


Abstract
Specialized DNA polymerases are involved in DNA synthesis during base-excision repair and translesion synthesis across a wide range of chemically modified DNA templates. Notable features of these enzymes include low catalytic efficiency, low processivity and low fidelity. Traditionally, in vitro studies of these enzymes have utilized radiolabeled substrates and gel electrophoretic separation of products. We have developed a simple homogeneous fluorescence-based method to study the enzymology of specialized DNA polymerases in real time. The method is based on fluorescent reporter strand displacement from a tripartite substrate containing a quencher-labeled template strand, an unlabeled primer and a fluorophore-labeled reporter. With this method, we could follow the activity of human DNA polymerases β, η, ι and κ under different reaction conditions, and we investigated incorporation of the aberrant nucleotide, 8-oxodGTP, as well as bypass of an abasic site or 8-oxoG DNA template lesion in different configurations. Lastly, we demonstrate that the method can be used for small molecule inhibitor discovery and characterization in highly miniaturized settings, and we report the first nanomolar inhibitors of Y-family DNA polymerases ι and η. The fluorogenic method presented here should facilitate mechanistic and inhibitor investigations of these polymerases and is also applicable to the study of highly processive replicative polymerases.
INTRODUCTION
Human cells posses at least 14 DNA polymerases (pols) (1). Pols α, δ and ε are involved in genome duplication. The remaining 11 DNA polymerases have specialized functions within the cell. For example, the X-family DNA polymerase, pol β, is primarily involved in base excision repair (BER) (2). In single-nucleotide BER (3), pol β acts downstream of the abasic endonuclease APE1 by adding a single nucleotide onto the 3′-end of an APE1-incised apurinic/apyrimidinic site and removing the resultant 5′ deoxyribose phosphate via its lyase activity to create a repaired strand with a single nick to be sealed by DNA ligase. The central role of pol β in BER has already been established and the protein appears to have a role in cancer progression (2,4).
Four of the specialized DNA polymerases (pols η, ι, κ and Rev1) belong to the Y-family of DNA polymerases (5) and participate in translesion DNA synthesis (TLS), a process whereby template DNA lesions which would normally block progression of the replication fork are bypassed, such that the damaged genome can be duplicated (6,7). The best-characterized Y-family DNA polymerase is pol η, which can bypass a cis-syn thymidine pyrimidine dimer with the same efficiency and accuracy as undamaged thymines (8,9). Indeed, such properties appear to help protect humans against the deleterious consequences of sunlight exposure, as humans lacking a functional pol η suffer from the sunlight-sensitive, cancer-prone variant form of xeroderma pigmentosum (10,11). Although defects and/or modifications in pols ι, κ or Rev1 have yet to be linked to a human disease, it is generally believed that like pol η, they also participate in the TLS of DNA lesions that would normally block the cell's replication machinery (6). In some cases, such as bypass of N2-dG adducts by pol κ, TLS is accurate and likely to be beneficial to cell survival (12,13). In other cases, it is likely to be error-prone and lead to mutagenesis and carcinogenesis in humans (14). In addition to their role in TLS, it is believed that the Y-family DNA polymerases also participate in a number of cellular processes. For example, pol η promotes A-T mutations during somatic hypermutation of immunoglobulin genes (15,16) as well as processing recombination intermediates (17); pol κ has been implicated in nucleotide excision repair (18); and pol ι has intrinsic 5′dRP lyase activity (19,20) and can extend nicked and gapped substrates (21), suggesting that it may have a specialized function in BER.
Unlike cellular replicases, which are endowed with high processivity, high catalytic efficiency and high fidelity, X- and Y-family DNA polymerases exhibit low processivity, low catalytic efficiency and low fidelity. To facilitate the ongoing studies of the enzymology and cellular roles of these polymerases, a robust and flexible method for monitoring their catalytic activity is needed. Traditionally, in vitro studies of low efficiency polymerases have utilized radiolabeled substrate constructs in conjunction with gel electrophoretic separation. In contrast, highly efficient and processive replicative polymerases are conveniently studied by a range of fluorescence-based methods such as staining of the dsDNA product with PicoGreen, donor–acceptor FRET-type reporter systems, or molecular beacon-based strand displacement (22–26). Methods such as these are poorly suited to measuring the enzymatic activity of the TLS and BER polymerases because the signal detection relies heavily on the production of long dsDNA products and the accumulation of product via multiple catalytic turnover cycles. Furthermore, assays relying on incorporation of fluorescently labeled nucleotide (akin to those used in sequencing) involve expensive reagents and separation steps and need to be extensively validated for each enzyme. Here, we present a universal homogeneous detection approach consisting of a novel fluorogenic substrate designed to report on the strand displacement triggered by the polymerase-catalyzed incorporation of just 1–2 nt. We demonstrate that the assay is capable of detecting the activity of pols β, η, ι and κ; it also permitted studies using uncommon substrates associated with damaged DNA. Furthermore, the assay was used to quantify the inhibitory effect of several small molecules against these specialized DNA polymerases.
MATERIALS AND METHODS
Reagents
Tris–HCl (1 M) was from Invitrogen, while Tween-20, KCl, MgCl2, MnCl2, ellagic acid, aurintriboxylic acid and dithiothreitol (DTT) were purchased from Sigma–Aldrich. Dimethyl sulfoxide (DMSO, certified ACS grade) was obtained from Fisher, Inc. Black 384-well and 1536-well plates were purchased from Greiner Bio-One (Monroe, NC).
Small molecule inhibitors
The compounds were initially prepared as 10 mM DMSO stock solutions and were arrayed for testing as serial 2-fold dilutions at 5 µl per well in 1536-well Greiner polypropylene compound plates following previously described protocols (27).
Enzymes
Recombinant human DNA pol β was either purchased from Trevigen Inc. (Gaithersburg, MD) or produced via published protocols (28). Equivalent performance was noted for the enzyme obtained from the two sources. Baculovirus expressed recombinant human DNA pols ι and η were expressed and purified as described previously (8,29). Recombinant human DNA pol κ was purchased from Enzymax, LLC (Lexington, KY).
Fluorogenic substrates
All oligodeoxynucleotides were purchased from Biosearch Technologies, Inc., (Novato, CA). Combinations of short complementary oligodeoxynucleotide primers and reporters labeled with carboxytetramethylrhodamine (TAMRA) were annealed to oligodeoxynucelotide templates labeled with BHQ-2 (sequences listed in Table 1) to create quenched double-stranded DNA substrates. The annealing mixture of unlabeled primer, TAMRA-labeled reporter and BHQ-2-labeled template in 50 mM Tris–HCl, pH 8.0, 100 mM NaCl, and with or without 5 mM MgCl2 was heated at 95°C for 5 min and allowed to cool gradually to room temperature. These constructs were then stored at −20°C as 50 µM stocks. The aberrant substrate 8-oxo-2′-deoxyguanosine 5′-triphosphate (8-oxodGTP) was purchased from Trilink Biotechnologies (San Diego, CA), while the oligonucleotides containing tetrahydrofuran (THF) as an abasic site mimetic or oxidized guanosine (8-oxoG) were synthesized by Biosearch Technologies.
Table 1.
Substrate constructs used in the present study
| Name | Tripartite substrate sequence |
|---|---|
| 0–17 | 5′-TC ACC CTC GTA CGA CTC TTT TTT TTT TTT TTT GC - 6 – TAMRA-3′ |
| 3′-AG TGG GAG CAT GCT GAG AAA AAA AAA AAA AAA CG - BHQ -2-5′ | |
| 6–17 | 5′-TC ACC CTC GTA CGA CTC TTT TTT TTT TTT TTT GC - 6 -TAMRA- 3′ |
| 3′-AG TGG GAG CAT GCT GAG AAA AAA AAA AAA AAA AAA AAA CG - BHQ -2-5′ | |
| 2–15 | 5′-TC ACC CTC GTA CGA CTC T TTT TTT TTT TTT GC - 6 - TAMRA-3′ |
| 3′-AG TGG GAG CAT GCT GAG AAA AAA AAA AAA AAA CG - BHQ - 2-5′ | |
| 0–12 | 5′-TC ACC CTC GTA CGA CTC T TTT TTT TTT GC - 6 - TAMRA -3′ |
| 3′-AGTGG GAG CAT GCT GAG A AAA AAA AAA CG - BHQ -2-5′ | |
| 0–10 | 5′-TC ACC CTC GTA CGA CTC T TTT TTT TTT GC - 6 - TAMRA -3′ |
| 3′-AG TGG GAG CAT GCT GAG A AAA AAA AAA CG - BHQ - 2-5′ | |
| 2–10 | 5′-TC ACC CTC GTA CGA CTC TT TTT TTT GC - 6 - TAMRA -3′ |
| 3′-AG TGG GAG CAT GCT GAG A AAA AAA AAA CG - BHQ - 2-5′ | |
| 0–9 | 5′-TC ACC CTC GTA CGA CTC T TTT TTT GC - 6 - TAMRA - 3′ |
| 3′-AG TGG GAG CAT GCT GAG A AAA AAA CG - BHQ - 2-5′ |
Names of tripartite DNA substrates and sequences of constituent unlabeled primer strands (underlined), 3′-end TAMRA labeled reporter strands and 5′-end BHQ-2 labeled template strands.
Assay buffers
All tests were performed in 50 mM Tris–HCl, pH 8.0, containing 0.01% Tween-20 and 2 mM DTT. After optimization, the following salt and divalent metal compositions were established for the respective enzyme: 10 mM KCl and 1 mM MgCl2 for DNA pol β; 40 mM KCl and 0.25 mM MnCl2 for DNA pol ι; 40 mM NaCl and 2 mM MgCl2 for DNA pols η and κ. Unless otherwise indicated, dTTP was used at 100 µM final concentration.
Reporter-strand displacement assay in 384-well format
The reporter-strand displacement assay was carried out in a 40-µl reaction mixture in the appropriate DNA pol buffer: 30 µl of DNA polymerase or buffer (no-enzyme control) was pipetted into a 384-well plate; subsequently, 10 µl of substrate was added to start the reaction. In order to demonstrate the stability of the end-point signal, reactions were stopped at time points 2, 4 and 6 min by adding 10 µl of 0.5 M EDTA. Kinetic fluorescence data were collected on ViewLux high-throughput CCD imager (Perkin Elmer, Waltham, MA) equipped with standard optics (excitation filter 525 nm and emission filter 598 nm).
Radiolabeled primer extension assay
Extension assays were performed in a 10 µl reaction mixture containing the same buffer as used in the fluorescent reporter-displacement assay. Radiolabeled 0-12-derived substrates I and II were prepared by 5′-end labeling of the primer-strand using [γ-32P]ATP and annealing to the BHQ-2 labeled template-strand with or without the corresponding TAMRA-labeled reporter-strand. Radiolabeled substrate (5 nM) and 10 nM of polymerase were used, and reactions were started by the addition of dTTP at 100 µM final concentration and incubated at 27°C for 30 min. To stop the reaction, 10 µl of 2× Sequencing gel running dye was added (95% formamide, 10 mM EDTA, 0.1% Xylene Cyanol and 0.1% Bromophenol Blue). Samples were heated to 95°C for 5 min and briefly chilled on ice. Five microliters of the samples were run on polyacrylamide, 8 M urea gels. The reaction products were visualized by autoradiography using PhosphorImager (Fujifilm FLA-5100).
Assay miniaturization in 1536-well format
Three microliters of reagents (buffer as negative control and DNA polymerase in the remainder of the plate) were dispensed by Flying Reagent Dispenser™ (FRD) (Beckman Coulter Inc., Fullerton, CA) into a 1536-well plate. Inhibitor compounds, pamoic, aurintricarboxylic and ellagic acid were delivered as 23 nl of DMSO solutions via pintool transfer as described elsewhere (30); vehicle-only control consisted of 23 nl DMSO (0.7% final concentration). The plate was incubated for 15 min at room temperature, and then 1 µl of substrate (50 nM final concentration) was added to start the reaction. The plate was immediately transferred into ViewLux reader for kinetic fluorescence data collection (27,30). IC50 values were calculated from the dose-response curve fits generated by GraphPad Prism using the fluorescence intensity change over the first 2 min of initial rates and relating it to uninhibited and no-enzyme controls, respectively.
RESULTS
Development of the strand displacement assay
DNA polymerases responsible for BER and TLS are characterized by low processivity. They tend to abort DNA synthesis and dissociate from their substrate/product DNA after having incorporated only a few bases. In order to study the in vitro enzymology of these polymerases, a detection strategy is needed to report on the extension of a primer strand by as few as 1–2 nt. The principle of the strand displacement assay designed and tested here is depicted in Figure 1A. A tripartite substrate was constructed by annealing the following three oligodeoxynucleotide components: (i) a template strand labeled with a BHQ-2 dark quencher on its 5′-end, (ii) a primer strand complementary to the 3′-portion of the template and (iii) a reporter strand complementary to the 5′-portion of the template and labeled with a rhodamine fluorophore on its 3′-end. Thus, in its unprocessed state, the tripartite substrate contains the rhodamine fluorophore in close proximity to the opposite-strand quencher and as a consequence the baseline fluorescence of the molecule is low. Signal generation entails polymerase-dependent extension of the primer and the resulting dissociation of the downstream reporter strand, relieving the quench effect. The primer sequence was random (Table 1), while the template featured a run of adenosines to allow for facile dissociation of the reporter and to also accommodate the preference of pol ι for dTTP (29,31). The 3′-CG-5′ terminus served a 3-fold purpose: it provided a stronger basepairing at the end of the reporter-template duplex to lower the overall length of the reporter, it helped minimize strand slippage during annealing, and with a G-base bearing the quencher, it allowed for a further reduction of the rhodamine signal (32).
Figure 1.

Strand displacement DNA synthesis assay. (A) Assay design. DNA polymerase incorporates dNTP thereby extending the primer strand and displacing the downstream reporter strand labeled with a 3′-fluorophore donor (F), leading to an increased fluorescence signal. F and Q denote 6-TAMRA and Black Hole Quencher-2 (BHQ-2), respectively. Chemical structures of 6-TAMRA and BHQ-2 used in the present study are also provided. (B) and (C) Assay demonstration. (B) DNA polymerase β concentration effect on reporter strand-displacement assay signal was tested using substrate 0–17 (Table 1). Time courses were run at room temperature using 50 nM of 0–17 and 100 μM of dTTP without (open square) or with pol β at different concentrations: 10, 20, 40 and 80 nM (filled squares, triangles, rhombs and circles, respectively). (C) dTTP concentration effect on the signal evolution was tested using 20 nM of pol β and 50 nM of 0–17 at different concentrations of dTTP : 0 (open squares), 25, 50, 100, 200 and 400 μM (filled triangles, inverted triangles, rhombs, circles and squares, respectively). (D) and (E) Template strand titration. Time course studies were performed at 50 nM substrate 0–10 with (filled squares) or without (open squares) 10 nM pol β (D) or 20 nM pol ι (E), respectively. When the fluorescence signal reached near saturation, 100 nM of template-strand labeled with BHQ-2 was added to the reaction mixture (arrow) and the fluorescence monitoring was resumed. Signal quenching upon addition of excess template strand is indicative of re-annealing of the intact displaced reporter strand.
In evaluating the proposed scheme, a series of candidate substrates was prepared via a standard annealing procedure; their sequences are shown in Table 1. To confirm that the tripartite construct is a viable polymerase substrate, the primer of the ‘0–12’ TAMRA/BHQ-2-labeled construct was 32P-end-labeled and pol β-, η- and ι-dependent primer extension monitored by gel electrophoresis and autoradiography analysis (Supplementary Figure 1). In the initial fluorescent assay test, when 50 nM solutions of substrate were treated with pol β (Figure 1B), a strong fluorescence increase was noted with time; no change was observed in the absence of polymerase. Consistent with the utilization of deoxyribonucleotide triphosphate as a co-substrate, the signal evolution was also strongly dependent on dTTP concentration (Figure 1C): decreasing dTTP below the optimal level of 100–200 μM resulted in simultaneous decrease of the reaction rate and an increase in the lag time before the onset of fluorescence increase. At a constant substrate concentration, the maximum signal reached was independent of the enzyme concentration used (Figure 1B), indicating that the reaction was reaching completion and that at the conditions of excess of substrate relative to enzyme the polymerase was undergoing multiple turnovers. Correspondingly, when the enzyme was held constant, the increase in substrate supplied was associated with a direct increase in signal amplitude (Supplementary Figure 2A). The endpoint signal obtained from the reaction was approximately 25% lower than the fluorescence associated with a free reporter strand in solution, likely due to re-equilibration of the released strand with the extension product. Indeed, we observed the same small decrease in fluorescence when a premade duplex corresponding to the reaction product was added to reporter strand (Supplementary Figure 2B). However, the said difference was not dependent on the enzyme concentration and as such should have no effect on the calculation of percent inhibition during assay application.
In some instances, a stopped reaction with an endpoint read might be preferable relative to a real-time kinetic read. To this end, we evaluated an EDTA-based reaction stopping protocol: to the polymerase reaction, which was allowed to proceed to a different percent completion, we added EDTA to a final concentration of 50 mM and monitored the reaction signal as a function of plate storage time. The signal was found to remain constant for at least 45 min (Supplementary Figure 3), thus confirming that a batch-mode processing of plates is possible with this assay.
To verify that the fluorescence signal increase was associated with a release of the reporter strand and not with a spurious nuclease-induced substrate degradation activity, an aliquot of template strand was added to pols β and ι reactions that were first allowed to proceed to near completion (high-fluorescence signal plateaus in Figure 1D and E). A dramatic drop in signal back to levels close to background was observed upon the addition of excess template strand (right half of time courses in Figure 1D and E). The quenching of signal upon addition of template was observed with both pols β and ι and strongly supports the strand displacement mechanism for signal generation in this assay.
Stability and dynamic range of substrates
The extent of fluorophore quenching within the tripartite substrate (‘dark fluorescence’) has a direct impact on the maximum fold signal increase expected from the corresponding polymerase extension reaction. The substrates listed in Table 1 were thus evaluated with respect to their maximum dynamic range by comparing the fluorescence intensity of 50 nM solutions of substrate with the intensity obtained from 50 nM corresponding free reporter strand (Figure 2A). All constructs tested exhibited strong degree of fluorophore quenching, with even the shortest reporters displaying ranges of at least 45-fold (substrates 2–10 and 0–9). The dependence of degree of quenching on reporter length was expected and matched the trend in melting temperature of the corresponding heteroduplex, with the longest reporters being quenched the strongest, close to 100-fold (substrate 0–17); we note that such dynamic ranges approach those of the best molecular beacons. Constructs bearing the shortest reporter strands displayed the lowest degree of quenching, consistent with a greater fraction of free unhybridized reporter as a function of weakened base pairing in those heteroduplexes (9mer and 10mer reporters versus their 15mer and 17mer counterparts). With the shortest reporters, a relationship between stability and presence of gap between the 3′-end of the primer and the 5′-end of the downstream reporter was also noted. For example, for both the 11mer and 10mer reporters, the fold quenching was the greatest for the zero-gap (i.e. nicked) constructs. In the case of the 9mer reporter, only the zero-gap construct could be prepared. The significant stabilization of the zero-gap constructs, such as 0–9, relative to the gap-containing substrates is in agreement with the previously noted coaxial base-stacking effect (33) operating when two oligonucleotides hybridize to a template directly adjacent to one another.
Figure 2.

Characterization of tripartite DNA substrates. (A) Maximum dynamic range, defined as the fluorescence signal ratio between free reporter strand and tripartite substrate (Table 1) measured at 50 nM. (B) Substrate stability. Substrate 0–10 was subjected to repeat freeze-thaw cycles and tested in the assay at days 0 (squares), 1 (triangles), 4 (rhombs), 7 (circles) and 10 (inverted triangles). Time courses were carried out using 50 nM substrate with (filled symbols) and without (empty symbols) 10 nM pol β.
The utility of the above substrates depends to a large degree on their ease of handling and their overall stability as reagents. During the course of our experiments, we routinely prepared the substrates following the simple annealing procedure and noted excellent batch to batch reproducibility (data not shown). To assess the effect of multiple freeze-thaw cycles on the substrate stock solutions, aliquots of the two shortest, and potentially least stable, substrates 0–12 and 0–10 were frozen at −20°C and thawed repeatedly. Both the background fluorescence intensity and their capacity to serve as substrates in the pol β assay were assessed. As demonstrated by the overlapping time-course trends for substrate 0–10 in Figure 2B, a series of 10 freeze-thaw cycles did not have an appreciable effect on the fluorescent properties or enzymatic reaction profile of the substrate. An additional demonstration of reagent stability was achieved when working stocks of pol β enzyme and substrate 0–12 were prepared and tested immediately after preparation (fresh) and after cold-room storage overnight (16 h); both the enzymatic and background reaction profiles were unchanged upon overnight reagent storage, with the Z′ factor (34) remaining above 0.7 (Supplementary Figure 4).
Effects of substrate structure and divalent metal ions on reaction progress
Because the different TLS polymerases have been reported to exhibit different substrate specificities, metal ion sensitivity and fidelity, we performed a series of evaluation experiments by using three polymerases (pols β, η and ι) in combination with six tripartite substrate constructs (Table 1). All three polymerases tested were able to at least begin to displace reporter strands in the corresponding tripartite substrates regardless of their structure when reaction mixtures were incubated for ∼25 min : however, the kinetic profiles of each polymerase relative to the substrates tested were quite different (Figure 3). A trend observed for all polymerases was that strand displacement DNA synthesis occurred at a faster rate when tripartite substrates had no gap or featured the shortest reporter strand. Pol β demonstrated high sensitivity to gaps, where introduction of a two-base gap with retention of the reporter length resulted in a significant delay in onset of fluorescence signal rise (substrates 2–10 versus 0–10, Figure 3A). Pol β, however, was less sensitive to the reporter length, processing longer-reporter substrates at rates comparable to those observed with the shorter counterparts (substrates 0–17 and 2–15 versus substrates 0–12 and 2–10), the latter feature being consistent with its recognized ability to continuously incorporate up to a dozen bases (35).
Figure 3.

Effect of DNA substrate structure on reaction profile. Time course studies with (A) pol β, (B) pol η and (C) pol ι were carried out at room temperature using 10 nM enzyme and 50 nM of substrates 0–17 (open squares), 2–15 (open triangles), 6–17 (open rhombs), 2–10 (filled squares), 0–12 (filled triangles) and 0–10 (filled rhombs).
Pol η did not show a preference from among the short gapped or nicked constructs utilizing efficiently 0–10, 0–12 and 2–10, but displayed progressively slower signal change as the reporter length increased (Figure 3B). Pol ι, on the other hand, displayed faster rates with 0–10 and 2–10 relative to 0–12 in this tested condition and the signal evolution was dampened significantly with the longer-reporter substrates (Figure 3C). Thus, pol ι appeared to be the most sensitive of the three enzymes with respect to both gap and reporter length of the substrate utilized. Consistent with these polymerases’ low processivity, substrate 6–17 (open rhombs, Figure 3), featuring the combination of the longest gap and reporter length, proved to be one of the most difficult to process by all three enzymes: negligible fluorescence signal change was observed with pols ι and β, and a slow onset of signal increase was detected with pol η with a 25-min incubation. This stood in marked contrast with the efficient Klenow exo-minus polymerase, an A family DNA polymerase, where 100% reaction completion was achieved rapidly with the same 6–17 substrate (Supplementary Figure 5).
The effects of salt and divalent metal cofactors Mg2+ and Mn2+ on TLS polymerase reaction could be studied conveniently with the present assay. Unlike replicative DNA polymerases (36), pols β, ι and η exhibited maximal activity under conditions of moderate KCl or NaCl concentrations of 20–60 mM (data not shown). The maximal activity of TLS polymerases was greatly influenced by the identity and exact concentrations of the divalent metal salts MgCl2 or MnCl2. For example, the previously noted strong preference for Mn2+ exhibited by pol ι (37) was easily detected here and stood in contrast with the profile presented by pol η (Figure 4). Pol ι displayed its highest activity in the presence of MnCl2 at low concentration, 0.05–0.1 mM; its activity also peaked at low Mg2+, but was considerably higher in the presence of Mn2+ (Figure 4A). In contrast to pol ι, pol η functioned best in the presence of 1–5 mM MgCl2, but exhibited dramatically lower activity in the presence of MnCl2 (Figure 4B). We note that lowering the divalent metal concentration to trace levels or zero resulted in lower stability of the short 0–10 substrate, as evidenced by the increase in baseline fluorescence intensity in the metal-free samples. Therefore, for more refined studies of salt or metal ion effects on pol ι and other similar polymerases, a more robust substrate featuring a longer reporter (such as 0–12) may be needed.
Figure 4.

Effect of divalent ions on the activity of pols ι and η. Reactions (20 min) were carried out in presence of increasing concentrations of either Mg2+ or Mn2+ ions using 10 nM enzyme [pol ι (A) and pol η (B)] and 50 nM substrate 0–12. Empty and solid bars indicate Mn2+ or Mg2+ ion concentrations, respectively.
Processing of aberrant substrates
Lesion bypass and incorporation of modified/unnatural nucleotides are important cellular responses to DNA damage during replication and have been studied almost exclusively by the use of radiolabeled DNA substrates (8,38–42). Thus, we investigated whether the current assay could be used in mechanistic studies of interactions of specialized DNA polymerases with such substrates. First, we tested abasic lesion bypass by pol β (43), pol η (39), pol ι (44–46) and pol κ (40). The sequence of substrate F-1-10 containing a tetrahydrofuran abasic site analog is shown in Table 2 (abasic site denoted by an F). All polymerases except pol β appeared able to bypass the abasic site using dTTP as a substrate, although their corresponding percent reaction (relative to maximum reporter displacement) achieved with a 20-min incubation were different: 73% for pol η, 25% for pol ι and 39% for pol κ, respectively. However, this is unlikely to reflect true TLS, but rather template misalignment; a common characteristic of Y family DNA polymerases (1,47).
Table 2.
Lesion bypass DNA constructs
| Lesion tested | Substrate name | Substrate sequence | Enzyme used | Percent reaction at 20 min |
|---|---|---|---|---|
| Abasic site | F-1-10 | 5′-TC ACC CTC GTA CGA CTC TT TTT TTT GC-6-TAMRA-3′ | Polβ | 0 |
| 3′-AG TGG GAG CAT GCT GAG F AA AAA AAA CG-BHQ-2-5′ | Polι | 25 | ||
| Polη | 73 | |||
| Polκ | 39 | |||
| 8-oxoG in template | 1X-10 | 5′-TC ACC CTC GTA CGA CTC TT TTT TTT GC-6-TAMRA-3′ | Polβ | 100 |
| 3′-AG TGG GAG CAT GCT GAG X AA AAA AAA CG-BHQ-2-5′ | Polι | 1 | ||
| Polη | 100 | |||
| Polκ | 47 | |||
| 0X2-10 | 5′-TC ACC CTC GTA CGA CTC TT TT TTT TTT GC-6-TAMRA-3′ | Polβ | 0 | |
| 3′-AG TGG GAG CAT GCT GAG XX AA AAA AAA CG-BHQ-2-5′ | Polι | 100 | ||
| Polη | 90 | |||
| Polκ | 49 | |||
| 0X-10G | 5′-TC ACC CTC GTA CGA CTC TC TTT TTT GC-6-TAMRA-3′ | Polβ | 100 | |
| 3′-AG TGG GAG CAT GCT GAG AX AAA AAA CG-BHQ-2-5′ | Polι | 84 | ||
| Polη | 100 | |||
| Polκ | 86 |
The sequences of tripartite substrates are shown with the template strand locations of an abasic site (tetrahydrofuran indicated by an F) or 8-oxoG (indicated by an X). Percent reaction was calculated as a fraction of complete displacement at 20 min of incubation. The primer strand is underlined.
Similarly, bypass activities of different polymerases were examined using three different constructs containing one or two oxidized guanosines [8-oxoG, also referred to as 8-OHdG (48), indicated by an X in Table 2] placed as either the first downstream base within the template opposite a gap (substrate 1X-10, Table 2), or as two mismatched bases in the template opposite the last two bases of the primer strand (substrate 0X2-10, Table 2), or as a single-base lesion within the template against the second base of the reporter strand (substrate 0X-10G, Table 2). Significant differences in processing efficiencies were observed for the four polymerases tested. Pol η efficiently bypassed the lesion regardless of the 8-oxoG position in the template, whereas pol κ bypassed all lesions at similar rates but less efficiently than pol η (47 and 49% pol κ activity with 1X-10 and 02X-10 substrates). In contrast to pols η and κ, and in agreement with previous reports (39,45,49), pol ι was unable to bypass the 8-oxoG lesion in the single gapped template (only about 1% reaction with substrate 1X-10, Table 2), but efficiently processed the primer-mismatch and the non-gapped substrates 0X2-10 and 0X-10G. Consistent with its generally high intolerance to primer-template mismatches, pol β failed to utilize the construct 0X2-10 featuring a double-8-oxoG mismatched primer but was capable of processing the other two lesion-containing substrates.
Finally, using the current assay we tested the utilization of oxidized guanosine dGTP (8-oxodGTP) by pols β and ι. In contrast to pol β, which was inactive with this substrate, pol ι partially incorporated 8-oxodGTP opposite template A, with 37% displacement at 14 min, compared with normal dTTP incorporation (Figure 5). Consistent with previous accounts (50), incorporation of 8-oxoG was also observed with pol η but at a lower efficiency (15%, data not shown). Importantly, the efficient utilization of 8-oxodGTP (misinsertion) by pol ι was dependent on the nature and level of divalent metal ion. Activity relied on the presence of 0.25 mM Mn2+, whereas no reaction was detected in the presence of Mg2+ (data not shown). These results highlight the potential utility of the current assay as a sensitive tool in lesion bypass studies.
Figure 5.

Utilization of a modified dNTP substrate. Pol β (A) and pol ι (B) reactions were conducted using 10 nM enzyme and 50 nM substrate 0–10 in the presence of 100 μM dTTP as control (squares) and 8-oxodGTP (circles). Filled and open symbols represent the presence or absence of enzyme, respectively.
Effect of small molecule inhibitors
In addition to probing the enzymology of specialized polymerases, the present assay was expected to be useful in high-throughput screening for the discovery and characterization of small molecule inhibitors of these enzymes. We easily adopted it to a highly miniaturized 1536-well format by direct volume scaling. Three microliters of enzyme were dispensed into the assay plate; addition of 1 μl of substrate started the reaction. The fluorescence signal increase was robust, with minimal well-to-well variation, as evidenced by the high Z′ factors of between 0.7 and 0.8 obtained with pols β, η, ι and κ (64 wells per condition tested, Figure 6). The excellent statistical performance of the assay in 1536-well format combined with the simple mix-and-read type of protocol make it especially relevant to high-throughput experiments. To probe the assay's ability to record polymerase inhibition, extension reactions involving these polymerases were carried out in the presence of three compounds previously noted for their inhibitory properties against either pol β [pamoic acid (51)] or other DNA-processing enzymes [aurintricarboxylic acid (52,53) and ellagic acid, PubChem AID 603] (structures and IC50 values in Table 3; example reaction time courses shown in Supplementary Figure 6). The inhibition experiments were performed in 1536-well format and the test compounds were arrayed as 12-point 2-fold dilutions in duplicate. With all three polymerases, a consistent concentration-dependent inhibition was observed for all three compounds. Pamoic acid, a previously reported pol β inhibitor (51), was the weakest of the three compounds: its IC50 against pol β was in the micromolar range [60 μM when tested in the 1536-well format (Table 3) and 45 μM in 384-well tests (data not shown)]. Interestingly, under the present assay conditions, pamoic acid was slightly more potent against pol ι (IC50 4.9 μM). Both aurintricarboxylic acid and ellagic acid displayed potent nanomolar activity against all three polymerases tested; they also exhibited almost identical inhibitory activities and appeared slightly more potent against pol β (IC50 ∼ 20 nM) than against pol ι (IC50 ∼ 90 nM).
Figure 6.

Assay miniaturization to 1536-well format. Time course assays for pol β (A), pol η (B), pol ι (C) and pol κ (D) were conducted in 4-μl reaction volumes with 10 nM of enzyme and 50 nM substrate 0–10. Filled and open symbols represent the presence or absence of enzyme, respectively. Averages and standard deviations shown are from 64 wells per reaction condition.
Table 3.
Small molecule inhibitors
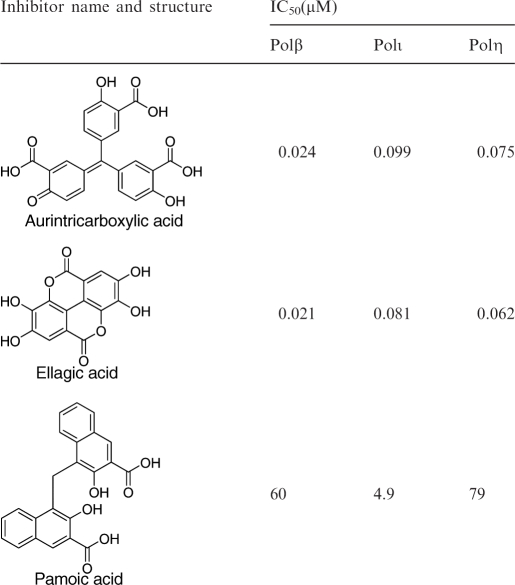 |
The structure of the compounds used in the current study. The compounds were tested as 12-point 2-fold dilutions in duplicate in 1536-well plates. IC50 values were calculated from the dose–response curve fits generated by GraphPad Prism.
DISCUSSION
We report the development and use of a real-time reporter-strand displacement assay for human DNA polymerases based on tripartite fluorogenic substrates. These constructs are only minimally fluorescent because a quencher present on the template strand attenuates the emission of the reporter-strand fluorophore. Upon polymerase-catalyzed reaction the fluorescence intensity increases as the primer strand is extended by addition of nucleotides and the downstream reporter strand is correspondingly displaced and released into solution leading to restoration of the reporter label fluorescence. Initial studies of the proposed assay included (i) the effect of the nature of the downstream reporter-template duplex, in terms of length and sequence that would maximize the quench, yet be amenable to polymerase strand separation and (ii) the presence of variable-length gap between the 3′ terminus of the primer and the 5′-end of the reporter strand. The primer, whose sequence and length were selected to ensure both robust duplex formation and minimal cross-hybridization with the reporter or the downstream portion of the template, was kept largely unchanged. We conveniently exploited the coaxial base stacking effect (33) to construct the shortest stable substrates which were at the same time the easiest to disrupt upon polymerase action. We note that in the present scheme both labels (fluorophore and quencher) are positioned as far away from the site of polymerase binding and catalysis as possible so as not to interfere with the binding and extension reactions.
TAMRA, a bright, pH-insensitive analogue of rhodamine, was utilized as the red-shifted fluorophore and was combined with BHQ-2, a spectrally matched non-emitting dark quencher which has been used in a variety of DNA/RNA detection schemes, such as molecular beacons (32,54). The need for shifting the fluorescence detection to longer wavelengths in comparison with the traditionally used fluorescein/DABCYL pair was demonstrated in our recent profiling study where up to 0.2% of the members of a diverse compound collection, the NIH Molecular Libraries Small Molecule Repository (MLSMR, https://mli.nih.gov/mli/mlsmr/), interfered with fluorescein-like detection (excitation near 480 nm, emission near 530 nm). The incidence of compounds capable of interfering with detection in the red-shifted light region (e.g. excitation around 550 nm and emission near 590 nm) diminished to well below 0.01% (55), thus making the rhodamine-type fluorophore a better choice for designing compound screening assays (56).
Another advantage of the current assay system is the modular nature of the substrate components which allows the construction of a wide variety of desired DNA synthesis-dependent reporter displacement substrates with or without lesions, thereby avoiding the generation and use of radiolabeled molecules and obviating the need for running time consuming gel assay protocols. While we have demonstrated that the present homogeneous assay can be useful to study the mechanism of single DNA polymerase enzymes in vitro, the method should also be applicable to lesion bypass studies involving a DNA polymerase in combination with auxiliary proteins (57) as the upstream portion of the substrate incorporating the primer can be extended significantly to accommodate potential binding regions for prospective co-recruited proteins.
The mechanistic investigation performed here revealed a number of significant differences among several members of the X- and Y-family DNA polymerases. We anticipated that the shortest reporters, such as in substrate 0–10, would be displaced upon incorporation of 1–2 bases while the longer reporters would require a more persistent sequential incorporation of bases by the polymerase before a signal increase is detected. Furthermore, it was anticipated that a gap introduced between the primer and reporter would represent an additional oligonucleotide tract, which the polymerase would need to fill before it encounters the reporter, and as such, a gap would have a slowing effect on the fluorescent signal evolution. These general dependencies were indeed observed with all polymerases tested: however, a spectrum of additional preferences and sensitivities was observed. Pol β was easily slowed down by the presence of gaps between the primer and the reporter but was less sensitive to increasing length of the reporter strand. Conversely, pol η was capable of processing same-length reporter substrates at similar rates regardless of the presence or absence of a gap, but displayed a pronounced slowdown when challenged with longer-reporter substrates. Pol ι, in turn, was the least efficient of the three enzymes and appeared to be sensitive to both the presence of gap and to the length of the reporter strand.
Our studies utilizing substrates containing abasic and oxidized-guanine lesions likewise demonstrated that key differences exist among the four polymerases tested with respect to their lesion-bypass abilities. Specifically, pol β was uniquely sensitive to mismatches between the terminal bases of the primer and template, while pol ι was particularly sensitive to gapped substrates but was otherwise capable of extending across lesions and mismatches; pols η and κ processed the different substrates at similar rates but pol κ was slower than pol η. The nature of the divalent metal ion was shown to have a significant effect on the enzymatic activities of TLS polymerases, at least in the cases of pols ι and η tested here. Because the above effects were all enzyme context-dependent and agreed with previous studies of these polymerases, we conclude that the present assay faithfully reports on the effects of reaction conditions on the enzyme activity and as such it represents a convenient tool for studying the reaction mechanisms of non-replicative polymerases.
The simple protocol and robust performance of the present assay allowed its miniaturization and application in 1536-well format. Tests performed here with several small molecules further validated the assay by recapitulating the previously noted inhibition of pol β by pamoic acid (51). While a recent study by Kimura et al. (58) claimed the identification of penicilliols as inhibitors specific for Y-family polymerases, we note that those natural products were of low potency (IC50 values in the 20–30 μM range). On the other hand, the present studies led to the identification of non-selective yet highly potent molecules: to the best of our knowledge, aurintricarboxylic acid and ellagic acid are the first reported nanomolar inhibitors of polymerases ι and η. The high potencies of aurintricarboxylic acid and ellagic acid have not been previously noted for these polymerases; however, the strong inhibitory power of aurintricarboxylic acid against a range of DNA-processing enzymes is well-documented (52,53,56,59,60). While ellagic has been noted mostly for its cancer-preventive antioxidant properties (61), a recent PubChem search revealed that it was among the top actives in a screen of Escherichia coli DNA polymerase III holoenzyme system (PubChem Assay ID 603). The present study adds pols β, ι and η to the list of molecular targets for this small molecule.
In summary, we have developed a simple homogeneous fluorescence-based method to study the enzymology of non-replicative polymerases in real time. Its use with several members of the X- and Y-family of human DNA polymerases (pols β, ι, η and κ), as well as a range of substrates, has highlighted significant differences in the mechanism of action of these enzymes. In addition to the four polymerases reported on here, our preliminary studies indicate that the assay can also be used to study pol μ and the archaeal Dpo4 enzyme from Sulfolobus solfataricus (data not shown). The real-time kinetic fluorescence acquisition makes the assay robust for the purposes of high-throughput screening and it makes possible the collection of initial-rate data for enzymological studies. The ability to report on weak, as well as on potent inhibitors, is an important feature of the assay making it suitable for screening of diverse compound collections and for use in the development of potent and selective small molecules directed against the individual polymerases. In addition to being suitable for inhibitor discovery and investigation, the present assay should find application for monitoring the progress of directed enzyme evolution where recently a SYBR Green-based technique was developed (62–64). Lastly, while the assay was developed to address specialized DNA polymerases, it can easily be utilized in studies of highly proficient and high-fidelity DNA polymerases, as demonstrated here with Klenow DNA polymerase.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research; Intramural Research Program of NIA, NIEHS, (Z01-ES050158 and Z01-ES050161), NICHD and NHGRI, NIH, and in association with NIH Grant 1U19CA105010. Funding for open access charge: Molecular Libraries Initiative of the NIH Roadmap for Medical Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv. Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 2.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem. Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 3.Wilson DM, 3rd, Bohr VA, McKinnon PJ. DNA damage, DNA repair, ageing and age-related disease. Mech. Ageing Dev. 2008;129:349–352. doi: 10.1016/j.mad.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase β and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 5.Ohmori H, Friedberg EC, Fuchs RPP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, et al. The Y-family of DNA polymerases. Mol. Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 7.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl Acad. Sci. USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase η. EMBO J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase η. J. Biol. Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 10.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of Xeroderma Pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 11.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 12.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Polκ protects mammalian cells against the lethal and mutagenic effects of polycyclic hydrocarbons. Proc. Natl Acad. Sci. USA. 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Wu X, Guo D, Rechkoblit O, Wang Z. Activities of human DNA polymerase κ in response to the major benzo[a]pyrene adduct: error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair. 2002;1:559–569. doi: 10.1016/s1568-7864(02)00055-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in Xeroderma Pigmentosum variant cells, which lack DNA polymerase η, DNA polymerase ι causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 15.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase η error spectrum. Nat. Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 16.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 17.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates (D-loops) of homologous recombination. Mol. Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Ogi T, Lehmann AR. The Y-family DNA polymerase κ functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006;8:640–642. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]
- 19.Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, Woodgate R, Kunkel TA. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase ι in vitro. Science. 2001;291:2156–2159. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- 20.Prasad R, Bebenek K, Hou E, Shock DD, Beard WA, Woodgate R, Kunkel TA, Wilson SH. Localization of the deoxyribose phosphate lyase active site in human DNA polymerase ι by controlled proteolysis. J. Biol. Chem. 2003;278:29649–29654. doi: 10.1074/jbc.M305399200. [DOI] [PubMed] [Google Scholar]
- 21.Frank EG, Tissier A, McDonald JP, Rapic-Otrin V, Zeng X, Gearhart PJ, Woodgate R. Altered nucleotide misinsertion fidelity associated with polι-dependent replication at the end of a DNA template. EMBO J. 2001;20:2914–2922. doi: 10.1093/emboj/20.11.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorjsuren D, Burnette A, Gray GN, Chen X, Zhu W, Roberts PE, Currens MJ, Shoemaker RH, Ricciardi RP, Sei S. Chemical library screen for novel inhibitors of Kaposi's sarcoma-associated herpesvirus processive DNA synthesis. Antiviral Res. 2006;69:9–23. doi: 10.1016/j.antiviral.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Seville M, West AB, Cull MG, McHenry CS. Fluorometric assay for DNA polymerases and reverse transcriptase. Biotechniques. 1996;21:664. doi: 10.2144/96214st04. [DOI] [PubMed] [Google Scholar]
- 24.Kozlov M, Bergendahl V, Burgess R, Goldfarb A, Mustaev A. Homogeneous fluorescent assay for RNA polymerase. Anal. Biochem. 2005;342:206–213. doi: 10.1016/j.ab.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 25.Ma C, Tang Z, Wang K, Tan W, Li J, Li W, Li Z, Yang X, Li H, Liu L. Real-time monitoring of DNA polymerase activity using molecular beacon. Anal. Biochem. 2006;353:141–143. doi: 10.1016/j.ab.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Summerer D, Marx A. A molecular beacon for quantitative monitoring of the DNA polymerase reaction in real-time. Angew. Chem. Intl. Ed. 2002;41:3620–3622. doi: 10.1002/1521-3773(20021004)41:19<3620::AID-ANIE3620>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 27.Yasgar A, Shinn P, Michael S, Zheng W, Jadhav A, Auld D, Austin C, Inglese J, Simeonov A. Compound management for quantitative high-throughput screening. J. Assoc. Lab Automat. 2008;13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 29.Tissier A, McDonald JP, Frank EG, Woodgate R. pol ι, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 30.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin C, Inglese J, Simeonov A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev. Technol. 2008;6:637–657. doi: 10.1089/adt.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Washington MT, Johnson RE, Prakash L, Prakash S. Human DNA polymerase ι utilizes different nucleotide incorporation mechanisms dependent upon the template base. Mol. Cell Biol. 2004;24:936–943. doi: 10.1128/MCB.24.2.936-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marras SAE, Kramer FR, Tyagi S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khrapko KR, Lysov Yu P, Khorlyn AA, Shick VV, Florentiev VL, Mirzabekov AD. An oligonucleotide hybridization approach to DNA sequencing. FEBS Lett. 1989;256:118–122. doi: 10.1016/0014-5793(89)81730-2. [DOI] [PubMed] [Google Scholar]
- 34.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 35.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- 36.Klenow H, Henningsen I. Effect of monovalent cations on the activity of the DNA polymerase of Escherichia coli B. Eur. J. Biochem. 1969;9:133–141. doi: 10.1111/j.1432-1033.1969.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 37.Frank EG, Woodgate R. Increased catalytic activity and altered fidelity of human DNA polymerase ι in the presence of manganese. J. Biol. Chem. 2007;282:24689–24696. doi: 10.1074/jbc.M702159200. [DOI] [PubMed] [Google Scholar]
- 38.Vaisman A, Warren MW, Chaney SG. The effect of DNA structure on the catalytic efficiency and fidelity of human DNA polymerase β on templates with platinum-DNA adducts. J. Biol. Chem. 2001;276:18999–19005. doi: 10.1074/jbc.M007805200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-prone lesion bypass by human DNA polymerase eta. Nucleic Acids Res. 2000;28:4717–4724. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank EG, Sayer JM, Kroth H, Ohashi E, Ohmori H, Jerina DM, Woodgate R. Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diolexpoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase ι. Nucleic Acids Res. 2002;30:5284–5292. doi: 10.1093/nar/gkf643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tissier A, Frank EG, McDonald JP, Iwai S, Hanaoka F, Woodgate R. Misinsertion and bypass of thymine-thymine dimers by human DNA polymerase ι. EMBO J. 2000;19:5259–5266. doi: 10.1093/emboj/19.19.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF. “Action-at-a-distance” mutagenesis. 8-oxo-7, 8-dihydro-2′-deoxyguanosine causes base substitution errors at neighboring template sites when copied by DNA polymerase β. J. Biol. Chem. 1999;274:15920–15926. doi: 10.1074/jbc.274.22.15920. [DOI] [PubMed] [Google Scholar]
- 44.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z. Response of human DNA polymerase ι to DNA lesions. Nucleic Acids Res. 2001;29:928–935. doi: 10.1093/nar/29.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tissier A, Frank EG, McDonald JP, Vaisman A, Fernandez de Henestrosa AR, Boudsocq F, McLenigan MP, Woodgate R. Biochemical characterization of human DNA polymerase iota provides clues to its biological function. Biochem. Soc. Trans. 2001;29:183–187. doi: 10.1042/0300-5127:0290183. [DOI] [PubMed] [Google Scholar]
- 47.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol. Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 48.Hidaka K, Yamada M, Kamiya H, Masutani C, Harashima H, Hanaoka F, Nohmi T. Specificity of mutations induced by incorporation of oxidized dNTPs into DNA by human DNA polymerase η. DNA Repair. 2008;7:497–506. doi: 10.1016/j.dnarep.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 49.Vaisman A, Woodgate R. Unique misinsertion specificity of pol ι may decrease the mutagenic potential of deaminated cytosines. EMBO J. 2001;20:6520–6529. doi: 10.1093/emboj/20.22.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu M, Gruz P, Kamiya H, Masutani C, Xu Y, Usui Y, Sugiyama H, Harashima H, Hanaoka F, Nohmi T. Efficient and erroneous incorporation of oxidized DNA precursors by human DNA polymerase η. Biochemistry. 2007;46:5515–5522. doi: 10.1021/bi062238r. [DOI] [PubMed] [Google Scholar]
- 51.Hu HY, Horton JK, Gryk MR, Prasad R, Naron JM, Sun DA, Hecht SM, Wilson SH, Mullen GP. Identification of small molecule synthetic inhibitors of DNA polymerase β by NMR chemical shift mapping. J. Biol. Chem. 2004;279:39736–39744. doi: 10.1074/jbc.M402842200. [DOI] [PubMed] [Google Scholar]
- 52.Blumenthal T, Landers TA. The inhibition of nucleic acid-binding proteins by aurintricarboxylic acid. Biochem. Biophys. Res. Comm. 1973;55:680–688. doi: 10.1016/0006-291x(73)91198-4. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez RG, Haxo RS, Schleich T. Mechanism of action of polymeric aurintricarboxylic acid, a potent inhibitor of protein—nucleic acid interactions. Biochemistry. 1980;19:4299–4303. doi: 10.1021/bi00559a023. [DOI] [PubMed] [Google Scholar]
- 54.Cook RM, Lyttle M, Dick D. 2006. USA patent no. 7019129. [Google Scholar]
- 55.Simeonov A, Jadhav A, Thomas C, Wang Y, Huang R, Southall N, Shinn P, Smith J, Austin C, Auld D, et al. Fluorescent spectroscopic profiling of compound libraries. J. Med. Chem. 2008;51:2363–2371. doi: 10.1021/jm701301m. [DOI] [PubMed] [Google Scholar]
- 56.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson III DM. Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS ONE. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 58.Kimura T, Takeuchi T, Kumamoto-Yonezawa Y, Ohashi E, Ohmori H, Masutani C, Hanaoka F, Sugawara F, Yoshida H, Mizushina Y. Penicilliols A and B, novel inhibitors specific to mammalian Y-family DNA polymerases. Bioorg. Med. Chem. 2009;17:1811–1816. doi: 10.1016/j.bmc.2009.01.064. [DOI] [PubMed] [Google Scholar]
- 59.Benchokroun Y, Couprie J, Larsen AK. Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem. Pharmacol. 1995;49:305–313. doi: 10.1016/0006-2952(94)00465-x. [DOI] [PubMed] [Google Scholar]
- 60.Bina-Stein M, Tritton TR. Aurintricarboxylic acid is a nonspecific enzyme inhibitor. Mol. Pharmacol. 1976;12:191–193. [PubMed] [Google Scholar]
- 61.Stoner GD, Mukhtar H. Polyphenols as cancer chemopreventive agents. J. Cell Biochem. Suppl. 1995;22:169–180. doi: 10.1002/jcb.240590822. [DOI] [PubMed] [Google Scholar]
- 62.Gloeckner C, Sauter KB, Marx A. Evolving a thermostable DNA polymerase that amplifies from highly damaged templates13. Angew. Chem. Int. Ed. 2007;46:3115–3117. doi: 10.1002/anie.200603987. [DOI] [PubMed] [Google Scholar]
- 63.Sauter KB, Marx A. Evolving thermostable reverse transcriptase activity in a DNA polymerase scaffold. Angew. Chem. Int. Ed. 2006;45:7633–7635. doi: 10.1002/anie.200602772. [DOI] [PubMed] [Google Scholar]
- 64.Summerer D, Rudinger N, Detmer I, Marx A. Enhanced fidelity in mismatch extension by DNA polymerase through directed combinatorial enzyme design. Angew. Chem. Int. Ed. 2005;44:4712–4715. doi: 10.1002/anie.200500047. [DOI] [PubMed] [Google Scholar]