


Abstract
We describe a Multiplex Primer Prediction (MPP) algorithm to build multiplex compatible primer sets to amplify all members of large, diverse and unalignable sets of target sequences. The MPP algorithm is scalable to larger target sets than other available software, and it does not require a multiple sequence alignment. We applied it to questions in viral detection, and demonstrated that there are no universally conserved priming sequences among viruses and that it could require an unfeasibly large number of primers (∼3700 18-mers or ∼2000 10-mers) to generate amplicons from all sequenced viruses. We then designed primer sets separately for each viral family, and for several diverse species such as foot-and-mouth disease virus (FMDV), hemagglutinin (HA) and neuraminidase (NA) segments of influenza A virus, Norwalk virus, and HIV-1. We empirically demonstrated the application of the software with a multiplex set of 16 short (10 nt) primers designed to amplify the Poxviridae family to produce a specific amplicon from vaccinia virus.
INTRODUCTION
Researchers employ numerous approaches for viral detection and discovery, including metagenomic sequencing (1–3), microarrays (4–9) or multiplex PCR followed by other methods of characterization such as mass spectrometry (10–14), suspension arrays (15,16) or amplicon sequencing (17). Multiplex PCR followed by analysis of amplified products fills a niche for viral identification when singleplex PCR has failed or there are a few dozen likely candidates but the expense of metagenomic sequencing or high-density microarrays is unwarranted (18). However, multiplex primer design for many highly divergent targets is challenging since no universally conserved primers may exist, and finding sets of primers likely to function well in multiplex (e.g. isothermal Tm’s, no primer dimers) adds to the complexity of finding conserved primer candidates. Primer design software that requires a multiple sequence alignment (MSA) as input can be problematic for diverse target sets, as MSAs can be difficult to construct, exhausting memory or available time before an alignment is completed. Even if an alignment does complete for divergent target sets such as all members of a family of RNA viruses or gene homologues across species, alignments may show little nucleotide sequence conservation, and multiple primers are required to amplify all targets. Considering the challenges of primer design for targets showing sequence variation, it is not surprising that many of the PCR-based assays in the literature are predicted to fail to detect desired targets when compared against available sequence data, and this problem is worst at higher taxonomic levels like family (19).
Most currently available multiplex primer prediction tools require an MSA (20–24). None of those tools build multiplex sets in which no primers in the set are predicted to form primer-dimers, although some avoid homodimers. We attempted to run a number of alternative software tools for multiplex or degenerate primer prediction for several species level target sets ranging in size from 41 to ∼6000 sequences: Primaclade (24), FastPCR (www.biocenter.helsinki.fi/bi/programs/fastpcr.htm), GeneUp (25), PDA-MS/UniQ software (26), Greene SCPrimer (20) and HYDEN (22). Only Greene SCPrimer and HYDEN could handle the two smallest, species-level target sets (Norwalk virus and FMDV), and none of the tools completed for the larger target sets that we attempted to run.
Therefore, we designed the MPP software to avoid the requirement of an MSA and to scale better for large and diverse target sets. MPP builds a multiplex-compatible set of primers capable of amplifying all target sequences, attempting to minimize the number primers in the set. We set out to determine a set of highly conserved ‘universal’ primers for viruses, akin to the highly conserved 16S rRNA universal primers for bacteria. Throughout, we use the term ‘detected’ to mean that there should be at least one PCR product. This is a loose definition of ‘detected’ adopted for convenience in the following discussions, and we recognize that a PCR product may prove insufficient for viral characterization. We predicted a set of ‘universal viral primers’ for all available complete genomes of all viruses, and found that the number of universal viral primers would be impractical to implement, even if short, highly conserved priming sequences were used. Then we predicted family-level primer sets for every viral family, as well as for several highly diverse species of RNA viruses, for primers of a traditional length as well as nontraditional, shorter, more highly conserved primers, which are more likely to amplify novel, unsequenced viruses and which could be an alternative to degenerate primers. While the software uses a greedy algorithm that may settle at a local minimum which is above the true minimal set, it generated primer sets for each viral family with fewer than half the number of primers that would be expected without optimization. Although family-level primer sets for some families are too large to be practical, 64% of the families had primer sets of no more than 20 primer pairs, quite feasible for multiplex PCR.
Finally, we empirically multiplexed a set of 16 short (10-mer) primers designed for the Poxviridae family. This demonstrated specific amplification of the expected viral fragment from vaccinia Lister strain. This preliminary demonstration suggests that specific amplification with family-level multiplexed sets of short primers might be feasible for viral detection and discovery, particularly as a means for selective enrichment of viral target for downstream amplicon characterization (e.g. sequencing or probe hybridization).
METHODS
MPP algorithm for calculating multiplexed primer sets
Here, we outline a greedy algorithm used to calculate conserved sets of multiplexed primers to amplify fragments from each member of a target set of sequences, and provide a more detailed description in the Supplementary Methods. First, we enumerate all candidate oligos fitting user-specified requirements for length, Tm, and lack of hairpin formation. We rank pairs of these by the number of targets in which that pair occurs within a distance range d1 bases to d2 bases of one another, where these might be specified so as to bound a reasonable range for a downstream characterization method such electrophoretic discrimination of bands, probe hybridization or sequencing. The most frequent pair is selected as primers. The process is repeated for the remaining targets that would not have an amplicon from the first pair, with the added consideration that new primers selected be predicted not to form dimers with other primers already selected, as well as can be predicted based on nearest neighbor thermodynamic predictions (27), although free energy calculations cannot predict with certainty that primer dimers will be excluded in practice. There is an option to bin primers into reaction subsets, if desired. With binning, primers are added to a bin until that bin contains b primers, at which point a new bin is begun, following the same process. Binning primers in smaller groups avoids exclusion of the most highly conserved oligos because of primer dimer free energy constraints. The universal set of primers is the set of selected primers to amplify all genomes in the target set. The primers within a bin should be multiplexed into a single PCR reaction, but each bin should be run separately. This is a simple binning strategy, and alternative strategies could be employed such as starting a new bin with any primer pair that dimerizes with other previously selected primers regardless of the number of primers in the bin, but this could have the possible disadvantage of bin explosion to numerous singleplex or small multiplex reactions. The graph-based algorithm of MuPlex (28,29) is another binning strategy that could be incorporated. Output on the number of primers rejected due to the various filters (Tm, hairpin free energy, etc.) is printed to standard output after each round of primer pairs are selected and also cumulatively, so a user can monitor if/which parameters might be too stringent. If an alternative set of primers is desired that does not overlap with the set selected, one can replace with N’s the subsequences matching the selected primers and their reverse complements in the target sequences and rerun the software with the modified input sequences.
The script find_amplicons.pl predicts all the amplicons that should be generated by a list of (multiplexed) primers mixed with a set of sequences. This PCR-simulating script lists amplicon sequences, their length and position, and the forward+reverse primer combination to yield each product, as well as a summary file of the fragment length distributions enabling a quick assessment of how well each target sequence can be discriminated based purely on amplicon length patterns. We used this script to check whether some viral primer sets are predicted to generate amplicons from the human genome.
For all Tm and free energy predictions, we used the following Unafold settings: [Na+] = 0.2 M, [Mg+] = 0.0015, annealing temperature = 30°C, and strand concentration of each strand = 10–7 M, making the total strand concentration of both strands = 2 × 10–7.
Imposing uniqueness requirements for target-specific priming
The MPP algorithm described here focuses on finding conserved primers, and does not require that the primers be family- or species-specific. For the runs predicting family-specific primers, we designed viral family primers that were unique relative to nontarget viral families by replacing any substring of 17 nt or longer that occurs in any non-target family with a substring of ‘N’’s, using the suffix array software vmatch (http://www.vmatch.de/). MPP eliminates oligos with N’s from consideration as primers. This approach is simple, although it risks being overly strict by eliminating some potentially successful candidate primers, since it disallows those cases where a single nonunique primer pairs with a unique primer, a pair of nonunique primers are too far apart to actually generate an amplicon in a nontarget sequence, or candidate oligos partially overlap a nonunique stretch of N’s.
In general, searching for primers from sequence that is specific to one set of targets and excluding candidate substrings present in nontarget sequence is a useful strategy to design signatures for pathogen detection. This can be done by replacing nonunique or nonspecific substrings with N’s in the sequences input to MPP using software such as vmatch.
Runs predicting universal viral primers
For runs predicting universal viral primers including all viruses at once in the target set, all viral complete genomes and segments downloaded from publicly available sequence databases (Genbank, Baylor, TIGR) as of 25 April 2007 were used. Draft sequences with multiple contigs were merged into a single sequence entry, with contigs separated by 1000 N’s, a stretch sufficiently long (>d2) so that primer pairs would not be designed to fall on different contigs, although there were very few draft sequences in contigs where this was necessary. Because of the large numbers of sequences in two families, only the MP segment sequences from Orthomyxoviridae and the L segment from Bunyaviridae were included, as these are the more conserved segments, reducing the number of targets by 23 017 sequences. The total number of target sequences for these all virus runs were 11 477. We predicted a multiplex set of universal 10-mers without binning the primers into separate reactions, but it was necessary to use a very low xdimer and xhomodimer of −11 kcal/mol to be possible to predict multiplexed primers to amplify every target. For the next calculations, we subdivided the primers into sub-reactions with 20 primers per reaction bin, to avoid excluding primers that were predicted to form primer dimers, and raised xdimer and xhomodimer to –7 kcal/mol. Primer sets of length 5–18 were predicted (Figure 1). To assess the effect of removing Tm constraints, we generated universal primer sets with length but no Tm requirements, and compared the primer counts to those with Tm constraints (Figure 2). Primer sets are available by contacting the authors.
Figure 1.

Hundreds to thousands of primer sequences are required to amplify all viruses. Percent of viral genomes detected versus number of primers required. Calculated for primers of different sizes, and based on sequence data as of 25 April 2007. Parameters used are given in Supplementary Tables S1 and S2.
Figure 2.

Effect of Tm constraints and sequence database size on size of universal primer set. The number of primers in the universal set for all viruses is shown, as a function of primer size. Calculations were based either on sequence data available April 2007 and imposing Tm constraints in primer selection, or without Tm or GC% constraints, or based on January 2004 sequence data with Tm constraints. For primer size of 5 with Tm constraints, a constraint of Tm > 0 was used, delivering primers with GC% of 80–100%.
We evaluated how the growth of sequence availability could affect the size of the universal set of viral primers, as well as our ability to detect unknown/unsequenced viruses. We predicted a universal viral primer set for all viral genomes and segments available as of 1 January 2004, totaling 9965 sequences, for primers of length 7–15 nt (Figure 2). It was not necessary to exclude any Orthomyxoviridae or Bunyaviridae segments, because these segments were not so deeply sequenced at that time. We then determined how much of the 2007 sequence data would have been detectable using the 2004 primer sets (Figure 3).
Figure 3.

Primers from old sequence data miss many newly sequenced viruses. Fraction of viral genomes available as of 25 April 2007 that would have been detected using primer sets developed based on the sequence data available as of 1 January 2004.
To predict how contamination with human DNA might affect the ability to detect specific amplification of viruses, the average number of amplicons for the human genome was also predicted based on these primer sets with 20 primers per bin (Figure 4). The effect of reducing the multiplex size to 10 primers per reaction on human genome amplification was calculated by dividing priming sets in half, with 10 primers per reaction.
Figure 4.

Average number of bands predicted from PCR against the human genome using the universal viral primers for the runs as described in Figure 2, average ± SD of the top 10 bins for each primer length. For bin size of 10, we divided the universal primer bins of 20 primers/bin into two bins each.
Viral family primers
Subdividing viral targets into families, we used an updated set of sequences, downloaded 5 February 2009. For family-level primers, we computed primer sets using three alternative parameter settings (Table 1): (i) 17–21-mer primers with Tm = 55–60°C, primers not checked for uniqueness; (ii) same length and Tm specifications as in (i) but eliminating from consideration as primers any oligos of at least 17 nt that occur in nontarget viral families, as described earlier; and (iii) 10–15-mer primers with Tm = 40–45°C. These primers are available as Supplementary Data.
Table 1.
Primer counts for viral family primer sets, as described in the methods
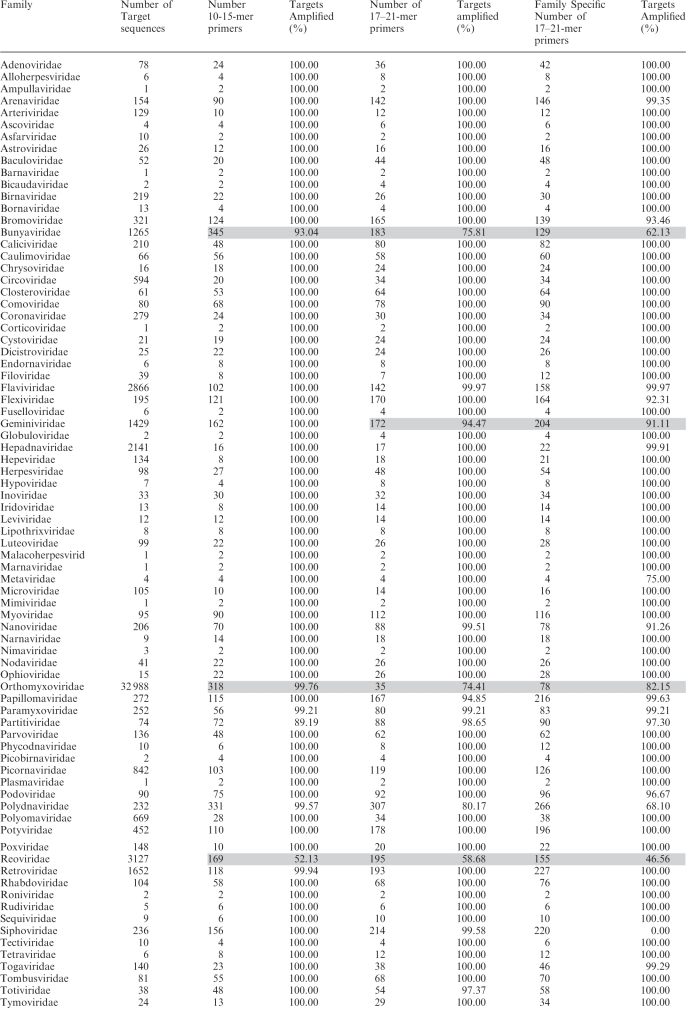 |
Grey shaded entries indicate where calculations were not run to completion. In other cases (not shaded) where fewer than 100% of targets were predicted to be amplified, the algorithm failed to find primer pairs that met all the required specifications for the remaining targets, that is, primers in the right length, Tm, and amplicon length range, with hairpin and dimer avoidance with other primers already selected to be in the set, could not be found.
For 10–15-mers, Tm = 40–45°C and for 17–21-mers, Tm = 55–60°C.
For both, the amplicon length range was 200–800 bp.
None of the calculations that completed required more than the 16 GB of RAM that was available.
Species level primers for several divergent species
We also generated primer sets for several species with high sequence diversity: HIV-1, FMDV, Norwalk virus and influenza A segments HA and NA (summary in Tables 2 and 3, primer sequences available as Supplementary Data). MPP parameters were the same as in Table S1, with Tm = 35–50°C for 10-mer primers and Tm = 55–70°C for 17–18-mer primers, and sequences were downloaded in 2007 or 2008 (influenza only). To illustrate the challenges of designing primers from an alignment, we aligned these organisms using MUSCLE (30) when possible. For the HIV-1 and influenza A segments HA and NA, MUSCLE ran out of memory before completing. An alternative alignment tool, ClustalW (31), had completed only a small fraction of the alignment after running for days. Therefore, for these large data sets, a random selection of ∼35 sequences for each target was aligned with MUSCLE, and this alignment was used to build a profile Hidden Markov Model (HMM) (hmmbuild) using HMMer (http://hmmer.wustl.edu/).(32) The full sequence set was then aligned to the HMM using hmmalign. For Norwalk virus and FMDV, we designed multiplex-degenerate primer sets using GreeneSCPrimer (20) and HYDEN (22), but the other three targets sets were too large for those programs. None of the other primer prediction programs we tried (as indicated in the ‘Results’ section) would scale to handle even these two smallest target sets.
Table 2.
Summary of results of primer set prediction for several diverse RNA virus species for primers of length 10 or 17–18 nt, the longest stretch of bases conserved among the targets in a multiple sequence alignment, illustrating the difficulty of selecting conserved primers, and the time required to perform the primer set selections
| Virus species | Number of sequences | Number of 10-mers | Number of 17–18-mer primers in set | Longest conserved region from MSA (nt) | Processing time on 1 cpu of an AMD Opteron to calculate 10-mer primers (hours) | Processing time on 1 cpu of an AMD Opteron to calculate 17–18-mer primers (hours) |
|---|---|---|---|---|---|---|
| NA segment of Influenza A* | 6375 | 52 | 120 | 5 | 5.7 | 28.9 |
| HA segment of Influenza A* | 5440 | 73 | 153 | 1 | 6 | 29.5 |
| HIV-1 | 1175 | 6 | 16 | 0 | 1.6 | 9.1 |
| FMDV | 187 | 4 | 6 | 9 | 0.18 | 0.7 |
| Norwalk | 41 | 7 | 20 | 6 | 0.1 | 0.5 |
*For Influenza A HA and NA segments, all complete sequences, including lab strains, from all hosts, countries and serotypes were downloaded from the NCBI Influenza Virus Resource database (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/multiple.cgi) on 18 January 2008.
Primer sequences are provided as Supplementary Data.
Table 3.
Summary of the number of 17–18-mer primers predicted to amplify several influenza A HA serotypes
| Influenza A HA Serotype | Number of sequences | Number of 17–18-mer primers in set |
|---|---|---|
| H1 | 1080 | 24 |
| H2 | 108 | 8 |
| H3 | 1972 | 15 |
| H5 | 1325 | 16 |
| H7 | 256 | 8 |
Primer sequences are provided as Supplementary Data.
Empirical testing of Poxviridae multiplex primers
For an empirical demonstration of our algorithm, we tested a multiplex set of 16 short, 10-nt primers designed for the Poxviridae viral family against commercially purified vaccinia virus extracts. The primer sequences are provided in Table 4 along with the predicted single amplicon for vaccinia Lister strain. We chose the Poxviridae family multiplex for these first empirical demonstrations because extracted viral nucleic acid was readily available for experiments. All primers were purchased from Integrated DNA Technologies (Coralville, IA, USA) and resuspended to 100 µM stock solutions in TE buffer (pH 8.0, Teknova, Hollister, CA, USA). Working solutions containing equimolar concentrations of each of the 16 primers were used in all experiments. Purified, quantitated vaccinia Lister strain DNA was purchased at a concentration of 1.3 × 104 copies/µl in nuclease-free water from Advanced Biotechnologies, Inc. (Columbia, MD, USA). All PCR experiments were prepared using the Superscript III RT-PCR kit from Invitrogen (Carlsbad, CA, USA). We selected the RT-PCR kit in order to establish a protocol that could later be readily applied to additional multiplex viral family reactions with viral DNA and/or RNA. Each 25 µl reaction contained 1 × SSIII buffer, 1 U of SSIII RT/Taq enzyme, 4.8 mM MgSO4, 0.1 µM each primer, and a viral template mass of 2.7 pg (∼104 copies). Tests were performed in triplicate and corresponding negative controls were run under identical conditions except that viral template was replaced with nuclease-free water (Ambion, Austin, TX, USA). All reactions were thermocycled on the Bio-Rad DNA Engine (Hercules, CA, USA) as follows: one cycle of 2 min at 94°C; 40 cycles of 15 s at 94°C, 30 s at 43.9°C, 1 min at 68°C; one cycle of 5 min at 68°C. In line with our goal to create a protocol applicable to both DNA and/or RNA templates, we verified that inclusion of an RT step (one cycle of 30 min at 45°C) does not alter the outcome of the subsequent PCR when working with DNA template (data not shown). As such, for all Poxviridae 16-plex results reported here, an RT step was not used. Another goal was to establish reaction conditions that can be applied to any multiplex viral primer set without the need for re-optimization, assuming the primer set is designed with the same general parameter contraints (Tms, etc.) used to compute the 16-plex presented here. As such, we first optimized master mix conditions, where it was determined that a 4.8 mM MgSO4 concentration resulted in most optimal amplification. Similarly, we determined the optimal annealing temperature based on results from annealing temperature gradient experiments. A more detailed discussion on the optimization of reaction conditions for amplification with our short primer viral multiplexes is the subject of a separate paper in preparation (Hiddessen and co-workers).
Table 4.
Sequence and size (nt) information for the 16 primers in the predicted family-level multiplex for Poxviridae viruses used in these experiments
| Primer sequence | Primer size (nt) |
|---|---|
| CGGAGACCAA (FP 617) | 10 |
| TCGTCGTCCA (RP 617) | 10 |
| TGTTGGTGTG | 10 |
| TCGTCTACGA | 10 |
| TGTGGTCCTT | 10 |
| CCATGTTCGC | 10 |
| TCGAGGAGAA | 10 |
| CCGGCTCCAG | 10 |
| TGAACCTGGT | 10 |
| GCGCACGTAC | 10 |
| TCACGCATCT | 10 |
| GGGAAACAGC | 10 |
| ACCGTTGTCA | 10 |
| TACCATCGTC | 10 |
| AACCTGTGCA | 10 |
| GGCGGAGGTA | 10 |
The forward and reverse primers (FP, RP) in bold are predicted to amplify vaccinia Lister with the indicated product length (617-bp amplicon size).
This multiplex was compared against the human genome, predicting 233 amplicons between 50 and 1000 bp from the Poxviridae 16-plex. For empirical tests against background human nucleic acids, we followed the same reaction conditions as above. In these tests, vaccinia DNA was held at a constant concentration of 2.7 pg (1.3 × 104 copies), and serial dilutions of human genomic DNA (Novagen, Madison, WI, USA), were added to the vaccinia PCR reaction mix, starting at 2.7 pg and titrating down over 4 orders of magnitude to 2.7 × 10–4 pg. These experiments were performed using mass ratios of vaccinia:human DNA at 1 : 1, 10 : 1, 100 : 1, 1000 : 1 and 10000 : 1. These correspond to copy number ratios of vaccinia:human genomes of 1.3 × 104 : 0.82, 1.3 × 104 : 0.082, 1.3 × 104 : 0.0082, 1.3 × 104 : 0.00082 and 1.3 × 104 : 0.000082, respectively.
All PCR experiments were analyzed on 3% agarose TBE gels containing ethidum bromide that were purchased from Bio-Rad (Hercules, CA, USA). Blue juice™ 10× loading dye was purchased from Invitrogen (Carlsbad, CA, USA) and diluted to 2× before use. A 50-bp DNA ladder was purchased from Novagen (Madison, WI, USA). For analysis, 15 μl from each separate 25 μl PCR reaction were combined with 2 μl of of loading dye and 15 μl of the loading-dye/product mixture was loaded per well and electrophoresed for 1 h 40 min at 85 V.
For confirmation of amplicon sequence, 9 μl of amplified PCR product was mixed with 4 μl of ExoSap-it (USB, Cleveland, OH, USA) and incubated at 37°C for 15 min followed by a denaturation step at 80°C for 15 min. Sanger sequencing was then performed with the BigDye V3.1 Terminator Kit (Applied Biosystems, Inc., Foster City, CA, USA). Single reactions contained 4 μl of Ready Reaction Mix, 0.2-µM primer, 2 μl 5× Sequencing Buffer, 11.5 µl of nuclease free water (Applied Biosystems, Inc.) and 2 μl of post-Exosap-it PCR product. The sequencing reaction used the following thermocycling profile: 1 cycle of 94°C for 1 min; 25 cycles of 94°C for 15 s, 38°C for 30 s and 60°C for 4 min. Two microliters of sequencing reaction product was combined with 18 μl of Hi-DiTm Formamide (Applied Biosystems, Inc.) and run on the ABI 3130 (Applied Biosystems, Inc.). The results were analyzed using Sequencing Analysis v5.2 (Applied Biosystems, Inc.).
RESULTS
Universal viral primers
Predicting a set of universal viral primers that are all mixed in a single large reaction, we predict that 1008 primers of length 10 nt would be required to ensure amplification of at least one fragment of length 80–620 bp from every viral genome. These are predicted to generate a mean and maximum number of amplicons per viral genome of 13.2 and 948, respectively. Binning the primers into smaller sets of 20 primers/bin doubled the number of primers required, as Figure 1 shows the fraction of viral genomes amplified versus the number of primers, for primers ranging from 6 to 18 nt in length. About 2000 binned 10-mers are required to amplify 100% of sequenced viruses, generating a mean of 1 and maximum of 2.9 amplicons per genome, on average. Using traditional-length 18-mer primers nearly doubles the number to ∼3700 primers. The incomplete curve for 7-mers was from inappropriate settings for this oligo length, since some genomes do not contain pairs of 7-mers with the required Tm, as given in the Supplementary Tables S1 and S2, within the desired amplicon length range. The concave curves showing diminishing returns reflect biased sequence availability rather than any particularly highly conserved primers: the primers that amplify the largest number of sequences are all from influenza, as far more sequences are available for this species than for any other.
Increasing availability of sequence data on universal primer sets
The increase in sequence data requires ∼700 more 10-mer primers to amplify all sequenced viruses in 2007 compared to 2004 (Figure 2). While the increase in the number of sequences used between the two dates was only ∼15%, the number of primers required increased by 48%, illustrating the substantial increase in diversity represented by the additional sequence data. Figure 2 also shows that removing all Tm constraints allows fewer primers to be used since no conserved primers are eliminated due to Tm, as some AT rich subsequences tend to be fairly conserved. Figure 3 shows that a universal primer set predicted using the 2004 sequence data would amplify only 35% of the 2007 sequences using primers of at least 10 nt in length. Shorter primers increase this fraction to over 60%, due to the higher likelihood of occurrence and conservation of shorter oligos, but even so, a multiplex of 7-mers is not guaranteed to amplify a fragment from every virus. The minor differences in the number of genomes detected between 12, 13, 14 and 15-mers can be attributed to the facts that a greedy but not necessarily optimal algorithm is used to select one solution from among many, that the primers in a particular set depend on the Tm ranges we used as well as length differences, and the unpredictable nature of novel viral sequences accumulated between 2004 and 2007, rather than any real difference in the ability to detect genomes among those primer sets.
Predicting effects of contamination from human DNA
If all human DNA cannot be removed from the sample, simulations indicate that on average, multiplexes of 10-mer primers are expected to produce hundreds of amplicons from the human genome, which would appear as a smear on a gel (Figure 4). With primers of length 11–14, there is a large variation among bins. While most short sequences do occur in the human genome (33), an amplicon requires that two occur in proximity, and primers ≥11 bases make this a sporadic event for bins with only 10 primers. Nonetheless, imperfect sample purification to eliminate eukaryotic nucleic acids could be problematic for universal viral priming using primers shorter than 15 bases, particularly for multiplexes of 10 or more primers.
Family identification by sequencing products from universal 10-mer primer set versus randomly amplified fragments
If universal viral primers amplified fragments from a newly emerged virus, would the product sequences show similarity to others in the same family? We predicted amplicon sequences from 10 newly emerged viruses (Australian bat lyssavirus, Crimean Congo hemorrhagic fever, Nipah, Hendra, Venezuelan equine encephalitis, West Nile, Ebola, Hanta, hepatitis C and Marburg viruses) using the universal 10-mer primer set and BLASTed (blastn) (34) those amplicon sequences against either other species in the same target family or viruses in other families. On average, 87% of the amplicons had BLAST hits in the correct family, with an average of 135 hits per amplicon. In contrast, only 11% of amplicons had hits in other, nontarget families, with an average of 2.5 hits per amplicon. For comparison, 72% of randomly selected fragments of 200 bp (or 65% and 78% for 100-bp and 600-bp fragments, respectively) had hits in the correct family with an average of 59 hits per amplicon, and 2.8% had hits in other families with an average of 2.4 hits per amplicon. Thus, universal primers do amplify more conserved regions than randomly selected fragments. These rough calculations depend heavily on available sequence data, and are likely to significantly overestimate our ability to characterize a truly novel virus, extrapolating from results of viral metagenomic sequencing in which over 60% of the sequences cannot be identified based on BLAST comparisons to existing sequence databases (2).
Viral family primers
The number of family-level primers for each family, and the number of genomes available for generating those primer sets, is given in Table 1, for three alternative settings: short primers of 10–15 nt with Tm 40–45°C, standard length primers of 17–21 nt and Tm 55–60°C, and the same but requiring that each primer subsequence of at least 17 nt be unique to the target viral family relative to other viral families. At least one amplicon of 200–800 bp was required from every genome. Hypothetically, the worst-case scenario to amplify a target set of N sequences would require 2N primers. MPP requires on average only 37% or 45% of this number, for primers of length 10–15 nt or 17–21 nt without the requirement for family specific primers, respectively (averaged across families, omitting those families for which the computations were not run to completion). The most diverse families, in particular Bunyaviridae, Geminiviridae, Polydnaviridae, Reoviridae, Retroviridae, Siphoviridae and Orthomyxoviridae require so many primers that actually applying family-level amplification is probably infeasible. For these, more restricted target sets may be necessary, such as limiting to a single segment for the segmented families or to subclades, and possibly the incorporation of primers with degenerate or inosine bases. Some families with many genomes can be amplified with relatively few primers, such as Coronaviridae, Hepadnaviridae, Poxviridae, Togaviridae, Microviridae and Polyomaviridae. Using primers of length 10–15-mers or 17–21-mers, 66% or 63%, respectively, of the viral families have primer sets of 40 or fewer primers (20 primer pairs), which is feasible for typical multiplexes. The sizes of the family-level primer sets show a trend for an increase with the number of available sequences in the family (Figure 5, P = 0.14). There is no clear relationship between the number of primers in the set and whether the genomes are single or double stranded RNA or DNA. All family primer sequences are available as Supplementary Data.
Figure 5.

Number of primers required to amplify fragments from each complete genome or segment in the family versus the number of available sequences in that family. Primer parameters were length 17–21-mers, Tm between 55°C and 60°C.
Species-level primers for divergent RNA viruses
Primer design with the MPP software indicates that relatively few primers are required to amplify all sequenced genomes of HIV-1, FMDV and Norwalk virus (Table 2), and these can be calculated in minutes. Influenza A HA and NA segments demand large numbers of 10-mer or 17–18-mer primers and hours to calculate, so one could break these into subgroups, possibly by serotype, as shown for several HA serotypes in Table 3. The percentage of genomes amplified versus the number of primers used, for primers of either 10-mers or 17–18-mers, is shown in Figure 6. This plot shows that a large fraction of targets are amplified with only 2 primers, and the addition of subsequent primers shows diminishing returns in amplifying fewer, more divergent targets not detected by the initial, more conserved, primer pair, although the true diminishing returns depend on the extent to which available sequence data is an unbiased representation of diversity.
Figure 6.

The percentage of genomes amplified versus the number of primers used, for primers of either 10-mers or 17-18-mers, for several highly diverse virus species, (A) HIV-1, FMDV and Norwalk; and (B) Influenza A HA and NA segments. The two most highly conserved primers amplify a large fraction of genomes, and additional primers show diminishing returns in detecting the remaining, more divergent sequences.
The more traditional method of attempting to find primers from a MSA would be problematic, probably requiring manually designed primer multiplexes or highly degenerate primers. For HIV-1, for example, there is not a single position with 100% conservation across all sequenced isolates. Dropping the required conservation down to 95% (58 of the 1175 genomes could disagree with a consensus base at any position), there are three conserved regions of at least 18 bases, with positions relative to the consensus: ACAGGAGCAGATGATACAGTA starting at position 3665; TATGGAAAACAGATGGCAGG starting at 7347; and CTATGGCAGGAAGAAGCG starting at 9071. These regions are too far apart to be used as primers for most polymerases used in diagnostic PCR protocols, where amplicons must typically be less than 300 bases long for efficient amplification. A recently published study (35) selected primers from the 5′ LTR U5 end to the Gag-Pol start (5′-TAGCAGTGGCGCCCGA-3′ and 5′-TCTCTCTCCTTCTAGCCTCCGC-3′), but a comparison against available genomic data indicates that 487 of the 1175 genomes (41%) do not contain a sequence match for this primer pair, so may fail to be amplified.
For influenza A segment HA, the size of the longest conserved region from the 95% consensus is only 5 bases, and for segment NA, only 6 bases, insufficient for even a single primer. For FMDV and Norwalk virus, the longest 100% conserved regions are 9 and 6 bases, respectively. The MPP software makes it straightforward for a nonexpert to predict a multiplex-compatible set of primers to amplify all targets, even for enormous and heterogeneous target sets that cannot be aligned.
Comparison with other software
For comparison, we considered other software options for designing primers for these heterogeneous viruses. FastPCR (www.biocenter.helsinki.fi/bi/programs/fastpcr.htm) and GeneUp (25) were the only programs that did not require an MSA as input. The FastPCR algorithm for group-specific PCR (i.e. universal amplification) designs PCR primer pairs individually for each target sequence without regard for primer conservation among targets, and then compares each primer pair to the other targets. This is a brute force strategy that is only suitable for small target sets and short target sequences (appropriate for gene lengths, but not for viral genome-length sequences). We ran the FastPCR software on our internal servers, but it did not complete ‘group-specific PCR’ for the smallest data set, Norwalk virus, after running for 18 h, and for ‘multiplex PCR’ gave the error message ‘No compatible combination of pair primers for multiplex PCR found’.
GeneUp simulates PCR with pairwise combinations of candidate primers which pass length, Tm, GC%, and palindrome filters against all target sequences, and uses a greedy algorithm to build a primer set to amplify all of the targets. Since testing all possible pairwise oligonucleotide combinations against each target explodes in time and memory for large target sets, a cap on the maximum number of candidate primers to be tested must be imposed, presumably using the most common oligos, although this is not explicit in the paper. Then a PCR simulation (performing a text search for the primer in the target sequence) of each pair versus each target is performed. Unfortunately, the most common oligos may not occur in the correct orientation or distance to serve as primer pairs, so that multiple iterations of the entire process must be performed before a set covering all targets is obtained. MPP, in contrast, uses an efficient ranking algorithm to favor pairs of primer candidates that will produce amplicons of the right size in the most targets. Because of the MPP hashing algorithm and data structures, those targets that are amplified is easily determined without simulating PCR. A copy of the GeneUp software for testing could not be obtained from the authors.
The PDA-MS/UniQ approach (26) uses a hash index of 4-mers and scoring heuristic to identify common regions in the target sequences with the most shared tetramers. Simulating combinations of candidate primers selected from these common regions is then performed using a genetic algorithm. Their genetic algorithm is a more scalable approach than those above, and the quality of the solution may be improved with more compute power, although a potential disadvantage of genetic algorithms is that they can be slow, particularly if there are very many possible combinations. For computational tractability, they limited primer size to 12 nt, and avoidance of hairpins and primer dimers was not modeled. This software was not available for download or on a public web server.
The other software all required MSA as input. While these tools could not be tested on larger (e.g. family level) primer prediction problems where alignments would not be possible or appropriate, we nevertheless attempted to run these tools on the alignable species-level target sets. The Primaclade (24) webserver timed out for the smallest alignments we tested, Norwalk and FMDV. CODEHOP (23) requires protein alignment as input so is not appropriate for whole-genome (nucleotide) alignments.
GreeneSCPrimer (20) did generate a number of degenerate primer candidates from the MSAs for Norwalk and FMDV, requiring the user to manually select a combination of forward and reverse groups from a set of options. We ran GreeneSCPrimer using length, Tm, etc. settings mirroring or more lenient than those we used for Table 3 (Tm = 55–65°C, GC% = 20–80%, length 17–25 bp, 100% coverage, product size 80–620 bp, allowed Tm difference 10°C, others left as defaults). HYDEN (22) also generated degenerate candidates from a MSA, although it does not check Tm and the length is limited to a single value rather than a range. For our tests using HYDEN, we used a length of 18 rather than 17 because of the lack of Tm control, and allowed 0 mismatches. The GreeneSCPrimer option requiring the fewest total primers for the Norwalk set required 18 primers, four of which had either 2-fold or 4-fold degeneracy so the actual number of priming sequences would be 26, compared to a total of 20 non-degenerate primers predicted by MPP (Supplementary Data). HYDEN generated four degenerate primers covering only 34 of 41 sequences, each with 3- or 4-fold degeneracy for Norwalk, which translates to 15 priming sequences in the reaction. One would need to find primers to amplify the remaining seven sequences. Small degenerate priming sets (e.g. four primers in this case) are less expensive to purchase, but because of dilution effects from the many sequence combinations actually present (15 priming sequences in the PCR), sensitivity may be reduced compared to nondegenerate priming. However, using a smaller set of degenerate signatures such as those from HYDEN may be preferable, and is a capability that could improve MPP in a future version.
For FMDV, MPP predicted six nondegenerate multiplex compatible primers to amplify all targets. GreeneSCPrimer generated a number of candidates, and manual inspection identified that the best of those primer combinations would require 6 primers, one of which had 2-fold and another had 3-fold degeneracy, totaling 9 actual priming sequences in a reaction (Supplementary Data). This compares with two primers each with 4-fold degeneracy using HYDEN (eight priming sequences in a reaction), to amplify 98% (183 of 187) targets. Again, the small number of signatures predicted by HYDEN is desirable for some applications, although the degeneracy is high and these must be supplemented to pick up the few outlying sequences
The aim of the MuPlex software (28,29) is to partition primer pairs into multiplex-compatible bins for SNP genotyping, and it does not employ any algorithm during primer selection to minimize the number of primers to amplify all targets, yielding a one-to-one correspondence between number of primer pairs versus number of targets. However, the MuPlex graph-based algorithm to partition primers into bins is more sophisticated than the simple binning scheme of MPP. In future work, MPP could be used to identify a universal set of primers and the backend of MuPlex used to optimize how they are binned into separate reactions.
In summary, no software except MPP was capable of predicting primer sets for the larger target sets we examined or generating a multiplexed primer set without a MSA for any of the target sets we examined, even the smallest ones containing only a single species. Only HYDEN and GreeneSCPrimer, both requiring MSA inputs, completed for the two smallest target sets, selecting smaller sets of degenerate primers than the nondegenerate MPP primers. If target sets are sufficiently conserved so that a reasonable MSA can be built, and degenerate primers are acceptable, these tools may be preferable over MPP. However, for larger and more diverse target sets, only MPP completed.
Poxviridae experimental results
Using the experimental conditions described in the ‘Methods’ section, we tested the Poxviridae 16-plex (Table 4) against vaccinia virus, Lister strain DNA. As assessed by agarose gel analysis, we achieved specific amplification of the predicted 617-bp amplicon for the target vaccinia genome (lane 3, Figure 7). The band does not appear in the no template control shown in lane 4 (Figure 7).
Figure 7.

PCR amplification of vaccinia DNA, Lister strain with the 16-plex Poxviridae primer set, with (lanes 6–10) or without (lanes 3 and 4) human DNA present in the reaction, for a reaction containing 4.8 mM MgSO4, 0.1 µM each primer and 2.7 pg (∼104 copies) of vaccinia Lister DNA, and using an annealing temperature of 43.9°C. Lane 3 shows specific amplification of vaccinia Lister DNA followed by the no template control in lane 4. Lanes 6–10 represent the experiments with mass ratios of vaccinia:human DNA of 1 : 1, 10 : 1, 100 : 1, 1000 : 1 and 10000 : 1, respectively. Lanes 12–14 represent amplification of human DNA only (no vaccinia present in reaction) for the same masses used at the 1 : 1, 100 : 1 and 10 000 : 1 ratios (or 2.7 pg, 0.027 pg and 2.7 × 10–4 pg human DNA, respectively). Lanes 5, 11 and 15 did not contain any sample. The arrow on the left points to the expected band (617 bp). The arrows on the right correspond to the 50-bp DNA ladder (lanes 1 and 16 contain the same ladder).
To confirm that the amplicon observed in gel analysis was a specifically amplified product from vaccinia virus, Lister strain, we sequenced the product according to the procedures described above. An analysis of the high-quality sequence read data taken from the electropherogram yielded a 95% identity (maximum) to vaccinia virus, Lister strain (AY678276.1) with a query coverage of 99% and an e-score of 0 (no data shown). These values indicate that the sequenced product was vaccinia DNA and not amplification from an exogenous nucleic acid source, e.g. host cell.
Notably, our multiplex reaction did not produce a smear (lane 3, Figure 7) that would be indicative of nonspecific priming of either the target or exogenous host cellular nucleic acids. While the exact manufacturer’s extraction protocol is proprietary, it is generally known that a standard sucrose gradient ultracentrifugation step is used to enrich for the viral capsids prior to viral nucleic acid extraction. However, to the best of our knowledge, no nuclease digestions are performed prior to viral capsid lysis. Furthermore, while the viral ‘extract’ contains a mixture of both viral DNA and cellular nucleic acids, the exact proportions of host cell and viral nucleic acids cannot be determined. Thus, our results indicate that a multiplex of 16 primers of 10 nt each amplifies only the specific predicted band from vaccinia.
In some applications, such as clinical or biodetection applications, the exogenous nucleic acids from other eukaryotic sources, notably human sources, may be present in varying and unknown concentrations. To test the effects of background human genomic DNA on the performance (amplification specificity) of the Poxviridae 16-plex primer set, we conducted PCR reactions across a series of mass ratios of vaccinia Lister DNA:human genomic DNA at 1 : 1, 10 : 1, 100 : 1, 1000 : 1 and 10000 : 1. These correspond to approximate copy number ratios of vaccinia : human genomes of 1.3 × 104 : 0.82, 1.3 × 104 : 0.082, 1.3 × 104 : 0.0082, 1.3 × 104 : 0.00082 and 1.3 × 104 : 0.000082, using genome sizes of 1.9 × 105 bp and 3 × 109 bp for vaccinia and human DNA, respectively. For context, there are 6.58 pg or two copies of the genome in one human cell. Our algorithm predicted 233 amplicons between 50 and 1000 bp from the human genome. In experiments, this would appear as a ‘smear’ on the agarose gel, which indeed was observed for the mass ratios of 1 : 1 and 10 : 1 (Figure 7, lanes 6 and 7, respectively). However, even at a ratio of 10 : 1 vaccinia:human DNA (lane 7), the 617-bp vaccinia amplicon is clearly visible on the gel, despite the numerous nonspecific amplicons. At ratios of 100 : 1, 1000 : 1 and 10000 : 1 (lanes 8, 9 and 10, respectively, Figure 7), the smear is drastically reduced to nonexistent (at the resolution of the agarose gel), and the vaccinia amplicon is readily visible. For comparison, we tested the 16-plex primers against the same mass of human DNA in the absence of vaccinia DNA at 2.7 pg (the 1 : 1 ratio mass), 0.027 pg (100 : 1 mass) and 2.7 × 10−4 pg (10000 : 1 mass) (lanes 12, 13, and 14, respectively, Figure 7). The data show a similar smear at 2.7 pg as was observed when vaccinia DNA was present (lane 6). However, the low-intensity 617-bp amplicon from vaccinia is visible in lane 6 (with vaccinia) while a similar amplicon is not present in lane 12 (without vaccina). These results provide evidence that amplification with these family level primer sets will depend on viral titers in the actual sample, and that there are cases where amplification will either not be possible or require additional sample purification steps. However, as discussed further in the next section, specific amplification with a short-primer multiplex could be used for selective enrichment of viral targets by generating amplicons with known sequence, which may be feasible for viral detection if combined with a probe-based amplicon detection method such as TaqMan® or Luminex bead based suspension arrays (http://www.luminexcorp.com/).
We compared recently published conserved Orthopoxvirus primers (36) to the 148 Poxviridae genomes, and computational predictions suggest that 67 of the available genomes might not be amplified by the two primers they designed for the Orthopox genus. These included a number of monkeypox, ectromelia, several vaccinia and a couple of variola strains. However, in permissive hybridization conditions it is possible that primers would anneal despite mismatches to target, allowing more of the targets to be amplified. Four conserved Orthopoxvirus primers from an earlier publication (37), before many of the Poxviridae genomes were available, do not match 53 genomes, including a number of monkeypox, ectromelia, camelpox and one variola minor genome. The primers in (37) included inosine bases, which we replaced with each possible A, T, G and C base in all possible combinations for our primer-target comparison. Thus, published Orthopoxvirus primers may fail to amplify many desired targets.
DISCUSSION AND CONCLUSIONS
Primer design for amplification and detection of divergent target sequences can be challenging, and this problem will only grow as sequencing technologies improve. Some methods are limited in scalability, particularly those requiring a MSA as input. Developing a PCR multiplex is often a tedious mix-and-match process from among primers originally designed to work in singleplex. We describe the MPP algorithm based on hashing of conserved k-mer subsequences that requires no MSA and where multiplex-compatible primer sets are built de novo to avoid primer dimer and hairpin formation to the extent that can be predicted based on free energy calculations.
The algorithm seeks to minimize the number of primers to amplify all targets, although because the algorithm is heuristic and not an exhaustive search of all combinations of primers, the smallest primer sets may not always be selected. This is an NP complete problem (38), so an exhaustive search for the global optimum is only practical for very small target sets. It is also beyond the scope of the current software to predict priming with mismatches between target and primer, which may occur under non-stringent hybridization conditions or for mismatches at the 5′ end of a primer. While allowing for such priming would reduce the number of primers needed to amplify all targets, it would substantially slow the algorithm and increase memory requirements. Nor do we claim to have taken all steps to optimize (e.g. through parallelization, see Supplementary Methods) for speed or memory, but instead present this software as a simple embodiment of one alternative to MSA for multiplex primer design.
The software handles a large number of input sequences, although for very diverse targets the predicted primer multiplex may be too large to be empirically feasible. For many target sets, such as those including all the genomes of a species, predicting a universal primer set requires only minutes up to a few hours, although for inputs with thousands of sequences a run may take days. We used MPP to design a universal primer set for a target set of all virus genomes, and showed that even if short primers are used, thousands of priming sequences would be required to amplify all sequenced viruses. We then applied MPP to design multiplex family level primers for every viral family, as well as for some diverse species target sets too large for other available primer prediction software.
In addition to finding primers for viral detection, another application of MPP could be to design multiplex primers for homologs in a gene family. The user can specify an appropriate product length range to ensure amplification of an adequate span across the gene. MPP could also be used to design multiplex primer sets for unrelated target sequences, for example, multiple bacterial and viral species or gene families, in a single reaction. Since no sequence alignment is required, there is no need for any sequence conservation among targets. Recent work showed that a large-scale multiplex of 800 primer pairs specifically designed to detect a diverse set of genes from nine pathogens improved sensitivity on a microarray by up to 1000-fold (39). The MPP software could be used to design multiplex primer sets for similar work.
We demonstrated the application of the algorithm experimentally using purified vaccinia DNA, amplifying the expected band with a Poxviridae short primer multiplex PCR, and confirmed the band’s expected sequence. We provided an example of the effect of background nucleic acids by spiking human DNA into the PCR reaction over a series of mass ratios. While a band for vaccinia was visualized by slab gel electrophoresis for all mass ratios, background DNA smears were also clear at the higher ratios. At a mass ratio of 1 : 1 vaccinia:human DNA, there are 2.7 pg or 0.82 copies of the human genome present in the reaction. To place this into context, there are ∼6.58 pg or two copies of genomic DNA in one human cell. Thus, the results from the 1 : 1 mass ratio reaction represents the impact that ∼41% of the total DNA content from one human cell could have on the results, when analyzed by slab gel electrophoresis. In a clinical sample, there could be a much greater number of human cells and DNA. Amplification with these family level primer sets will depend on viral titers in the actual sample, and these experiments show that there are cases where amplification will either not be possible or require additional sample purification steps. While short primer PCR multiplexes may enable amplification of diverse target sets, they will not match the specificity of longer primers. Combination of these short primer multiplexes with digitized, microfluidic-based picoliter reactions (40) and/or next-generation microfluidic-based sample purification technologies that have the ability to isolate target from contaminating nucleic acids may help to overcome this limitation. However, even in the absence of such technologies, a highly conserved short primer multiplex could enrich amplification products for members of a particular viral family compared to randomly amplified or unamplified sample. Then it would need to be followed by product sequencing or array hybridization, such as TaqMan® or Luminex bead-based suspension arrays (http://www.luminexcorp.com/), to provide more specific information about the organism(s) present, since electrophoretic banding patterns may contain unexpected bands, particularly if the sample contains a significant amount of nonviral nucleic acids.
Previous studies have shown the utility of short primer singleplex PCR using 9-mers or 10-mers followed by gel electrophoresis for genetic fingerprinting of eukaryotes and bacteria (41,42). For viruses, with much smaller and more diverse genomes, the large numbers of 9-mer or 10-mer primers required to generate at least one band from every virus as predicted by our analyses implies that primer size would need to be as short as 5-mers to rely on a gel banding pattern using only one priming sequence for fingerprinting viruses (unpublished data). However, the analyses here predict that imperfect sample purification to eliminate eukaryotic nucleic acids could be problematic for universal viral priming using primers shorter than 15 bases, particularly for multiplexes of 10 or more primers. Nanda et al. (43,44) were able to achieve sufficient viral isolation from cell culture samples to allow viral identification using viral PCR with priming sequences as short as pentamers, so the problem of contaminating host nucleic acids for specific, short primer PCR of viruses is not insurmountable. They found specific pentamer PCR to be several logs more sensitive than nonspecific amplification, provided that they purified encapsidated viral nucleic acids prior to PCR. Another method that has been used for virus discovery is VIDISCA (Virus discovery cDNA-AFLP) using restriction enzyme digestion, adaptor ligation, and PCR by priming with the adaptor sequence (45,46). This method, like the pentamer priming used by (43,44), requires prior separation of encapsidated viral nucleic acids, as it generates fragments from any DNA present, viral, host or otherwise. Multiplex PCR with primers 10–15 nt in length may be yet another alternative strategy lying between these nonspecific methods and PCR with standard primers of at least 18 nt, as we have shown that it can add some measure of specificity for a viral family.
In summary, we applied the MPP software to generate multiplex-compatible primer sets for every viral family and several divergent viral species and experimentally demonstrated application of one multiplex set to show nonrandom amplification with a set of short primers.
Code and Primer Availability
Primer sets are available as supplemental material or by request from the authors. The MPP software is freely available for academic and nonprofit use at http://mpp.llnl.gov.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Laboratory Directed Research and Development (LDRD) and Computations TechBase awards from the Lawrence Livermore National Laboratory. Funding for open access charge: Lawrence Livermore National Laboratory.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank anonymous reviewers for helpful comments. Auspices statement (required by LLNL): This work performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
REFERENCES
- 1.Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. Metagenomic analyses of viruses in the stool of children with acute flaccid paralysis. J. Virol. 2009 doi: 10.1128/JVI.02301-08. doi:10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards RA, Rohwer F. Viral metagenomics. Nat. Rev. Micro. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura S, Yang C-S, Sakon N, Ueda M, Tougan T, Yamashita A, Goto N, Takahashi K, Yasunaga T, Ikuta K, et al. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS ONE. 2009;4:e4219. doi: 10.1371/journal.pone.0004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kistler A, Avila P, Rouskin S, Wang D, Ward T, Yagi S, Schnurr D, Ganem D, DeRisi J, Boushey H. Pan-viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. J. Infect. Dis. 2007;196:817–825. doi: 10.1086/520816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin B, Blaney KM, Malanoski AP, Ligler AG, Schnur JM, Metzgar D, Russell KL, Stenger DA. Using a resequencing microarray as a multiple respiratory pathogen detection assay. J. Clin. Microbiol. 2007;45:443–452. doi: 10.1128/JCM.01870-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin B, Wang Z, Vora GJ, Thornton JA, Schnur JM, Thach DC, Blaney KM, Ligler AG, Malanoski AP, Santiago J, et al. Broad-spectrum respiratory tract pathogen identification using resequencing DNA microarrays. Genome Res. 2006 doi: 10.1101/gr.4337206. gr.4337206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Coscoy L, Zylberberg M, Avila P, Boushey H, Ganem D, DeRisi J. Microarray-based detection and genotyping of viral pathogens. PNAS. 2002;99 doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quan PL, Palacios G, Jabado OJ, Conlan S, Hirschberg DL, Pozo F, Jack PJM, Cisterna D, Renwick N, Hui J, et al. Detection of respiratory viruses and subtype identification of influenza A viruses by GreeneChipResp oligonucleotide microarray. J. Clin. Microbiol. 2007;45:2359–2364. doi: 10.1128/JCM.00737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palacios G, Quan P, Jabado O, Conlan S, Hirschberg D, Liu Y, Zhai J, Renwick N, Hui J, Hegyi H, et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 2007;13:73–81. doi: 10.3201/eid1301.060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ecker D, Drader J, Gutierrez J, Gutierrez A, Hannis J, Schink A, Sampath R, Blyn LB, Eschoo MW, Hall TA, et al. The Ibis T5000 universal biosensor: an automated platform for pathogen identification and strain typing. JALA. 2006;11:341–351. [Google Scholar]
- 11.Sampath R, Hall TA, Massire C, Li F, Blyn LB, Eshoo MW, Hofstadler SA, Ecker DJ. Rapid identification of emerging infectious agents using PCR and electrospray ionization mass spectrometry. Ann. NY Acad. Sci. 2007;1102:109–120. doi: 10.1196/annals.1408.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamson D, Renwick N, Kapoor V, Liu Z, Palacios G, Ju J, Dean A, St.George K, Briese T, Lipkin WI. Mass tag polymerase chain reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York State during 2004–2005. J. Infect. Dis. 2006;194:1398–1402. doi: 10.1086/508551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briese T, Palacios G, Kokoris M, Jabado O, Zhiqiang L, Renwick N, Kapoor V, Casas I, Pozo F, Limberger R, et al. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerg. Infect. Dis. 2005;11:310–313. doi: 10.3201/eid1102.040492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominguez SR, Briese T, Palacios G, Hui J, Villari J, Kapoor V, Tokarz R, Glodé MP, Anderson MS, Robinson CC, et al. Multiplex MassTag-PCR for respiratory pathogens in pediatric nasopharyngeal washes negative by conventional diagnostic testing shows a high prevalence of viruses belonging to a newly recognized rhinovirus clade. J. Clin. Virol. 2008;43:219–222. doi: 10.1016/j.jcv.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox JD. Nucleic acid amplification tests for detection of respiratory viruses. J. Clin. Virol. 2007;40:S15–S23. doi: 10.1016/S1386-6532(07)70005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenhoff RJ, Naraghi-Arani P, Thissen JB, Olivas J, Carillo AC, Chinn C, Rasmussen M, Messenger SM, Suer LD, Smith SM, et al. Multiplexed molecular assay for rapid exclusion of foot-and-mouth disease. J. Virol. Methods. 2008;153:61–69. doi: 10.1016/j.jviromet.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths D, Kellam P, Weiss R. International patent no. 2002 WO/2002/099130. [Google Scholar]
- 18.Quan PL, Briese T, Palacios G, Lipkin WI. Rapid sequence-based diagnosis of viral infection. Antivir. Res. 2008;79:1–5. doi: 10.1016/j.antiviral.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemmon GH, Gardner SN. Predicting the sensitivity and specificity of published real-time PCR assays. Ann. Clin. Microbiol. Antimicrob. 2008;7:18. doi: 10.1186/1476-0711-7-18. doi:10.1186/1476-0711-1187-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jabado OJ, Palacios G, Kapoor V, Hui J, Renwick N, Zhai J, Briese T, Lipkin WI. Greene SCPrimer: a rapid comprehensive tool for designing degenerate primers from multiple sequence alignments. Nucleic Acids Res. 2006;34:6605–6611. doi: 10.1093/nar/gkl966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarman SN. Amplicon: software for designing PCR primers on aligned DNA sequences. Bioinformatics. 2004;20:1644–1645. doi: 10.1093/bioinformatics/bth121. [DOI] [PubMed] [Google Scholar]
- 22.Linhart C, Shamir R. The degenerate primer design problem. Bioinformatics. 2002;18:S172–S181. doi: 10.1093/bioinformatics/18.suppl_1.s172. [DOI] [PubMed] [Google Scholar]
- 23.Rose T, Henikoff J, Henikoff S. CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) PCR primer design. Nuclei Acids Res. 2003;31:3763–3766. doi: 10.1093/nar/gkg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadberry MD, Malcomber ST, Doust AN, Kellogg EA. Primaclade–a flexible tool to find conserved PCR primers across multiple species. Bioinformatics. 2005;21:1263–1264. doi: 10.1093/bioinformatics/bti134. [DOI] [PubMed] [Google Scholar]
- 25.Pesole G, Liuni S, Grillo G, Belichard P, Trenkle T, Welsh J, McClelland M. GeneUp: a program to select short PCR primer pairs that occur in multiple members of sequence lists. BioTechniques. 1998;25:112–123. doi: 10.2144/98251bc02. [DOI] [PubMed] [Google Scholar]
- 26.Huang YC, Chang CF, Chan Ch, Yeh TJ, Chang YC, Chen CC, Kao CY. Integrated minimum-set primers and unique probe design algorithms for differential detection on symptom-related pathogens. Bioinformatics. 2005;21:4330–4337. doi: 10.1093/bioinformatics/bti730. [DOI] [PubMed] [Google Scholar]
- 27.Markham N, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rachlin J, Ding C, Cantor C, Kasif S. MuPlex: multi-objective multiplex PCR assay design. Nucleic Acids Res. 2005;33:W544–W547. doi: 10.1093/nar/gki377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rachlin J, Ding C, Cantor C, Kasif S. Computational tradeoffs in multiplex PCR assay design for SNP genotyping. BMC Genomics. 2005;6:102. doi: 10.1186/1471-2164-6-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 33.Herold J, Kurtz S, Giegerich R. Efficient computation of absent words in genomic sequences. BMC Bioinformatics. 2008;9:167. doi: 10.1186/1471-2105-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul SF, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casabianca A, Gori C, Orlandi C, Forbici F, Federico Perno C, Magnani M. Fast and sensitive quantitative detection of HIV DNA in whole blood leucocytes by SYBR green I real-time PCR assay. Mol. Cell Probes. 2007;21:368–378. doi: 10.1016/j.mcp.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Putkuri N, Piiparinen H, Vaheri A, Vapalahti O. Detection of human Orthopoxvirus infections and differentiation of smallpox virus with real-time PCR. J. Med. Virol. 2009;81:146–152. doi: 10.1002/jmv.21385. [DOI] [PubMed] [Google Scholar]
- 37.Olson VA, Laue T, Laker MT, Babkin IV, Drosten C, Shchelkunov SN, Niedrig M, Damon IK, Meyer H. Real-time PCR system for detection of Orthopoxviruses and simultaneous identification of smallpox virus. J. Clin. Microbiol. 2004;42:1940–1946. doi: 10.1128/JCM.42.5.1940-1946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson WR, Robbins G, Wrege DE, Zhang T. A new approach to primer selection in polymerase chain reaction experiments. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995;3:285–291. [PubMed] [Google Scholar]
- 39.Palka-Santini M, Cleven B, Eichinger L, Kronke M, Krut O. Large scale multiplex PCR improves pathogen detection by DNA microarrays. BMC Microbiol. 2009;9:1. doi: 10.1186/1471-2180-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beer NR, Hindson BJ, Wheeler EK, Hall SB, Rose KA, Kennedy IM, Colston BW. On-chip, real-time, single-copy polymerase chain reaction in picoliter droplets. Anal. Chem. 2007;79:8471–8475. doi: 10.1021/ac701809w. [DOI] [PubMed] [Google Scholar]
- 41.Caetano-Anolles G, Brant JB, Peter MG. DNA amplification fingerprinting using very short arbitrary oligonucleotide primers. Nat. Biotech. 1991;9:553–557. doi: 10.1038/nbt0691-553. [DOI] [PubMed] [Google Scholar]
- 42.Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990;18:6531–6535. doi: 10.1093/nar/18.22.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nanda S. ASM Biodefense and Emerging Diseases Research Meeting. DC: Washington; 2007. p. 53. [Google Scholar]
- 44.Nanda S, Jayan G, Voulgaropoulou F, Sierra-Honigmann AM, Uhlenhaut C, McWatters BJP, Patel A, Krause PR. Universal virus detection by degenerate-oligonucleotide primed polymerase chain reaction of purified viral nucleic acids. J. Virol. Methods. 2008;152:18–24. doi: 10.1016/j.jviromet.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyrc K, Jebbink MF, Berkhout B, Hoek L. SARS – and Other Coronaviruses. 2008. pp. 1–17. [Google Scholar]
- 46.de Vries M, Pyrc K, Berkhout R, Vermeulen-Oost W, Dijkman R, Jebbink MF, Bruisten S, Berkhout B, van der Hoek L. Human Parechovirus Type 1, 3, 4, 5, and 6 Detection in Picornavirus Cultures. J. Clin. Microbiol. 2008;46:759–762. doi: 10.1128/JCM.02009-07. [DOI] [PMC free article] [PubMed] [Google Scholar]