculate long-range nonbonded interactions (default parameters include grid spacing of 1.0 Å combined with fourth-order B-spline interpolation). Constant-pressure simulations were performed at an average pressure of 1 atm, employing an isotropic position scaling algorithm and a relaxation time of 2 ps. All simulations were done with explicit TIP3P water. SHAKE constraints with the tolerance parameter of 10−5 Å were applied to eliminate bond-stretching freedom for all bonds involving hydrogen. The simulation jobs were run in parallel using 8 and 16 processor clusters at the University of Arizona High Performance Computing Center. All trajectories were processed with ptraj and visualized with VMD (33,43). Graphical presentations were prepared using Pymol and LigPlot (44,45).
Solution simulations
For the MD simulations, a neutralized P51G-m4-CVN:di-mannose complex was solvated with 10,722 water molecules using a truncated octahedron box with a buffer distance of 14 Å in all three directions. The initial energy minimization was performed in two steps. The first step included 500 steepest descent minimization cycles followed by 500 conjugate gradient minimization cycles. Coordinates of the protein complex were “fixed” at the starting positions using harmonic restraints with a force constant of 500 kcal/(mol · Å2), whereas solvent molecules (water and Na+) were allowed to relax the unfavorable contacts between each other and with the solute. The second step consisted of 1000 steepest descent and 1500 conjugate gradient cycles of energy minimization for the entire system with no positional restraints. Next, the system was heated up from 0 to 300 K during a 20 ps MD simulation run with a 0.002 ps time step and weak positional restraints on the solute (force constant of 10 kcal/(mol · Å2)). Finally, a 52 ns production run in the NVP ensemble was performed with a time step of 0.002 ps; the first 200 ps of the run included equilibration. There were no harmonic restraints on the system. Output coordinate files were written every 0.5 ps. The same protocol was used for all solution simulations. These conditions produced stable trajectories as structures of the protein and ligand were maintained throughout the simulations, as evidenced by comparing the root mean-square displacement (RMSD) against the starting minimized complexes (see Fig. S1).
Crystal simulations
The P51G-m4-CVN:di-mannose complex was crystallized in the monoclinic unit cell with the space group P21 and parameters a = 49.205, b = 38.452, c = 55.953 Å, and β = 99.94° (PDB ID: 2RDK). A corresponding triclinic unit cell contained four protein-ligand pairs. To solvate the system, a 3 × 3 × 3 crystal block was constructed from 27 triclinic unit cells. This crystal block was “submerged” into a rectangular solvation box with a buffer distance of 6 Å around the solute. The box was filled with 135,897 water molecules to a final density of 1 g/cm3 for the entire system. The energy of the 27-unit cell solvated system was minimized in two steps. First, the solvent was relaxed though 2000 steepest descent and 2000 conjugate gradient energy minimization cycles. Coordinates of solute atoms were restrained to their initial values with a force constant of 500 kcal/(mol · Å2). Next, the energy of the solvent, ligands, and protein side chains was minimized in 3000 steepest descent and 4000 conjugate gradient cycles, whereas the main-chain atoms of the solute remained restrained with the same force constant. After minimization, the system was heated up to 300 K during a 20 ps simulation followed by a 40 ps equilibration run in the NVT ensemble with the same positional restraints to main-chain atoms. This preliminary simulation was performed to allow water molecules to evenly fill up the space between protein molecules.
For the crystal simulations, the central unit cell was “cut out” from the 27-unit cell block. This system included four protein chains with ligands, 12 Na+ counterions, and 1557 water molecules. Periodic boundary parameters were set to be equal to the parameters of the triclinic unit cell. The density of the system was equal to 1.16 g/cm3. An evaluation of our solvation model is presented in detail in the Supporting Material. The energy of the system was again minimized in 2000/2000 steepest descent/conjugate gradient cycles to relax water molecules, followed by 3000/4000 steepest descent/conjugate gradient cycles to relax unfavorable contacts between solvent molecules and side chains of the solute. Then the system was heated up to 300 K during a 20 ps run with positional harmonic restraints on the main-chain atoms (force constant of 500 kcal/(mol · A2)). A 16 ns production run in the NVT ensemble was performed with no positional restraints. Output coordinate files were produced every 1 ps of simulation. The initial crystal arrangement was preserved throughout the simulations, as evidenced by comparing the RMSD against the starting model derived from the experiment (Fig. S2).
Clustering and network representation
Network representations and clustering were used for analyses of the conformational ensembles of Arg-76 (33,34). Clustering and network construction were both based on the RMSD pairwise matrix. Nonhydrogen atoms for the Arg-76 residue and a di-mannose residue adjacent to Arg-76 were included in RMSD calculations. For the network representation, we employed a force-directed layout algorithm that is a part of the Cytoscape program (46). In brief, the network is a graph consisting of nodes that represent sampled configurations of the system, and links (edges) that represent similarity between configurations. The links are established by a cutoff applied to the RMSD matrix. Results from a series of trials indicated that an RMSD cutoff of 0.75 Å distinguished well between different clusters. Upon construction of the force-directed layout, the potential energy of the graph undergoes minimization as a virtual physical system in which links are modeled as springs (attractive forces) and all nodes have an electrical repulsion between them. For clustering, we utilized the average linkage algorithm implemented in ptraj. The uniqueness or equivalence of different clusters was assessed based on visual comparison of representative structures. More details on the use of clustering and network analysis are presented in the Supporting Material.
The most effective way to use unweighted force-directed network constructions is to 1), reveal clusters and their content; and 2), establish connections between clusters. A network representation of ensemble configurations is helpful for visualizing the topology of the conformational space in a way that cannot be easily done with clustering analysis (especially for a flexible residue such as arginine, which has >30 rotameric states (47)). However, network processing is memory-demanding and limits the number of nodes that can be computationally handled and represented in a visually informative way to ∼1000. In our case, to represent a 50 ns trajectory, we had to sample ∼500 configurations, corresponding to the wide sampling rate of 100 ps. Nevertheless, in our preliminary investigation (see the Supporting Material) we found that the 500-frame sampling set reasonably covered the most populated part of the conformational space of the Arg-76 residue in solution (Fig. 1 c). The clustering was performed on a 5000-frame reference set (10 ps sampling rate).
Results
Comparison of Arg-76 conformational sampling in solution and crystal
Fig. 1 c shows the assignment of a network constructed from 500 Arg-76 conformations from the solution simulation to the six most-populated clusters from the 5000-frame reference sampling set. This figure primarily demonstrates the high mobility of the Arg-76 residue in the “free” protein complex, and the existence of three major preferable configurations and two minor ones. The highest-rank clusters (clusters 1–3) from the MD simulation correspond to Arg-76 conformations identified by x-ray crystallography, as is evident from Fig. 1 d. This remarkable agreement indicates the quality of the structural information from the x-ray data, as well as the reliability of the model from the MD simulation.
Further analysis illuminates the effects of crystal packing on the Arg-76 conformational ensemble. Fig. 1 d represents a network consisting of 950 frames from the P51G-m4-CVN:di-mannose complex simulations in both solution and crystal. These 950 nodes include 500 frames from the solution simulation and 150+300 frames from the crystal simulation of chains a and b. As can be seen from the figure, the a chain includes only one conformation for the Arg-76 residue, which corresponds to cluster 3 from the solution simulation (Fig. 1 c). Analysis of the crystal packing indicates that Arg-76 of the a chain is trapped in this experimentally observed conformation because of direct interactions with a neighboring molecule (Fig. S4). In contrast, in the b chain the Arg-76 residue is exposed to solvent, and samples almost the same conformational space as in solution (Table S2). Nonetheless, in agreement with our calculations, two conformations observed experimentally in the crystal (b1 and b2) correspond to the two most occupied clusters (clusters 1 and 2), and the b1 conformation is more populated than b2 (Table S2). Yet, the simulation indicates that the Arg-76 residue in the b chain is disordered at room temperature at least by five positions, and the precision of the experiment does not allow resolution of all these conformations. MD simulation also complements the experiment with more details about the nature of the Arg-76 disorder in the b chain, providing information on the timescale of Arg-76 dynamics. Arg-76 remained in one conformation for less than ∼2.3 ns and changed its conformation numerous times during the 50 ns simulation. We note that the B-factor of the residue is not very high, but that does not contradict our observation. A detailed analysis of disordered fragments is beyond the capabilities of a single-temperature routine x-ray diffraction experiment because of strong correlations between positional, thermal, and occupational parameters (48). However, the fact that this residue is disordered indicates that it is flexible.
The MD simulations show that the conformation observed in the a chain is trapped and probably should not be treated as experimental evidence of Arg-76 participation in the binding-site gating mechanism. Additionally, the “trapped” Arg-76 conformation in the a chain appears to be shifted off of the centroid of cluster 3 from the solution simulation. The presence of this extra packing effect can hardly be noted on Fig. 1 d because of limitations of the network presentation (see the Supporting Material for details). To elucidate the effect, we calculated distributions of Arg-76 RMSDs from experimentally observed conformations (Fig. 2). A range of 0–0.75 Å on the histograms corresponds to cluster 1 or cluster 3 from the solution simulation of the P51G-m4-CVN:di-mannose complex (Fig. 1, c and d). As can be seen from the upper histogram, the experimentally observed conformation “a” lies on the slope of the distribution. Thus, this configuration is shifted from the cluster 3 centroid, which corresponds to the closest rotameric state. In contrast, the experimentally observed configurations b1 and b2 (not shown) appear to be very close to centroids of clusters 1 and 2, since the RMSD distributions have maxima in the range of 0–0.75 Å (Fig. 2). These results are in line with the fact that Arg-76 in the b chain does not have direct interactions with neighboring proteins that could otherwise affect the position and conformational sampling of this residue. In conclusion, the crystal environment of the P51G-m4-CVN:di-mannose complex not only traps Arg-76 of the a chain in a single conformation, it also distorts it from its closest rotamer geometry.
Figure 2.
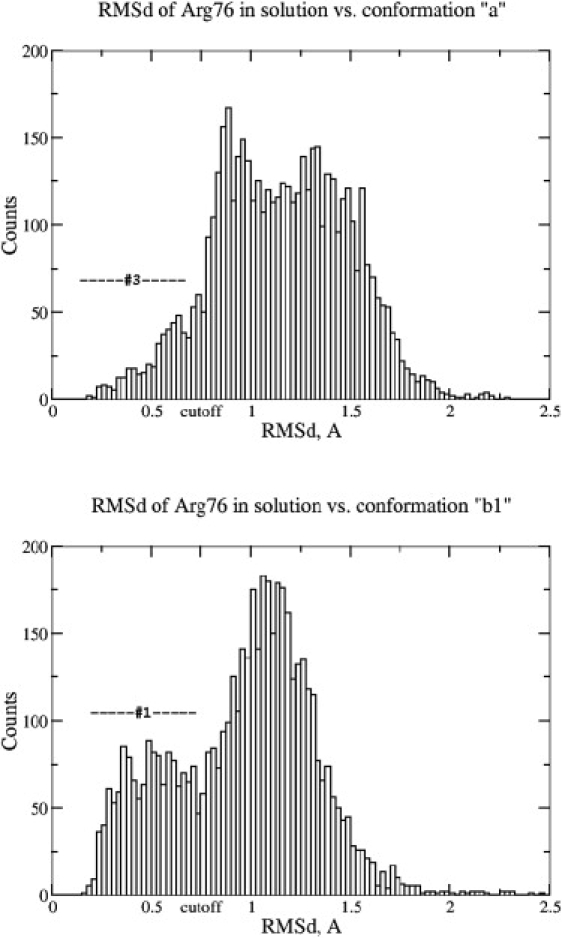
Distributions of Arg-76 RMSD in solution against experimentally observed conformations in chains a and b. The upper and bottom panels demonstrate two situations in which the experimentally observed conformations stay, respectively, off of and close to centroids of the corresponding clusters. “Cutoff” denotes the 0.75 Å cutoff applied to assign links between nodes (configurations) for force-directed network constructions (Fig. 1, c and d).
Conformational sampling of Arg-76 from P51G-m4-CVN and m4-CVN solution simulations
Fig. 3 represents conformations of the Arg-76 residue from representative structures for the six most-populated clusters observed in the last 50 ns of simulation of the P51G-m4-CVN:di-mannose complex in solution. The first five clusters are mt(t/m/p)X rotamers, according to the nomenclature of Penultimate Rotamer Library, i.e., they are related to each other through rotation around Cγ-Cδ and Cδ-Nɛ bonds (47). Cluster 6 is close to ttm−85° rotamer. All of these conformations, except for cluster 5, are in the top list of the most frequently observed arginine conformations. These results demonstrate no pronounced preference for Arg-76 to adopt conformations specific for gating-and-locking of di-mannose in the binding site, and bring into question the relevance of this mechanism for high oligomannose specificity. Additionally, we did not observe the statistically most favorable mtt180° conformation (according to the Penultimate Rotamer Library) as a stable one, in contradiction to the previous conclusion from MD simulations of native CVN (16). Also, formation of the double H-bond between the guanidinium fragment of Arg-76 and two oxygens of di-mannose, related to this mtt180° configuration (Fig. 1 b), took place only occasionally and at best for only a few consecutive frames (for ∼1–2 ps). This result was quite unexpected, so we decided to investigate whether a P51G mutation could cause such a difference in Arg-76 conformational sampling.
Figure 3.
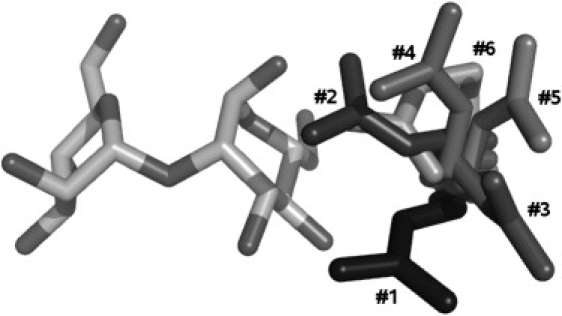
Conformations of the Arg-76 residue from representative structures for the six most populated clusters from the last 50 ns of simulation of the P51G-m4-CVN:di-mannose complex in solution (sampling rate: 10 ps). The closest rotamers are mtt−85° for cluster 1, mtt+85° for cluster 2, mtm180° for cluster 3, mtp−105° for cluster 4, mtp180° for cluster 5, and ttm−85° for cluster 6, according to the nomenclature adopted in the Penultimate Rotamer Library (47). In the rotameric state notation, t, m, and p are used to describe the sequence of trans-, minus, and plus gauche-conformations, respectively, in the aliphatic part of arginine, and the last number describes the orientation of the guanidinium fragment.
First, we performed a solution simulation of the m4-CVN:di-mannose complex that did not have a G51P mutation and therefore had the same binding site as native CVN. We found that Arg-76 samples the same conformational space as in the P51G-m4-CVN:di-mannose complex (Fig. S5 and Table S2). Once again, during a 52 ns simulation of m4-CVN:di-mannose, we did not observe formation of the double H-bond between Arg-76 and di-mannose as a stable configuration. Moreover, in the control 16 ns solution simulation of native CVN, we encountered no such configuration at all. The last simulation was performed on exactly the same system as in previous work, and could be expected to reproduce the published results. The origin of this contradiction remains undetermined. At another extreme, Fujimoto et al. (15) mentioned that in their simulations of CVN, the Arg-76 residue did not make any specific interactions with the sugar. Such variations in computational results may be caused by the use of different force fields and have to be resolved by experiment. Considering the remarkable agreement between the conformations observed in our MD simulation and x-ray structure, we believe our MD protocol is sufficiently reliable. In any case, our results oppose the suggestion of a special role for Arg-76 in di-mannose binding by CVN.
Discussion
Importance of Arg-76
The results of the P51G-m4-CVN:di-mannose complex simulation in solution and crystal, as well as m4-CVN:di-mannose simulation in solution, bring into question the importance of Arg-76 as the residue that locks the ligand in the bound state (the lid). They demonstrated no conformational preferences for Arg-76 toward tight interaction with the di-mannose ligand. Thus, other factors that could provide high specificity of CVN for oligo-mannose should be analyzed. To address this issue, we compared BM binding sites in P51G-m4-CVN, m4-CVN and homologs from the CVN Homology (CVNH) family (49,50).
Sequence alignment of homologs from the CVNH family was previously performed by Percudani et al. (49). Experimental solution structures of three of these homologs were also recently established (50). For the purpose of our study, two important observations can be derived from the sequential comparative analysis. First, Arg-76 is not a well-conserved residue. The conservation rate is only 31% among 16 homologs. The majority of homologs (56%) contain small and/or hydrophobic residues (Gly, Ala, Val, Ser, Thr, Cys, and Pro), which are not the functional analogs of the Arg-76 residue. In line with our previous conclusion, this observation again casts doubt on the importance of Arg-76 for CVN specificity. Second, the majority (75%) of CVN homologs have a “P51G” mutation in the binding-site BM. Thus, the P51G-m4-CVN mutant appears to be more representative of the BM binding site in the CVNH protein family than m4-CVN. To obtain more details about the possible influence of the P51G alteration on the structure of the binding site, we decided to compare the structures of three known homologs and two CVN mutants (P51G-m4-CVN and m4-CVN).
Fig. S6 presents a superposition of five structures: P51G-m4-CVN:di-mannose and m4-CVN:di-mannose complexes, and experimentally determined solution structures of Cr_CVN (after Ceratopteris richardii, PDB ID: 2JZJ), Tb_CVN (Tuber borchii, PDB ID: 2JZK) and Nc_CVN (Neurospora crassa, PDB ID: 2JZL). The structures of the CVN mutants correspond to their energetically minimized geometries, but are very close to the experimental structures used as initial models for the simulations. The RMSD for all nonhydrogen atoms of the original PDB coordinates of the P51G-m4-CVN:di-mannose complex (PDB ID: 2RDK) and the minimized structure is 0.486 Å. The analogous value for m4-CVN:di-mannose structures is 0.566 Å (the initial model was created from PDB IDs 1IIY and 2RDK; see Materials and Methods). Experimental structures for CVN homologs are in apo forms and thus do not have any ligand. Because of this feature, one can note pronounced flexibility of the 74–79 loop, which contains the Arg-76 residue in CVN (Fig. S6). Based on this observation, we speculate that the whole loop (rather than one residue) may participate in accommodation of the ligand in the binding-site BM.
Our conclusion that the Arg-76 residue does not appear to be critically important for keeping di-mannose in the bound state probably should be considered as a hypothesis to be tested experimentally. Indirect experimental support for this conclusion comes from the fact that at least two CVN homologs, Cr_CVN and Nc_CVN (which have Ala and Cys, respectively, in place of Arg-76), bind to di-mannose, as evidenced by 1H-15N heteronuclear single quantum coherence NMR spectroscopic experiments (9). Also, it has been shown that one of the CVN mutants, CV-NmutDB, which has four mutations in the binding site BM (E41A, N42A, T57A, and R76A), does not bind mannose oligomers (11). This result does not contradict our conclusion, since the Thr-57 residue (but not Glu-41 and Arg-76) seems to be very important for binding of di-mannose, as discussed below. Finally, in our trial 16 ns MD simulation of the m4-CVN:di-mannose complex with Glu-41 and Arg-76 “mutated” to Ala, the ligand remained in the bound state for the entire run. It is still possible that polar residues such as Glu-41 and Arg-76 may play a role in the initial attraction and orientation of di-mannose before binding, but that process is beyond the scope of our study.
Another interesting finding from the structural alignment of CVNH family members is that the 49–52 loop also adopts several different conformations (Fig. S6). We investigated the effect of the P51G mutation to the binding site BM and made the following observations (see the Supporting Material for details): 1), P51G-m4-CVN and m4-CVN represent two distinctive and stable main-chain pathways for loop 49–52 among structurally characterized members of the CVNH family (Fig. S7); 2), in the m4-CVN:di-mannose complex, Ser-52 is directed toward the di-mannose ligand, causing a 0.86 Å ligand shift with respect to its position in P51G-m4-CVN:di-mannose; and 3) despite that, the ligands remained in the bound state for the entire duration (>50 ns) of the simulations (Fig. S1). In our further search for an explanation for the stability of these protein-ligand complexes, we decided to analyze the entire H-bonding network as a factor that could provide strong and stable attractive interactions between the protein and ligand.
H-bond network of the ligand-binding site BM
Di-mannose ligand forms an extended H-bond network with the protein. Based on the global analysis of all direct interactions between two mutants and the ligand, we found that discussion of this network could be reduced to eight essential H-bonds. By “essential”, we mean that a particular H-bond is present most of the time (>74%) during the simulation. It is interesting that all eight essential components of this network are the same in both P51G-m4-CVN and m4-CVN (Fig. 4). Three out of four di-mannose oxygen atoms simultaneously play the roles of donor and acceptor of H-bonds, and the average time during which the ligand is involved in this H-bonding is 94% and 97% for P51G-m4-CVN and m4-CVN, respectively (Fig. S8). The di-mannose ligand donates H-bonds to the main-chain oxygen atoms of the Asn-42, Lys-74 (or Val-43), Asn-53, and Ser-52 residues while accepting H-bonds from main-chain nitrogen atoms of residues Asn-42 and Asp-44, and side-chain Oγ of Thr-57. This H-bond net occasionally expands by direct H-bonds with side chains of residues Glu-56, Glu-41, Thr-75, Arg-76, and Gln-78. Concerning Glu-41, it is interesting that all other members of the CVNH family have absolutely conserved Gly instead of the Glu residue. Additionally, an O-H…O H-bond with Oɛ of Glu-41, and an N-H…O H-bond with the terminal amino group of Arg-76 occur only 9% and 6% of the time, respectively. This again brings into question the critical importance of these residues for CVN specificity to di-mannose (Fig. S8). In contrast, based on the multiple character and stability of the “essential” H-bond pattern in our simulations, we suggest that these interactions are a major factor in the specificity and high affinity of CVN toward mannose oligomers.
Figure 4.

“Essential” H-bond network in the P51G-m4-CVN:di-mannose complex as revealed by the explicit water simulation. Three out of four di-mannose oxygen atoms simultaneously play the roles of donor and acceptor of H-bonds; the average time during which the ligand is involved in this H-bonding is 94% and 97% for P51G-m4-CVN and m4-CVN, respectively. Hydrogen atoms are removed for clarity.
Our simulations provide a fully atomistic picture of H-bonding between CVN and di-mannose in the binding site BM. This picture is in agreement with Sandstrom et al.'s (9) conclusion that 3′- and 4′-hydroxyl groups play a key role in high-affinity binding, based on the results of their NMR study of CVN interactions with di-mannose deoxy analogs. Regarding our set of eight “essential” H-bonds, only the interaction with Oγ of Thr-57 was mentioned as a potential H-bond in the first CVN:di-mannose solution structure (10). The presence of the “essential” H-bonds could be deduced from the crystal structures of the P51G-m4-CVN:di-mannose complex, but none were discussed in any detail (18,22). The H-bonding descriptions in these structural investigations were at best limited to interactions with amino residue side chains. To our knowledge, our study provides the first full description of direct H-bonding with di-mannose in the binding-site BM. In particular, the MD simulations indicate that almost all of the interactions with side chains were very transient. On the other hand, taking into account that seven out of eight “essential” interactions occurred with main-chain atoms, and the remaining one interaction occurred with a conserved Thr/Ser-57 residue, we could extrapolate that these H-bonds would also play a key role in the oligomannose specificity in other members of the CVNH family.
Conclusions
We performed MD simulations of a cyanovirin P51G-m4-CVN mutant to investigate the conformational flexibility of Arg-76, which has been considered to be important for ligand binding. The results from the MD simulations revealed a high flexibility of Arg-76 and identified its three stable conformers, which are in remarkable agreement with a recent high-resolution x-ray structure. However, a comparison of the MD simulations in solution and in the crystal environment indicates that one of the three conformers observed in the x-ray structure is influenced by crystal packing. For the second symmetrically independent polypeptide chain, the MD simulation complements the experiment and provides more details about the dynamic nature of Arg-76.
The combination of network analysis and the clustering method proved to be useful for analyzing conformational ensembles. Network representation can visualize relationships between clusters, and differences in conformational ensemble in solution and crystal were clearly illustrated as changes in the topology of the network.
Our results bring into question the importance of the Glu-41 and Arg-76 residues for cyanovirin's high specificity to di-mannose. Both residues participated in H-bonding with di-mannose for only a small fraction of time during the simulations. In addition, these residues are not conserved among homologs. Analysis of specific protein-ligand interactions revealed a strong and stable H-bond network between O3 and O4 hydroxyl groups of mannose residues and predominantly main-chain oxygen and nitrogen atoms. The finding that these eight essential H-bonds may play a key role in the high specificity of cyanovirin to oligomannose is in agreement with recent experimental evidence. However, our conclusion that the conformations of Glu-41 and Arg-76 likely do not relate to binding stability needs to be evaluated experimentally.
Acknowledgments
We thank Dr. R. Fromme and Dr. G. Ghirlanda for providing us with structural data and for helpful discussions. Calculations were performed at the University of Arizona High Performance Computing Center.
This study was supported by the University of Arizona startup fund.
Supporting Material
References
- 1.Boyd M.R., Gustafson K.R., McMahon J.B., Shoemaker R.H., O'Keefe B.R. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob. Agents Chemother. 1997;41:1521–1530. doi: 10.1128/aac.41.7.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrientos L.G., O'Keefe B.R., Bray M., Sanchez A., Gronenborn A.M. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res. 2003;58:47–56. doi: 10.1016/s0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- 3.Barrientos L.G., Lasala F., Otero J.R., Sanchez A., Delgado R. In vitro evaluation of cyanovirin-N antiviral activity, by use of lentiviral vectors pseudotyped with filovirus envelope glycoproteins. J. Infect. Dis. 2004;189:1440–1443. doi: 10.1086/382658. [DOI] [PubMed] [Google Scholar]
- 4.Helle F., Wychowski C., Vu-Dac N., Gustafson K.R., Voisset C. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 2006;281:25177–25183. doi: 10.1074/jbc.M602431200. [DOI] [PubMed] [Google Scholar]
- 5.Shan M., Klasse P.J., Banerjee K., Dey A.K., Iyer S.P. HIV-1 gp120 mannoses induce immunosuppressive responses from dendritic cells. PLoS Pathog. 2007;3:e169. doi: 10.1371/journal.ppat.0030169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai C.C., Emau P., Jiang Y., Agy M.B., Shattock R.J. Cyanovirin-N inhibits AIDS virus infections in vaginal transmission models. AIDS Res. Hum. Retroviruses. 2004;20:11–18. doi: 10.1089/088922204322749459. [DOI] [PubMed] [Google Scholar]
- 7.Tsai C.C., Emau P., Jiang Y., Tian B., Morton W.R. Cyanovirin-N gel as a topical microbicide prevents rectal transmission of SHIV89.6P in macaques. AIDS Res. Hum. Retroviruses. 2003;19:535–541. doi: 10.1089/088922203322230897. [DOI] [PubMed] [Google Scholar]
- 8.Hu Q., Mahmood N., Shattock R.J. High-mannose-specific deglycosylation of HIV-1 gp120 induced by resistance to cyanovirin-N and the impact on antibody neutralization. Virology. 2007;368:145–154. doi: 10.1016/j.virol.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandstrom C., Hakkarainen B., Matei E., Glinchert A., Lahmann M., Oscarson S., Kenne L., Gronenborn A.M. Atomic mapping of the sugar interactions in one-site and two-site mutants of cyanovirin-N by NMR spectroscopy. Biochemistry. 2008;47:3625–3635. doi: 10.1021/bi702200m. [DOI] [PubMed] [Google Scholar]
- 10.Bewley C.A. Solution structure of a cyanovirin-N:Man α1–2Manα complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 2001;9:931–940. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
- 11.Barrientos L.G., Matei E., Lasala F., Delgado R., Gronenborn A.M. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Eng. Des. Sel. 2006;19:525–535. doi: 10.1093/protein/gzl040. [DOI] [PubMed] [Google Scholar]
- 12.Botos I., O'Keefe B.R., Shenoy S.R., Cartner L.K., Ratner D.M. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J. Biol. Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 13.Botos I., Mori T., Cartner L.K., Boyd M.R., Wlodawer A. Domain-swapped structure of a mutant of cyanovirin-N. Biochem. Biophys. Res. Commun. 2002;294:184–190. doi: 10.1016/S0006-291X(02)00455-2. [DOI] [PubMed] [Google Scholar]
- 14.Ziolkowska N.E., Wlodawer A. Structural studies of algal lectins with anti-HIV activity. Acta Biochim. Pol. 2006;53:617–626. [PubMed] [Google Scholar]
- 15.Fujimoto Y.K., Terbush R.N., Patsalo V., Green D.F. Computational models explain the oligosaccharide specificity of cyanovirin-N. Protein Sci. 2008;17:2008–2014. doi: 10.1110/ps.034637.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margulis C.J. Computational study of the dynamics of mannose disaccharides free in solution and bound to the potent anti-HIV virucidal protein cyanovirin. J. Phys. Chem. B. 2005;109:3639–3647. doi: 10.1021/jp0406971. [DOI] [PubMed] [Google Scholar]
- 17.Chang L.C., Bewley C.A. Potent inhibition of HIV-1 fusion by cyanovirin-N requires only a single high affinity carbohydrate binding site: characterization of low affinity carbohydrate binding site knockout mutants. J. Mol. Biol. 2002;318:1–8. doi: 10.1016/S0022-2836(02)00045-1. [DOI] [PubMed] [Google Scholar]
- 18.Fromme R., Katiliene Z., Giomarelli B., Bogani F., McMahon J. A monovalent mutant of cyanovirin-N provides insight into the role of multiple interactions with gp120 for antiviral activity. Biochemistry. 2007;46:9199–9207. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 19.Barrientos L.G., Lasala F., Delgado R., Sanchez A., Gronenborn A.M. Flipping the switch from monomeric to dimeric CV-N has little effect on antiviral activity. Structure. 2004;12:1799–1807. doi: 10.1016/j.str.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Mori T., Barrientos L.G., Han Z., Gronenborn A.M., Turpin J.A. Functional homologs of cyanovirin-N amenable to mass production in prokaryotic and eukaryotic hosts. Protein Expr. Purif. 2002;26:42–49. doi: 10.1016/s1046-5928(02)00513-2. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y., Carroll J.R., Holt L.A., McMahon J., Giomarelli B. Multivalent interactions with gp120 are required for the anti-HIV activity of cyanovirin. Biopolymers. 2009;92:194–200. doi: 10.1002/bip.21173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fromme R., Katiliene Z., Fromme P., Ghirlanda G. Conformational gating of dimannose binding to the antiviral protein cyanovirin revealed from the crystal structure at 1.35 A resolution. Protein Sci. 2008;17:939–944. doi: 10.1110/ps.083472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anselmi M., Brunori M., Vallone B., Di Nola A. Molecular dynamics simulation of the neuroglobin crystal: comparison with the simulation in solution. Biophys. J. 2008;95:4157–4162. doi: 10.1529/biophysj.108.135855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neugebauer A., Klein C.D., Hartmann R.W. Protein-dynamics of the putative HCV receptor CD81 large extracellular loop. Bioorg. Med. Chem. Lett. 2004;14:1765–1769. doi: 10.1016/j.bmcl.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 25.Cerutti D.S., Le Trong I., Stenkamp R.E., Lybrand T.P. Simulations of a protein crystal: explicit treatment of crystallization conditions links theory and experiment in the streptavidin-biotin complex. Biochemistry. 2008;47:12065–12077. doi: 10.1021/bi800894u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malek K., Coppens M.O. Molecular simulations of solute transport in xylose isomerase crystals. J. Phys. Chem. B. 2008;112:1549–1554. doi: 10.1021/jp069047i. [DOI] [PubMed] [Google Scholar]
- 27.Hu Z., Jiang J. Molecular dynamics simulations for water and ions in protein crystals. Langmuir. 2008;24:4215–4223. doi: 10.1021/la703591e. [DOI] [PubMed] [Google Scholar]
- 28.Bond P.J., Faraldo-Gomez J.D., Deol S.S., Sansom M.S. Membrane protein dynamics and detergent interactions within a crystal: a simulation study of OmpA. Proc. Natl. Acad. Sci. USA. 2006;103:9518–9523. doi: 10.1073/pnas.0600398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meinhold L., Smith J.C. Fluctuations and correlations in crystalline protein dynamics: a simulation analysis of staphylococcal nuclease. Biophys J. 2005;88:2554–2563. doi: 10.1529/biophysj.104.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 31.Baucom J., Transue T., Fuentes-Cabrera M., Krahn J.M., Darden T.A. Molecular dynamics simulations of the d(CCAACGTTGG)2 decamer in crystal environment: comparison of atomic point-charge, extra-point, and polarizable force fields. J. Chem. Phys. 2004;121:6998–7008. doi: 10.1063/1.1788631. [DOI] [PubMed] [Google Scholar]
- 32.Babin V., Baucom J., Darden T.A., Sagui C. Molecular dynamics simulations of DNA with polarizable force fields: convergence of an ideal B-DNA structure to the crystallographic structure. J. Phys. Chem. B. 2006;110:11571–11581. doi: 10.1021/jp061421r. [DOI] [PubMed] [Google Scholar]
- 33.Shao J., Tanner S.W., Thompson N., Cheatham T.E. Clustering molecular dynamics trajectories: 1. characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 34.Rao F., Caflisch A. The protein folding network. J. Mol. Biol. 2004;342:299–306. doi: 10.1016/j.jmb.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 35.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R. The AMBER biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Case D.A., Darden T.A., Cheatham T.E., 3rd, Simmerling C.L., Wang J. University of California; San Francisco: 2008. AMBER 10. [Google Scholar]
- 37.Case, D.A., editor. 2008. AmberTools Users' Manual.
- 38.Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 39.Kirschner K.N., Yongye A.B., Tschampel S.M., Gonzalez-Outeirino J., Daniels C.R. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponder J.W., Case D.A. Force fields for protein simulations. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 41.DeMarco M.L., Woods R.J. 2008. Structural glycobiology: a game of snakes and ladders. Glycobiology. 2004;18:426–440. doi: 10.1093/glycob/cwn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerutti D.S., Duke R., Freddolino P.L., Fan H., Lybrand T.P. Vulnerability in popular molecular dynamics packages concerning Langevin and Andersen dynamics. J. Chem. Theory Comput. 2008;4:1669–1680. doi: 10.1021/ct8002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 44.DeLano W.L. DeLano Scientific; Palo Alto, CA, USA: 2008. The PyMOL Molecular Graphics System. [Google Scholar]
- 45.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 46.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lovell S.C., Word J.M., Richardson J.S., Richardson D.C. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 48.Vorontsov I.I., Coppens P. On the refinement of time-resolved diffraction data: comparison of the random-distribution and cluster-formation models and analysis of the light-induced increase in the atomic displacement parameters. J. Synchrotron. Radiat. 2005;12:488–493. doi: 10.1107/S0909049505014561. [DOI] [PubMed] [Google Scholar]
- 49.Percudani R., Montanini B., Ottonello S. The anti-HIV cyanovirin-N domain is evolutionarily conserved and occurs as a protein module in eukaryotes. Proteins. 2005;60:670–678. doi: 10.1002/prot.20543. [DOI] [PubMed] [Google Scholar]
- 50.Koharudin L.M., Viscomi A.R., Jee J.G., Ottonello S., Gronenborn A.M. The evolutionarily conserved family of cyanovirin-N homologs: structures and carbohydrate specificity. Structure. 2008;16:570–584. doi: 10.1016/j.str.2008.01.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.