

Abstract
Rationale
Prepulse inhibition (PPI) of startle is a measure of sensorimotor gating that is heritable and deficient in certain psychiatric disorders, including schizophrenia. Sprague–Dawley (SD) rats are more sensitive to PPI disruptive effects of dopamine (DA) agonists at long interstimulus intervals (60–120 ms) and less sensitive to their PPI-enhancing effects at short (10–30 ms), compared with Long–Evans (LE) rats. These heritable strain differences in sensitivity to the PPI disruptive effects of DA agonists must ultimately reflect neural changes "downstream" from forebrain DA receptors.
Objective
The current study evaluated the effects of the DA agonist, apomorphine (APO), on ventral pallidal (VP) gamma-aminobutyric acid (GABA) and glutamate efflux and PPI in SD and LE rats.
Methods
PPI was tested in SD and LE rats after vehicle or APO (0.5 mg/kg, subcutaneously (s.c.)) in a within-subject design. In different SD and LE rats, VP dialysate was collected every 10 min for 120 min after vehicle or APO (0.5 mg/kg, s.c.) and analyzed for GABA and glutamate content by capillary electrophoresis (CE) coupled with laser-induced fluorescence (LIF).
Results
As predicted, SD rats exhibited greater APO-induced PPI deficits at long intervals and less APO-induced PPI enhancement at short intervals compared to LE rats. APO significantly reduced VP GABA efflux in SD but not in LE rats; glutamate efflux was unaffected in both strains.
Conclusion
Heritable strain differences in PPI APO sensitivity in SD vs LE rats parallel, and may be mediated by, strain differences in the VP GABA efflux.
Keywords: Gamma-aminobutyric acid (GABA), Microdialysis, Dopamine, Prepulse inhibition, Schizophrenia, Strain, CE-LIF
Introduction
Prepulse inhibition (PPI) of the startle reflex is a cross-species measure of sensorimotor gating, in which a startle response is blunted by a weak lead stimulus (Graham 1975; Hoffman and Ison 1980). PPI is reduced in individuals with specific brain disorders and in rats after manipulations of limbic cortex, striatum, pallidum, or pontine tegmentum (cf. Swerdlow et al. 2008). This limbic cortico-striato-pallido-pontine circuitry has been studied in rats to reveal the neurochemical and neuroanatomical substrates regulating PPI at a high level of resolution. In translational cross-species research, this detailed circuit information in rats is used as a “blueprint” to identify substrates that may lead to PPI deficits in humans (Swerdlow et al. 2001a).
PPI in rats is regulated by forebrain dopamine (DA) receptors, with perhaps strongest evidence supporting a primary role for the “D2-like” family of receptors in the nucleus accumbens (NAC; Swerdlow et al. 1986; Mansbach et al. 1988; Caine et al. 1995; Wan and Swerdlow 1997). One major mesolimbic “output” pathway arising within NAC is a dense GABAergic projection into the subpallidal regions that include the ventral pallidum (VP; Jones and Mogenson 1980a, b). This striatopallidal projection forms the next segment of a pervasive neural circuit regulating central inhibitory mechanisms in mammals (Mogenson 1987). Decreased PPI after NAC DA activation might thus reflect reduced activity in GABAergic neurons projecting from the NAC to the VP. Consistent with this notion, the PPI disruptive effects of NAC DA infusion or NAC cell lesions in rats are reversed by infusion of the GABA agonist muscimol into the VP (Swerdlow et al. 1990; Kodsi and Swerdlow 1994) or ibotenic acid lesions of the VP (Kretschmer and Koch 1998) and are reproduced by subpallidal infusion of the GABA antagonist, picrotoxin (Swerdlow et al. 1990; Kodsi and Swerdlow 1995). We also reported that systemic administration of the D3/D2 agonist, quinelorane, disrupts PPI and decreases VP GABA release (Qu et al. 2008). These studies suggest that NAC-mediated decreases in PPI might result from reduced VP GABA release.
Baseline and drug-induced changes in PPI exhibit robust and heritable differences across rat strains. For example, Sprague–Dawley (SD) rats are significantly more sensitive than Long–Evans (LE) rats to the PPI disruptive effects of both direct and indirect DA agonists (cf. Swerdlow et al. 2001a; Swerdlow et al. 2008), but not to the PPI disruptive effects of serotonin agonists and N-methyl-D-aspartate receptor antagonists (Swerdlow et al. 2003). This strain difference in PPI sensitivity to DA agonists is heritable (Swerdlow et al. 2004a, b), independent of fostering conditions or differences in maternal–pup interactions (Swerdlow et al. 2004a), stable across testing and breeding facilities (Swerdlow et al. 2001b), and detectable as early as postnatal day 18 (Swerdlow et al. 2004b). These strain differences appear to be linked to differences in NAC DA-stimulated signal transduction: They are accompanied by, and correlate significantly with, strain differences in NAC [35S]GTPγS binding (Swerdlow et al. 2006), apomorphine (APO)-induced changes in NAC phosphorylation of cyclic adenosine monophosphate response element binding protein (Saint Marie et al. 2007), FOS expression (Saint Marie et al. 2006), and gene expression (Shilling et al. 2008). Differences in DA agonist sensitivity in SD vs LE rats are also detected in behaviors other than PPI (Swerdlow et al. 2006).
Strain differences in sensitivity to the PPI disruptive effects of DA agonists must ultimately reflect changes “downstream” from forebrain DA receptors, in efferent projections that converge onto PPI-mediating circuitry in the pons. By localizing the neural substrates responsible for this heritable phenotype, we can develop models for mechanisms by which genes modify brain circuitry to generate phenotypes of relevance to brain disorders such as schizophrenia. Evidence reviewed above indicates that the PPI disruptive effects of DAergic activation of NAC projection neurons are dependent on reduced GABAergic activity in the VP. Thus, because strain differences in PPI APO sensitivity appear to be mediated via differences in DA-dependent signal transduction in the NAC, there is strong inference that they should be accompanied by differences in the effects of DA agonists on VP GABA release. In fact, any other outcome would force a re-evaluation of evolving models for the neural regulation of PPI. In this study, we compared APO effects on VP GABA release in SD vs LE rats. To assess the neurochemical specificity of any finding, the effects of APO on VP glutamate dialysate were also compared across strains.
Materials and methods
Animals
Male SD and LE rats (n = 30/strain; 225–300 g; Harlan laboratories) were housed in a temperature-controlled vivarium (22°C), maintained on a 12-h reversed light/dark cycle (lights off 7:30am, lights on 7:30pm), and given ad. libitum access to food and water. All testing occurred between 8am to 4pm. Rats were handled within 48 h of arrival and allowed to acclimate to the laboratory for 7 days prior to behavioral testing or surgery. Studies were conducted in accordance with the “Guide for Care and Use of Laboratory Animals” provided by the National Institutes of Health.
Drugs and reagents
Apomorphine (0.5 mg/kg) or vehicle (0.1% ascorbate in saline) was administered subcutaneously (s.c.) to rats.
In vivo microdialysis
Rats (n = 15/strain) received 1 ml/kg (sc) atropine sulfate solution (0.054 mg/ml, Vedco. Inc., St. Joseph, MO 64507, USA) before being anesthetized with 1.4 ml/kg (intraperitoneally (i.p.)) Nembutal sodium solution (50 mg/ml, Ovation Pharmaceuticals Inc., Deerfield, IL 60015, USA) and placed in a Kopf stereotaxic instrument (tooth bar at −3.3 mm). Intracerebral microdialysis guide cannulae (1.0 mm OD; MAB 2.20.G, SciPro Inc, Sanborn, NY, USA) were stereotaxically implanted to terminate 1 mm above the VP (when inserted, the dialysis probe extended ~2 mm from the cannula tip, and the distal 1 mm contained the dialysis membrane (0.6 mm OD; MAB 6.20.1, SciPro Inc)). Probes were unilateral, with sides balanced across strain and treatment groups. Coordinates were predetermined based on the distribution of substance P immunoreactivity in the VP for each strain (see Fig. 1). Dialysis began 7–10 days after surgery.
Fig. 1.

Probe placement in a Nissl-stained section of a dialyzed rat (left) and in a substance P immunostained section (right). Dark substance P staining clearly outlines the VP (right). From the base of the guide cannula tracts (double headed arrows), the dialysis probes extend ~2 mm, with the ventralmost 1 mm containing the dialysis membrane (0.6 mm OD). Substance P staining and the presence of the decussating anterior commissure (ac) were used initially to determine the appropriate AP coordinates for probe placement in SD (−0.30) and LE (+0.20) rats. Other coordinates (from Bregma) were the same for both strains: ML ± 2.5 mm; DV –6.3 mm (after Paxinos and Watson 1998)
Dialysis experiments were conducted between 8am and 3am in a sound-controlled environment. Rats remained in their home cage with bedding, food, and water bottle throughout the experiment. Each rat was lightly anesthetized (0.5 ml/kg sodium pentobarbital solution (50 mg/ml, i.p.) Ovation Pharmaceuticals Inc., Deerfield, IL 60015, USA). The microdialysis probe was inserted and secured to the guide cannula in the afternoon before sample collection and perfused with artificial cerebrospinal fluid (pH 7.2–7.4; 149 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 5.4 mM d-glucose, 0.25 mM ascorbate) delivered via a liquid swivel at 0.2 μl/min overnight. The next morning, the perfusion flow rate was increased to 0.6 μl/min, and background noise was maintained at 60 dB(A). After 1-h equilibration and habituation, dialysate samples were collected in polypropylene tubes at 10-min intervals over a 60-min baseline period. Rats then received either 0.5 mg/kg APO or vehicle (s.c.), and dialysate sampling continued at 10-min intervals for an additional 2 h. This active dose of APO yields the maximal difference in PPI APO sensitivity in SD vs LE rats, based on several published reports from our group of full dose-response characteristics (e.g., Swerdlow et al. 2001b, 2002, 2004b, c). After collection, the samples were frozen on dry ice and stored at −70°C.
Probe placement verification and substance P immunohistochemistry
After the dialysis session, rats were given an overdose of pentobarbital and perfused transcardially with saline, followed by fixation with 10% buffered formalin (pH 7.0). Brains were dissected and cryoprotected for 3 days in the buffered formalin plus 30% sucrose. Frozen brain sections (40 μm) were cut in the coronal plane with a sliding microtome, and mounted sections were then Nissl stained to confirm probe placement (e.g., Figs. 1 and 2). Placement was determined by comparing stained sections to a standard rat brain stereotaxic atlas (Paxinos and Watson 1998). Only two rats had placements that fell outside of the VP, and their results were excluded from the final analysis.
Fig. 2.

Schematic representation of probe placements for all dialysis cases. Regardless of the side dialyzed, which was balanced for each strain, Sprague–Dawley (SD) rats are represented on the left, Long–Evans (LE) on the right. Illustrations are adapted from Paxinos and Watson (1998). VP ventral pallidum, ac anterior commissure
Additional rats (n = 3/strain) had cannulae and probes implanted to verify placement based on substance P immunoreactivity of the ventral pallidum. These rats were perfused immediately after surgery, with the probe in place, and the fixation solution was 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). In these cases, frozen brain sections (40 μm) were collected in commercial Tris-buffered saline (TBS, pH 7.4), and all subsequent solutions were made with this TBS. Endogenous peroxidase activity was suppressed by pre-exposure of the sections to 0.525% H2O2 for 20 min, and this was followed by 2 h in a blocking solution containing 10% normal horse serum (Invitrogen, Carlsbad, CA, USA) and 0.33% Triton X-100. Sections were then incubated overnight with a substance P antiserum (Invitrogen, Carlsbad, CA, USA), diluted 1:1000 with the blocking solution. After intervening rinses, the tissue was sequentially exposed to (1) biotinylated donkey antirabbit serum (Jackson ImmunoResearch, West Grove, PA, USA) for 4 h, diluted 1:800 with the blocking solution; (2) avidin-biotin-peroxidase reagent (Standard Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA) overnight, diluted 1:100 with TBS plus 0.33% Triton X; and (3) reacted with a diaminobenzidine/NiCl peroxidase substrate kit solution (Vector Laboratories). An example is presented in Fig. 1b.
Digital images were acquired with a Leitz Laborlux S microscope equipped with a Polaroid DMC-1 digital camera and software. Digital micrographs presented in this report (Fig. 1a and b) were processed to correct for background illumination and contrast only.
Sample analysis
Microdialysate GABA and glutamate content were determined using CE-LIF detection as previously described (Caille and Parsons 2004, 2006; O’Dell et al. 2006; O’Dell and Parsons 2004; Roberto et al. 2004a, b). GABA and glutamate were derivatized by mixing 4–6 μl of microdialysate with 9 μl of 40 mM borate buffer (pH 10.5) containing 3.8 mM KCN and 1.5 μl of 5 mM naphthalene-2,3-dicarboxaldehyde in methanol. This mixture was reacted at room temperature in the dark for 60 min before loading into the CE instrument (Agilent Technologies, Wilmington, DE, USA) by 50 millibars of pressure for 10 s. GABA and glutamate were separated on a 90-cm fused silica capillary (30 μm inner diameter) by using 15 kV and a background electrolyte solution consisting of 200 mM borate buffer, pH 9.2, containing 34 mM SDS and 2.5 mM 2-hydroxypropyl)-β-cyclodextrin. GABA and glutamate were detected using a LIF detector (Zetalif, Picometrics) that employs a 442 nm HeCd laser (30 mW, Melles Griot). External calibration standards were run in duplicate throughout the sample run. The limits of quantification are approximately 1 nM for each of the analytes, which is well below the concentration of GABA and glutamate typically found in these microdialysis samples (Caille and Parsons 2004, 2006; O’Dell et al. 2006; O’Dell and Parsons 2004; Roberto et al. 2004a, b).
PPI testing
Separate experimental groups (n = 12/strain) were used for behavioral and neurochemical measures. All startle experiments utilized four startle chambers (SR-LAB; San Diego Instruments, San Diego, CA, USA) housed in a sound-attenuated room with a 60-dB ambient noise level. Each startle chamber consisted of a Plexiglas cylinder, 8.7 cm in internal diameter, resting on a 12.5 × 25.5-cm Plexiglas stand. Acoustic stimuli and background noise were presented via a Radioshack Supertweeter mounted 24 cm above the Plexiglas cylinder. Startle magnitude was detected and recorded as transduced cylinder movement via a piezoelectric device mounted below the Plexiglas stand. Acoustic stimulus intensities and response sensitivities were calibrated to be nearly identical in each of the four startle chambers (maximum variability <1% of stimulus range and <5% of response ranges) by using an SR-LAB Startle Calibration System. Chambers were also balanced across all experimental groups. Sound levels were measured and calibrated with a sound level meter (Quest electronics: Oconomowoc, WI, USA), A scale (relative to 20 N/M2), with microphone placed inside the Plexiglas cylinder.
Rats were exposed to a brief ‘matching’ startle session used to assign rats to balanced drug groups according to their average level of PPI. Four days later, rats were treated with APO (0.5 mg/kg) or vehicle (0.1% ascorbate in saline) and placed immediately into the startle chambers. As noted above, this active dose was determined in several previous full dose-response studies to yield the maximal difference in SD vs LE PPI APO sensitivity. Each session was approximately 65 min long, began with a 5-min acclimation to a 70 dB(A) background noise followed by six 10-min trial blocks containing 6 trial types: PULSE (120 dB(A) 40-ms noise bursts); PULSE preceded 10, 30, or 120 ms by a 5-ms noise burst, 12 dB above background; PREPULSE (5-ms noise bursts 12 dB above 70-dB background); and a NOSTIM trial (no stimulus delivery). Each block began with a PULSE trial and in total consisted of five PULSE trials, 15 PREPULSE + PULSE trials (five of each PREPULSE type), 3 PREPULSE trials, and 24 NOSTIM trials. Trials other than NOSTIMs were presented in pseudorandom order with a variable intertrial interval (average of 15 s). NOSTIM trials were interspersed between each consecutive stimulus trials and were used to assess gross motor activity during the test session, but were not included in the calculation of intertrial intervals. Each block ended with a stimulus-free period, lasting 4 min 15 s.
Data collection and analysis
Between-group differences in baseline microdialysate GABA concentrations were first compared by analysis of variance (ANOVA). After confirmation that strains did not differ in baseline VP GABA efflux, post-injection data for each rat were converted to a value corresponding to a percent of the average baseline concentration obtained for 60 min prior to the injection. The effects of drug on VP dialysate levels of GABA and glutamate were then evaluated using ANOVA with repeated measures over time, with dose and strain as between-subjects factor.
Startle magnitudes were calculated for each trial in the 20–100-ms time window following startle stimulus onset. Startle magnitude was examined separately for the PULSE trials that were interspersed among the PREPULSE + PULSE trials, vs those that were presented at the onset of each trial block. PPI was calculated as a percent reduction in startle magnitude on PREPULSE + PULSE trials compared to PULSE trials, i.e., 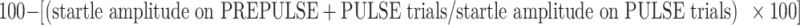 . Any drug effects on %PPI prompted separate analyses to assess the relationship of these effects to drug-induced changes in startle magnitude on PULSE trials. All startle magnitude data were analyzed using ANOVAs with dose and strain as the between-subject factor, respectively. Post hoc comparisons of significant interaction effects and relevant main factor effects were conducted using Fisher’s protected least significant difference and one-factor ANOVA tests.
. Any drug effects on %PPI prompted separate analyses to assess the relationship of these effects to drug-induced changes in startle magnitude on PULSE trials. All startle magnitude data were analyzed using ANOVAs with dose and strain as the between-subject factor, respectively. Post hoc comparisons of significant interaction effects and relevant main factor effects were conducted using Fisher’s protected least significant difference and one-factor ANOVA tests.
Results
GABA efflux
A summary of probe placements used in the following analyses are presented in Fig. 2. Baseline GABA concentrations of SD and LE rats in VP dialysates were stable over 60 min, did not differ between strains, and did not differ between rats subsequently randomized to vehicle vs APO treatment groups in each strain (SD, vehicle vs APO: 57.14 ± 13.04 nM vs 58.22 ± 11.76 nM; LE, vehicle vs APO: 51.67 ± 8.95 nM vs 52.38 ± 8.31 nM). ANOVA of baseline GABA revealed no significant effect of strain, group assignment, strain x group assignment interaction, time block, time block x strain interaction, time block x group assignment interaction, or time block x strain x group assignment interaction. Dialysate data were then expressed as a percent of baseline level, based on the mean levels across the 60-min pre-drug sampling period (Fig. 3a, b). ANOVA of post-injection GABA efflux revealed no significant effect of strain or APO dose, but a significant dose x strain interaction (F = 5.90, df 1, 26, p < 0.022). There was a significant effect of time (F = 2.07, df 11, 286, p < 0.023), but no significant interactions of time x dose, time x strain, or time x dose x strain. GABA efflux in SD rats after 0.5 mg/kg APO treatment declined rapidly to almost 60% baseline levels (F = 10.92, df 1, 11, p = 0.007), where it remained for 120 min. By contrast, APO treatment did not significantly change GABA efflux in LE rats. There was no significant interaction of dose x time for SD or LE rats.
Fig. 3.

VP GABA efflux (% Baseline) in SD (a) and LE (b) rats after vehicle or 0.5 mg/kg APO s.c. (at Time = 0 min). GABA efflux after APO in SD rats declined rapidly to almost 60% baseline levels, where it stayed for 120 min. * Significant reduction in VP GABA efflux compared to vehicle dose (all p values < 0.05). GABA efflux after APO in LE rats remained relatively stable. VP glutamate efflux (% Baseline) in SD (c) and LE (d) rats after vehicle or 0.5 mg/kg APO s.c. (at Time = 0 min). No significant effects of drug or strain on VP glutamate efflux were detected, though an injection artifact was noted after vehicle injection in two LE rats (d)
Glutamate efflux
Baseline VP glutamate concentrations of SD and LE rats dropped during the initial 10 min of sampling but were stable over the subsequent 50 min of baseline collection (effect of time block, minutes 0–60: F = 4.20, df 5, 120, p < 0.002; minutes 10–60: NS), did not differ between strains, and did not differ between rats subsequently randomized to vehicle vs APO treatment groups in each strain (SD, vehicle vs APO: 1.33 ± 0.28 μM vs 1.41 ± 0.22 μM; LE, vehicle vs APO: 1.97 ± 0.45 μM vs 1.89 ± 0.51 μM). ANOVA revealed no significant effect of strain, group assignment, or strain x group assignment interaction. Glutamate efflux was then calculated as a percent of baseline values, based on the mean levels across the 60-min predrug sampling period (Fig. 3c, d). ANOVA of %baseline efflux revealed no significant effect of strain, APO dose, or strain x dose interaction. There was a significant effect of time block (F = 1.96, df 11, 264, p < 0.04) reflecting a postinjection spike in two LE rats injected with vehicle, but no significant interactions of time block x strain, time block x dose, or time block x strain x dose.
Behavior
PPI in SD and LE rats is seen in Fig. 4, for the same postdrug time period assessed in the above microdialysis studies. ANOVA of PPI revealed a significant effect of strain (F = 10.08, df 1, 22, p < 0.005), no main effect of APO dose (F = 3.17, df 1, 22, p < 0.09), a significant effect of prepulse interval (F = 83.12, df 2, 44, p < 0.0001), and significant interactions of dose x strain (F = 7.54, df 1, 22, p < 0.015), dose x interval (F = 26.67, df 2, 44, p < 0.0001), and dose x strain x interval (F = 4.33, df 2, 44, p < 0.02). There was no significant effect of time block, but significant interactions of time x dose (F = 2.75, df 5, 110, p < 0.025), time x interval (F = 3.07, df 10, 220, p < 0.002), and time x dose x strain x interval (F = 2.34, df 10, 220, p < 0.015). Post hoc comparisons confirmed patterns identical to those reported previously: compared to LE rats, SD rats exhibited a greater APO-induced reduction in PPI at long prepulse intervals and a weaker APO-induced increase in PPI at short prepulse intervals (specific statistical comparisons are identified in Fig. 4). APO effects on PPI in both strains were prominent for 30–40 min and then, waned by 60-min post-injection.
Fig. 4.

%PPI in SD and LE rats for 60 min after vehicle or 0.5 mg/kg APO (s.c.). Inset shows data collapsed across 60 min; main figure shows data for 10, 30, and 120 ms prepulse intervals, across the 60-min test session. As predicted, APO markedly disrupted long interval PPI in SD (but not LE rats), and APO enhanced short interval PPI in LE (but not SD) rats. Both effects waned prior to the change in SD VP GABA efflux, as seen in Fig. 3. * Significant PPI-reducing effect of APO (all p values < 0.05); # significant PPI-enhancing effects of APO (all p values < 0.05)
Analysis of startle magnitude on PULSE trails during PPI testing (as shown in Fig. 5a) revealed significant main effects of strain (LE > SD; F = 5.25, df 1, 22, p < 0.032) and time block (F = 16.99, df 5, 110, p < 0.0001) and a significant interaction of strain x time block (F = 2.56, df 5, 110, p < 0.026; Fig. 5). However, there was no significant main effect of APO dose, or significant interactions of dose x strain, or dose x strain x time block (F = 1.09, df 5, 110, NS). Therefore APO effects on startle magnitude were clearly dissociated from those on PPI in SD and LE rats. Similarly, startle habituation (measured from the initial to final startle trials in the session, as shown in Fig. 5b) was not significantly impacted by APO dose or by an interaction of dose x strain.
Fig. 5.

Startle magnitude during PPI testing (a) and on the first trial of each block (b) in SD and LE rats
Analysis of cage displacement after prepulses presented alone, and gross motor activity (NOSTIM levels) during this 60-min period (Table 1) revealed significant effects of strain on both measures (p < 0.04 and p < 0.05, respectively) and a significant effect of APO dose on gross motor activity (p < 0.01). Most critically, the interaction of strain x APO dose was not significant for either of these measures.
Table 1.
APO effects on startle habituation, cage displacement after prepulses alone, and gross motor activity (NOSTIM levels)
| Main effect/interaction | Startle habituation | Cage displacement after prepulses presented alonea | Gross motor activityb (NOSTIM levels) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F value | df | P | F value | df | P | F value | df | P | |
| Strain | 7.27 | 1, 22 | 0.013 | 4.91 | 1, 22 | 0.037 | 4.97 | 1, 22 | 0.04 |
| APO | <1 | <1 | 8.41 | 1, 22 | 0.008 | ||||
| Strain x APO | <1 | <1 | 3.38 | 1, 22 | NS | ||||
| Time blocks | 25.8 | 5, 110 | <0.0001 | 1.17 | 5, 110 | NS | 3.38 | 5, 110 | 0.007 |
| Time blocks x strain | 3.07 | 5, 110 | 0.012 | 2.81 | 5, 110 | 0.02 | 2.35 | 5, 110 | <0.05 |
| APO x time blocks | <1 | <1 | 4.98 | 5, 110 | 0.0004 | ||||
| APO x time blocks x strain | <1 | <1 | 2.48 | 5, 110 | 0.04 | ||||
APO apomorphine
aMean (SEM) cage displacement after prepulse presented alone: Sprague–Dawley rats = 0.76 (2.5), Long–Evans rats = 0.18 (1.2)
bPost hoc comparisons: APO increased NOSTIM activity in SD rats in block 1 (p < 0.03), block 2 (p < 0.03), block 3 (p < 0.02), block 4 (p < 0.04), and decreased NOSTIM activity in LE rats in block 3 (p < 0.05), but increased NOSTIM activity in block 5 (p < 0.05)
Discussion
The major finding in the present study is that systemic administration of APO causes a sustained reduction of VP GABA efflux in SD, but not LE rats. Strain differences in APO effects on PPI reproduced previous findings (e.g., Swerdlow et al. 2004a; Shilling et al. 2008) that compared with LE rats, SD rats are more sensitive to the PPI disruptive effects of APO at long prepulse intervals and less sensitive to the PPI-enhancing effects of APO at short prepulse intervals.
The most important implication of this finding is that it appears to localize the neural circuit basis for this strain difference in PPI APO sensitivity at, or “upstream” from, the NAC medium spiny neuron (MSN) efferent projection. Thus, a neural event known to reduce PPI—reduced VP GABAergic transmission—is triggered by APO in SD but not LE rats. While this finding does not preclude other strain differences in “downstream” circuitry in the VP or pons, there appears to be a strain-based difference in NAC MSN efferent activity that could account for the heritable behavioral phenotype. This finding supports the possibility that reported strain differences in intracellular mechanisms in NAC MSNs—such as DA-linked signaling (Swerdlow et al. 2005; Saint Marie et al. 2006, 2007) or gene expression (Shilling et al. 2008)—might be “upstream” events responsible for the phenotypic differences in APO effects on both VP GABA efflux and PPI. To the degree that the present model reflects a heritable “vulnerability” to a pathological DA-mediated deterioration in sensorimotor gating, these findings suggest that therapeutic targets for such pathology lie upstream from the VP GABA synapse, within the MSN signaling pathways.
The aim of this study was relatively narrow: to test the falsifiable hypothesis that SD vs LE differences in PPI APO sensitivity should be accompanied by differences in VP GABA efflux. A single dose of APO was selected, based on evidence from numerous past dose-response studies that it would yield maximal strain differences in PPI sensitivity (e.g., Swerdlow et al. 2001b, 2002, 2004b, c). Had this dose yielded the expected strain difference in PPI APO sensitivity, absent the predicted difference in VP GABA efflux, the hypothesis would have been falsified. This was not the case. Certainly, a full dose-response characterization of the effects of APO on VP GABA efflux would provide additional layers of information; based on the differential sensitivity and dynamic ranges of very distinct measures (startle and CE-LIF), it would not be surprising if these measures were to diverge at either end of the dose-response range. The goal of this study, however, was not to provide a comprehensive physiological profile of this brain-behavior relationship: rather, it was to select a dose that yields a maximal behavioral signal and to test the clear prediction that it should be accompanied by a specific neurochemical signal.
The VP is also innervated by glutamatergic projections, arising from the frontal cortex, amygdaloid, and subthalamic nuclei (Delgado-Martinez and Vives 1993; Fuller et al. 1987; Groenewegen and Berendse 1990; Kretschmer et al. 2000). The present findings suggest that strain differences in PPI APO sensitivity are not associated with differences in the effect of APO on these VP glutamate projections as a group, but do not preclude the possibility that one or more of these projections might differ across strains, in a manner that is either diluted or otherwise masked by other converging glutamate inputs.
Conversely, striatal GABAergic projections innervate the full extent of the globus pallidus (GP), among many other structures. Others have reported changes in GP GABAergic efflux in response to systemic administration of DA agonists and antagonists (e.g., Robertson et al. 1991; Opacka-Juffry et al. 1998), and it is fully expected that the systemic administration of APO in this study resulted in changes in GABAergic and other forms of neurotransmission throughout the rat brain. Some of these changes might also contribute to the PPI disruptive effects of APO; indeed, our group first reported on the importance of dorsal striatopallidal GABAergic projections in the regulation of PPI (Kodsi and Swerdlow 1995). As noted above, our narrow focus on VP GABA efflux in the present study was designed to test a specific hypothesis based on reports that identify the NAC-VP projection as a potent mediator of the PPI disruptive effects of DA agonists and as a candidate neural substrate for heritable differences in the sensitivity to these effects. The outcome of this study could have falsified this hypothesis, but it did not.
Across the 60-min time course of this PPI session, there was a clear temporal dissociation (implying a functional dissociation) of APO effects on PPI from those on startle magnitude, prepulse-elicited motor activity, and baseline “NOSTIM” activity. Furthermore, APO effects on PPI in both strains are fully resolved after 60 min of testing, while those on VP GABA efflux in SD rats persist for at least 120 min. We reported a similar pattern of persistent changes in VP GABA efflux despite a resolution of PPI changes, in response to quinelorane (Qu et al. 2008). Conceivably, the waning of the APO effect on PPI may reflect changes “downstream” from GABA release, e.g., via an upregulation of VP GABA receptors or comparable forms of neuroadaptation. It is also possible that APO effects on VP GABA efflux (like those on PPI) might be curtailed after 60 min of repeated startle-evoking stimuli; this hypothesis could be tested via the delivery of startling stimuli to rats during VP GABA dialysis.
In summary, strain differences in the PPI disruptive effects of APO are accompanied by predicted, parallel changes in VP GABA efflux. Heritable and presumably genetic mechanisms mediating an enhanced “vulnerability” to the gating-disruptive effects of DA activity appear to be acting upstream from the VP GABA synapse, most likely within intracellular signaling pathways in medium spiny GABAergic efferents from the NAC. Molecules in these pathways are being explored as potential targets of strong inference for therapeutic interventions in disorders of heritable gating deficits.
Acknowledgments
Supported by MH068366 (NRS), MH53484 (NRS), AG00216 (YQ), AA014619 (LHP), and DA024194 (LHP). The authors acknowledge the expert technical advice of Dr. Martin Weber and the administrative assistance of Ms. Maria Bongiovanni.
Disclosures/Conflict of interest
In the past 3 years, NRS has had grant support from Allergan, Inc. and was a paid Consultant to Sanofi/Aventis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Caille S, Parsons LH. Intravenous heroin self-administration decreases GABA efflux in the ventral pallidum: an in vivo microdialysis study in rats. Eur J Neurosci. 2004;20:593–596. doi: 10.1111/j.1460-9568.2004.03497.x. [DOI] [PubMed] [Google Scholar]
- Caille S, Parsons LH. Cannabinoid modulation of opiate reinforcement through the ventral striatopallidal pathway. Neuropsychopharmacology. 2006;31:804–813. doi: 10.1038/sj.npp.1300848. [DOI] [PubMed] [Google Scholar]
- Caine SB, Geyer MA, Swerdlow NR. Effects of D3/D2 dopamine receptor agonists and antagonists on prepulse inhibition of acoustic startle in the rat. Neuropsychopharmacology. 1995;12:139–145. doi: 10.1016/0893-133X(94)00071-7. [DOI] [PubMed] [Google Scholar]
- Delgado-Martinez AD, Vives F. Effects of medial prefrontal cortex stimulation on the spontaneous activity of the ventral pallidal neurons in the rat. Can J Physiol Pharmacol. 1993;71:343–347. doi: 10.1139/y93-053. [DOI] [PubMed] [Google Scholar]
- Fuller TA, Russchen FT, Price JL. Sources of presumptive glutamergic/aspartergic afferents to the rat ventral striatopallidal region. J Comp Neurol. 1987;258:317–338. doi: 10.1002/cne.902580302. [DOI] [PubMed] [Google Scholar]
- Graham FK. Presidential Address, 1974. The more or less startling effects of weak prestimulation. Psychophysiology. 1975;12:238–248. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Berendse HW. Connections of the subthalamic nucleus with ventral striatopallidal parts of the basal ganglia in the rat. J Comp Neurol. 1990;294:607–622. doi: 10.1002/cne.902940408. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Ison JR. Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev. 1980;87:175–189. doi: 10.1037/0033-295X.87.2.175. [DOI] [PubMed] [Google Scholar]
- Jones DL, Mogenson GJ. Nucleus accumbens to globus pallidus GABA projection subserving ambulatory activity. Am J Physiol. 1980;238:R65–69. doi: 10.1152/ajpregu.1980.238.1.R65. [DOI] [PubMed] [Google Scholar]
- Jones DL, Mogenson GJ. Nucleus accumbens to globus pallidus GABA projection: electrophysiological and iontophoretic investigations. Brain Res. 1980;188:93–105. doi: 10.1016/0006-8993(80)90559-4. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Quinolinic acid lesions of the ventral striatum reduce sensorimotor gating of acoustic startle in rats. Brain Res. 1994;643:59–65. doi: 10.1016/0006-8993(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Prepulse inhibition in the rat is regulated by ventral and caudodorsal striato-pallidal circuitry. Behav Neurosci. 1995;109:912–928. doi: 10.1037/0735-7044.109.5.912. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD, Koch M. The ventral pallidum mediates disruption of prepulse inhibition of the acoustic startle response induced by dopamine agonists, but not by NMDA antagonists. Brain Res. 1998;798:204–210. doi: 10.1016/S0006-8993(98)00424-7. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD, Goiny M, Herrera-Marschitz M. Effect of intracerebral administration of NMDA and AMPA on dopamine and glutamate release in the ventral pallidum and on motor behavior. J Neurochem. 2000;74:2049–2057. doi: 10.1046/j.1471-4159.2000.0742049.x. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology. 1988;94:507–14. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ. Limbic-motor integration. In: Epstein AN, Morrison AR, editors. Progress in Psychobiology and Physiological Psychology. Academic Press: New York; 1987. pp. 117–170. [Google Scholar]
- O’Dell LE, Parsons LH. Serotonin1B receptors in the ventral tegmental area modulate cocaine-induced increases in nucleus accumbens dopamine levels. J Pharmacol Exp Ther. 2004;311:711–719. doi: 10.1124/jpet.104.069278. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Manzardo AM, Polis I, Stouffer DG, Parsons LH. Biphasic alterations in serotonin-1B (5-HT1B) receptor function during abstinence from extended cocaine self-administration. J Neurochem. 2006;99:1363–1376. doi: 10.1111/j.1471-4159.2006.04163.x. [DOI] [PubMed] [Google Scholar]
- Opacka-Juffry J, Ashworth S, Ahier RG, Hume SP. Modulatory effects of L-DOPA on D2 dopamine receptors in rat striatum, measured using in vivo microdialysis and PET. J Neural Transm. 1998;105:349–364. doi: 10.1007/s007020050063. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4. San Diego, CA, USA: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Qu Y, Swerdlow NR, Weber M, Stouffer D, Parsons LH. Quinelorane, a dopamine D3/D2 receptor agonist, reduces prepulse inhibition of startle and ventral pallidal GABA efflux: Time course studies. Pharmacol Biochem Behav. 2008;90:686–690. doi: 10.1016/j.pbb.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RG, Graham WC, Sambrook MA, Crossman AR. Further investigations into the pathophysiology of MPTP-induced Parkinsonism in the primate: an intracerebral microdialysis study of gamma-aminobutyric acid in the lateral segment of the globus pallidus. Brain Res. 1991;563:278–80. doi: 10.1016/0006-8993(91)91545-C. [DOI] [PubMed] [Google Scholar]
- Saint Marie RL, Neary AC, Shoemaker J, Swerdlow NR. Apomorphine effects on CREB phosphorylation in the Nucleus Accumbens of rat strains that differ in PPI sensitivity. Biol Psychiatry. 2007;61:37S. [Google Scholar]
- Saint Marie RL, Neary AC, Shoemaker J, Swerdlow NR. Apomorphine effects on CREB phosphorylation in the Nucleus Accumbens of rat strains that differ in PPI sensitivity. Biol Psychiatry. 2007;61:37S. [Google Scholar]
- Shilling PD, Saint Marie RL, Shoemaker JM, Swerdlow NR. Strain differences in the gating-disruptive effects of apomorphine: relationship to gene expression in nucleus accumbens signaling pathways. Biol Psychiatry. 2008;63:748–758. doi: 10.1016/j.biopsych.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA, Koob GF. Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry. 1986;21:233033. doi: 10.1016/0006-3223(86)90005-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff D, Geyer MA. GABAergic projection from nucleus accumbens to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res. 1990;532:146–150. doi: 10.1016/0006-8993(90)91754-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Platten A, Kim YK, Gaudet I, Shoemaker J, Pitcher L, Auerbach P. Sensitivity to the dopaminergic regulation of prepulse inhibition in rats: evidence for genetic, but not environmental determinants. Pharmacol Biochem Behav. 2001;70:219–226. doi: 10.1016/S0091-3057(01)00598-6. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Pitcher L, Platten A, Kuczenski R, Eleey CC, Auerbach P. Genetic differences in startle gating-disruptive effects of apomorphine: evidence for central mediation. Behav Neurosci. 2002;116:682–690. doi: 10.1037/0735-7044.116.4.682. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Platten A, Pitcher L, Goins J, Crain S. Heritable differences in the effects of amphetamine but not DOI on startle gating in albino and hooded outbred rat strains. Pharmacol Biochem Behav. 2003;75:191–197. doi: 10.1016/S0091-3057(03)00078-9. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Auerbach PP, Pitcher L, Goins J, Platten A. Heritable differences in the dopaminergic regulation of sensorimotor gating. II. Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology. 2004;174:452–462. doi: 10.1007/s00213-003-1480-4. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Crain S, Goins J, Onozuka K, Auerbach PP. Sensitivity to drug effects on prepulse inhibition in inbred and outbred rat strains. Pharmacol Biochem Behav. 2004;77:291–302. doi: 10.1016/j.pbb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Platten A, Pitcher L, Goins J, Auerbach PP. Heritable differences in the dopaminergic regulation of sensorimotor gating. I. Apomorphine effects on startle gating in albino and hooded outbred rat strains and their F1 and N2 progeny. Psychopharmacology. 2004;174:441–451. doi: 10.1007/s00213-003-1481-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Kuczenski R, Goins JC, Crain SK, Ma LT, Bongiovanni MJ, Shoemaker JM. Neurochemical analysis of rat strain differences in the startle gating-disruptive effects of dopamine agonists. Pharmacol Biochem Behav. 2005;80:203–211. doi: 10.1016/j.pbb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Krupin AS, Bongiovanni MJ, Shoemaker JM, Goins JC, Hammer RP., Jr Heritable differences in the dopaminergic regulation of behavior in rats: relationship to D2-like receptor G-protein function. Neuropsychopharmacology. 2006;31:721–729. doi: 10.1038/sj.npp.1300877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) 2008;199:331–88. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan FJ, Swerdlow NR. The basolateral amygdala regulates sensorimotor gating of acoustic startle in the rat. Neuroscience. 1997;76:715–724. doi: 10.1016/S0306-4522(96)00218-7. [DOI] [PubMed] [Google Scholar]