


Abstract
In the yeast Saccharomyces cerevisiae, the Rad1–Rad10 protein complex participates in nucleotide excision repair (NER) and homologous recombination (HR). During HR, the Rad1–Rad10 endonuclease cleaves 3′ branches of DNA and aberrant 3′ DNA ends that are refractory to other 3′ processing enzymes. Here we show that yeast strains expressing fluorescently labeled Rad10 protein (Rad10-YFP) form foci in response to double-strand breaks (DSBs) induced by a site-specific restriction enzyme, I-SceI or by ionizing radiation (IR). Additionally, for endonuclease-induced DSBs, Rad10-YFP localization to DSB sites depends on both RAD51 and RAD52, but not MRE11 while IR-induced breaks do not require RAD51. Finally, Rad10-YFP colocalizes with Rad51-CFP and with Rad52-CFP at DSB sites, indicating a temporal overlap of Rad52, Rad51 and Rad10 functions at DSBs. These observations are consistent with a putative role of Rad10 protein in excising overhanging DNA ends after homology searching and refine the potential role(s) of the Rad1–Rad10 complex in DSB repair in yeast.
INTRODUCTION
DNA double-strand break repair (DSBR) is highly conserved between the yeast Saccharomyces cerevisiae and Homo sapiens (1,2). In both organisms, double-strand breaks (DSBs) are resected, generating 3′ single-stranded regions that are coated with replication protein A (RPA). Subsequent strand invasion of single-stranded end(s) into a homologous duplex requires that Rad51 recombinase displaces RPA and forms a nucleoprotein filament. In mammalian cells, displacement of RPA and activation of Rad51 is mediated by BRCA2 (3). In budding yeast, Rad52 protein facilitates formation of Rad51 filaments by displacing RPA protein and recruiting proteins of the RAD52 epistasis group (2,4). Among these, Rad52 functions downstream of the Mre11–Rad50–Xrs2 (MRX) complex (5). Hence, Rad52 function is required in advance of the functions of many proteins participating late in homologous recombination (HR) (downstream of key nuclease processing events), including those required for homology searching, and excision of extraneous DNA following homology searching and DNA ligation.
The Rad1–Rad10 endonuclease complex in S. cerevisiae participates both in HR and in nucleotide excision repair (NER), recognizing junctions between single- and double-stranded DNA, and cleaving in the double-stranded region a few nucleotides 5′ to the junction to remove 3′ nonhomologous ends (6,7). Recent genetic studies have demonstrated that the complex is also required for microhomology-mediated end joining (MMEJ), a mode of DSB repair that functions independently of Rad52. MMEJ is also Ku-independent (Yku70–Yku80), and is thus distinct from nonhomologous end-joining (NHEJ) (8–10). In human cell lines ERCC1 protein, the human homolog of Rad10, also facilitates a mode of homology-mediated DSB repair that is Ku86-independent (11,12). On the basis of existing evidence, the most likely model is that the Rad1–Rad10 endonuclease functions downstream of Rad52, cleaving nonhomologous DNA ends prior to gap filling and DNA ligation of the recombination site. However, the precise timing of Rad1–Rad10 recruitment to DSB sites relative to other DSBR proteins has not been directly demonstrated. Additionally, recent work has revealed that Saw1 recruits the Rad1–Rad10 complex to DSBs in the context of single-strand annealing (SSA), one mode of DSB repair (13). Other than for SSA, the mechanism by which Rad1–Rad10 is recruited to DSBs remains largely unclear (13).
Recent studies have demonstrated the utility of fluorescence microscopy in monitoring biochemical events in live yeast cells (5,14). Three advantages of this experimental approach are especially noteworthy. First, the gene that encodes a given fluorescent protein under study is in the same chromosomal location as the native gene, and is also under control of its endogenous promoter. Thus, physiologically irrelevant overexpression associated with the use of ectopic genes expressed from non-native promoters is avoided. Second, this technique avoids fixation of cells prior to staining with fluorescent antibodies, as is typically the case in mammalian fluorescence microscopy experiments. Thus, the dynamics of the proteins under study are observed in real time in live cells, allowing for the accurate analysis of temporal relationships (14). Third, HR is monitored at the single-cell level, which allows for the analysis of individual repair events separately. Using this technology, the present study demonstrates that yeast Rad10 tagged with yellow fluorescent protein (Rad10-YFP) is recruited directly to DSB repair foci.
When a site-specific DSB is induced by the restriction endonuclease I-SceI in a DNA sequence context lacking direct repeat sequences, recruitment of Rad10-YFP to DSB foci requires functional RAD51 and RAD52 genes, but not MRE11. In contrast, when DSBs are induced by ionizing radiation (IR) during which DNA damage can occur in random DNA sequence contexts including those with direct repeats (6), we find that recruitment of Rad10-YFP to Rad52-CFP foci is RAD51-independent. These observations provide the first direct evidence that Rad1–Rad10 functions downstream of Rad52 during DSB repair and that its recruitment to DSB sites has different genetic requirements depending on the method by which DSB induction occurs and therefore, probably, the location of the DSB within the genome and the level of DNA sequence homology flanking the DSB. Moreover, the results support a model of HR in which the primary role of Rad1–Rad10 is cleavage of DNA ends following homology searching.
MATERIALS AND METHODS
Engineering of yeast strains Rad10-YFP and rad10Δ
The S. cerevisiae RAD10 gene was genetically fused in frame at the chromosomal locus with the DNA coding region of YFP to prepare the Rad10-YFP strain in the haploid W303-1A genetic background by the adaptamer-mediated PCR method as described previously (15). The resulting strain was backcrossed to the W3646-11D strain to produce WPF006-4C, which was used in microscopy experiments. The presence and identity of the YFP tag were confirmed by PCR, fluorescence microscopy and DNA sequencing which showed no mutations.
Expression of the full-length Rad10-YFP polypeptide was confirmed by immunoblotting of yeast whole cell extracts (WCEs) from appropriate strains using an α-Rad10 antibody. The Rad10 polyclonal antibody was prepared by overexpression of Rad10 in E. coli, purification and rabbit immunization. SDS PAGE was conducted with NuPAGE® Novex 4–12% Bis-Tris (Invitrogen) protein gels with 2-(N-Morpholino)ethanesulfonic acid (MES) running buffer used according to manufacturer’s recommendations to separate proteins prior to immunoblotting. Bands were sized with BenchmarkTM protein standards (Invitrogen).
An isogenic disruption mutant of RAD10 (PF033-6C) was prepared by PCR amplification of the KanMX deletion cassette from the YML095C strain (#6492) from the BY4741 deletion library and transformation into the W303-1A background. Transformants were selected on YPD plates containing 400 mg/ml G418 (Gibco) and backcrossed to wild-type strain W1588-4C to give PF033-6C. The presence of the deletion cassette was confirmed by PCR. All other strains used in this study were prepared by genetic crosses. The rad51Δ and rad52Δ genotypes were confirmed by gamma-irradiation (400 Gy) sensitivity.
UV survival studies
The UV sensitivity of the Rad10-YFP strain was tested using standard methodologies in parallel with isogenic wild-type and deletion mutant controls (16). Triplicate experiments were conducted and the results averaged and reported along with the corresponding standard error.
SSA/gene conversion assays
SSA/gene conversion (GC) assays were conducted essentially as previously described (17,18). Strains were co-transformed with the pJF6 reporter plasmid and a plasmid containing the HO endonuclease under the control of the GAL promoter, pFH800. Transformants were cultured in YEP-Lactate to a cell density of 5 × 106 to 1 × 107 cells/ml, induced by addition of 1/10 volume of 20% aqueous galactose (w/w) and incubated with shaking (2–4 h). Plasmid DNA was isolated, digested with HindIII, PstI and SmaI restriction enzymes, resolved on a 0.8% agarose gel (1× TAE, 50 V, 21 h), and transferred to Immobilon-Ny+ Transfer Membrane (Millipore). Following transfer, the membrane was hybridized (12 h, 68°C) with a digoxigenin-labeled (Roche) DNA probe prepared by PCR amplification of an ∼800 bp fragment using primers 5′-d(CGTCATAGCGATAACGAG) and 5′-d(CGGTCGGGATAGTTTTCTTGCG). The probe was detected using a standard α-digoxigenin/CSPD protocol and manufacturer’s instructions (Roche). Quantity One image analysis software (BioRad) was used for densitometric calculations.
General microscopy
Cultures for microscopy experiments were propagated in SC medium supplemented with 200 µg/ml adenine (SC + ade) at 23°C unless otherwise indicated. Stationary phase cultures were freshly diluted (to 0.1 OD600) and incubated (3 h). Aliquots of the cultures were then centrifuged (3600g), resuspended in residual medium and admixed on glass microscope slides with an equal volume of identical medium at 42°C containing 1% (w/v) low-melting agarose (Fischer). Microscopy was performed on a Zeiss AxioImager M1 microscope with a Plan–Apochromat 100×, 1.4 numerical aperture (NA) objective oil immersion lens, a motorized Z-drive and automated shutters. The fluorescent light source was an X-Cite 120 IRIS Fluorescent Light Source. IR experiment images for Figure 6 were acquired on a DM5500B Leica fluorescence microscope with a 100×, 1.46 NA objective and a mercury fluorescence light source. For both microscopes, images were captured with a Hamamatsu ORCA-ER digital camera (CA4742-80-12AG). Images at different focal planes through the entire thickness of the cells were captured at 0.3 µm intervals along the Z-axis (a Z-stack). Filter sets employed were Yellow GFP, Chroma #41028, (λex = 500/20, λdichroic = 515, λem = 535/30), CFP, Chroma #31044V2 (λex = 436/20, λdichroic = 455, λem = 480/40) and NAR/EX DsRed, Chroma #41035, (λex = 546/11, λdichroic = 560, λem = 605/75). Integration time for image acquisition was 800 ms for Rad10-YFP, 400 ms for Rad52-CFP and 400 ms for TetR-RFP. For both setups, microscopes and cameras were controlled by the Volocity software package (Improvision v 3.7.0 for the Zeiss or v 5.0.2 for the Leica). Foci were counted by inspecting images from each focal plane of the Z-stack. As is commonly the case in fluorescence microscopy work, with each fluorophore it was necessary to contrast enhance the original images, which was optimized using the gain, offset and gamma contrast parameters of Volocity. The same contrast enhancement settings were used for all images of a given fluorophore. For publication quality images, the settings were re-optimized so that foci could be seen when printed and, for merged images, the opacity and fill settings were changed so that the various colors could be viewed in a single image.
Figure 6.

Rad10-YFP foci are induced in response to gamma-irradiation. Strains containing both Rad10-YFP and Rad52-CFP (WPF019-26C and W8757-12A) were cultured, induced with gamma-irradiation (40 Gy) as described in ‘Materials and Methods’ section and imaged by fluorescence microscopy. Rad10-YFP and Rad52-CFP foci in fluorescent images were counted. Data corresponding to Rad10-YFP/Rad52-CFP colocalized foci are shown for RAD51 wild-type and rad51Δ strains. Black bars show results from irradiated cells; white bars show results corresponding to non-irradiated controls. Results shown are the summation of two independent experiments in which at least 100 cells were counted over the course of the two experiments and error bars report exact binomial confidence intervals at the 95% confidence level.
Cells were classified with respect to phase in the cell cycle based on their appearance in both the fluorescence images and in differential interference contrast (DIC) images. Unless otherwise noted, for each trial of each strain, at least 100 cells were counted. Graphs of the data report averages of at least three independent experiments with the corresponding standard error. In the case of mre11Δ data, P-values were calculated using a two-tailed Student’s t-test, to determine whether mre11Δ data were statistically significantly different from wild-type (19). Images were prepared for publication using the Adobe Photoshop and Adobe Illustrator software packages (Adobe Systems, Mountain View, CA). For images showing triple colocalization, only single-plane images were captured and the exposure time for Rad10-YFP was adjusted to 3 s. For single-plane image experiments, at least 600 galactose-induced cells (total) were analyzed for the appearance of colocalized foci from two independently conducted experiments of at least 300 cells each.
Induction of DSBs by I-SceI endonuclease
Strains for DSB induction experiments bear chromosomally integrated copies of the Tetracycline repressor protein (TetR) fused to RFP (TetR-RFP) and 224 copies of the Tetracycline operator (tetO) repressor binding site abutted with one copy of the I-SceI cut site at the iYER186 intergenic region on chromosome V as described (20). The site is cut by the I-SceI endonuclease with 60–70% efficiency in asynchronously growing cells (20,21). The strains used in I-SceI DSB induction experiments included PF025-7A, PF030-49A, PF023-15A, WPF019-26C and PF034-11D. Strains were transformed with plasmid pWJ1320, containing the I-SceI gene under control of the GAL1 promoter and containing the ADE2 selectable marker as described (4). Transformants were plated on SC–ade medium containing 2% raffinose (SC–ade w/raff). Cultures for DSB induction experiments were propagated in SC–ade w/ raff liquid medium at 23°C. Overnight cultures were diluted to 0.1 OD600 and incubated (3 h). DNA DSBs were subsequently induced by adding galactose to the medium to a final concentration of 2% and incubating the cultures (4 h) prior to processing cells for microscopy as described above.
DAPI-stained images
Cell cultures were grown as described under ‘General Microscopy’ and centrifuged. The cell pellet was resuspended in fixative solution (2.5% formaldehyde, 30 mM sodium phosphate, pH 7) and incubated (5 min). Cells were centrifuged (3600g), washed twice with water, resuspended in 1 µl residual water and admixed on glass microscope slides with DAPI mounting medium [SC + ade, 1% [w/v] low-melting agarose (Fischer), 50 ng/ml 4′,6-diamidino-2-phenylindole (DAPI)]. In addition to DIC and YFP images, cells were imaged using the YFP filter set and a DAPI/Hoechst/AMCA filter set, Chroma #31000v2, (λex = 350/50, λdichroic = 400, λem = 460/50). YFP exposures were 3 s and DAPI exposures were 20 ms.
Induction of DSBs by gamma irradiation
Gamma induction experiments were conducted as described above for I-SceI induction experiments except for the following experimental alterations. Strains WPF019-26C and W8757-12A which did not contain the plasmid pWJ1320 were cultured in SC + ade medium, diluted to an OD600 = 0.1 and grown (3 h), irradiated with gamma radiation (40 Gy, 60Co source), incubated (2 h) and imaged using the same conditions as for I-SceI experiments.
Table 1.
Strains used in this study
| Name | Genotype | Published in |
|---|---|---|
| SX46a | MATa ade2-1 trp1-289 his3-532 ura3-52 | (34) |
| BJRad10Δ | MATa rad10::URA3 leu2 trp1 ura3-52 pep4-3 prb1-1122 prc1-407 | From EC Friedberg laboratory strain collection |
| W303-1A | MATa ade2-1 lys2Δ trp1-1 can1-100 his3-11,15 leu2-3,112 ura3-1 rad5-535 | (35) |
| W1588-4C | MATa ade2-1 lys2Δ trp1-1 can1-100 his3-11,15 leu2-3,112 ura3-1 | (36) |
| W3646-11D | MATα lys2Δ TRP1 can1-100 his3-11,15 bar1::LEU2 ura3-1 | (35) |
| WPF006-4C | MATa lys2Δ trp1-1 can1-100 his3-11,15 leu2-3,112 ura3-1 Rad10-YFP | This manuscript |
| PF033-6C | MATa trp1-1 his3-11,15 leu2-3,112 ura3-1 rad10::KanMX | This manuscript |
| PF044-24A | MATa ADE lys2Δ trp1-1 can1-100 his3-11,15 leu2-3,112 ura3-1 | This manuscript |
| WPF033-6C | MATa ade2-1 rad10::KanMX his3-11,15 leu2-3,112 trp1-1 ura3-1 | This manuscript |
| ML47-1D | MAT::HIS3 ade2-1 trp1-1 can1-100 his3-11,15::YFP-lacI-R197L::HIS3 leu2-3, TetR-mRFP(iYGL119W) URA3::tetOx224(iYER187W) I-SceI(iYER186C) Rad52-CFP | A derivative of strains described in supplementary section of (14) |
| PF025-7A | MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP | This manuscript |
| WPF021-4A | MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP rad52::HIS5 | This manuscript |
| PF030-49A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI(iYER186C) Rad10-YFP rad51Δ | This manuscript |
| PF023-15A | MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP mre11::LEU2 | This manuscript |
| WPF019-26C | MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP Rad52-CFP | This manuscript |
| PF034-11D | MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP Rad51-CFP | This manuscript |
| W8757-12A | MATa ade2-1 TRP1 can1-100 his3-11,15 leu2-3,112 TetR-mRFP (iYGL119W) URA3::tetOx224(iYER187W) I-SceI (iYER186C) Rad10-YFP Rad52-CFP rad51Δ | This manuscript |
All strains in this study are haploid and derivatives of W303-1A and W303-1B (35) except SX46a and BJRad10Δ. Additionally, all strains are wild-type for the ADE2 and RAD5 genes unless otherwise noted.
RESULTS
Rad10 protein in the yeast S. cerevisiae was tagged at its C-terminal end by integrating the gene encoding the yellow-shifted version of green fluorescent protein from Aequorea victoria in-frame at the RAD10 chromosomal locus, using adaptamer-mediated PCR as previously described (15). The resulting yeast strain expresses a Rad10-YFP fusion protein (Rad10-YFP) that migrates as a 54 kDa polypeptide during SDS PAGE, whereas the untagged protein migrates as a 24 kDa band (compare lanes 2 and 4 with lanes 5–7 in Figure 1A). A control lane containing GFP protein alone (lane 1) exhibits no signal with α-Rad10 antibody, and extracts from a rad10 deletion mutation (rad10Δ, lane 3) exhibit only background signal of the antibody. As expected, by fluorescence microscopy, the Rad10-YFP strain exhibits yellow fluorescence in the nucleus as indicated by colocalization of the YFP signal with DAPI-stained DNA in the nucleus (Figure 1B).
Figure 1.
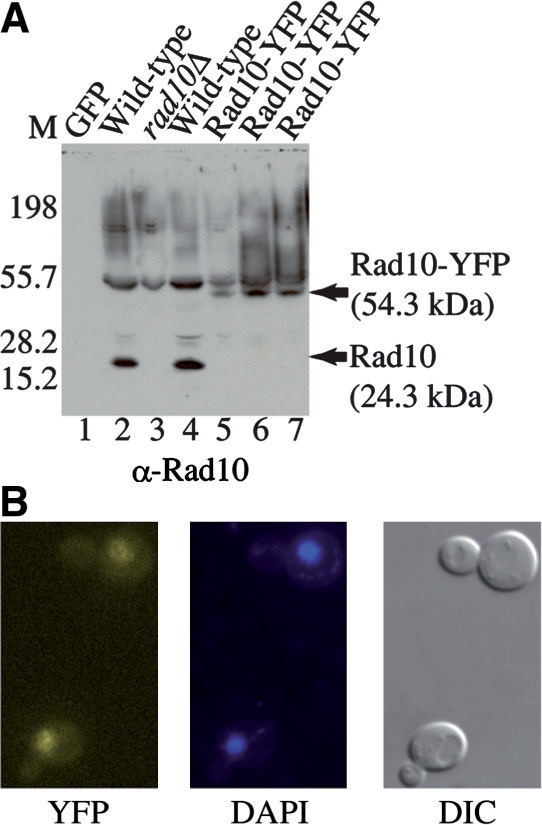
(A) The Rad10-YFP strain exhibits an increased polypeptide size consistent with Rad10 expressed as a YFP fusion protein. Immunoblot of WCE from the indicated yeast strains probed with a Rad10 antibody. 1 µg total protein was loaded to each lane. Lane 1: GFP protein (Santa Cruz Biotechnology); lane 2: SX46a WILD-TYPE; lane 3: BJRad10Δ; lane 4: W303-1A wild-type (W1588-4C); lanes 5 and 7: Rad10-YFP (WPF006-4C); lane 6: Rad10-YFP Rad52-CFP (WPF019-26C). The wild-type strains (lanes 2 and 4) show a 24 kDa Rad10 band while the Rad10-YFP strains (lanes 5–7) show a 54 kDa Rad10-YFP band. A rad10 deletion mutant (rad10Δ, lane 3) shows only background. Markers were run on the left. (B) The Rad10-YFP protein exhibits nuclear localization. YFP fluorescence (left panel) colocalizes with the DAPI fluorescence signal (center panel) confirming the nuclear localization of the YFP fluorescence. A DIC image of the same cells is also shown (right panel).
Next, the functionality of the Rad10-YFP protein in vivo was examined. A plasmid-based assay was used to test Rad10-YFP function in SSA and GC (Figure 2A). Densitometric analysis of genome blot results (Figure 2B) shows that the efficiencies of SSA and GC are somewhat lower (86 and 54%, respectively) in the YFP-tagged strain compared with wild-type. However, the fluorescent label does not compromise the function of Rad10 protein during NER, as evidenced by examining the UV radiation sensitivity of the Rad10-YFP strain. Indeed, the Rad10-YFP strain exhibits wild-type levels of survival following exposure to UV-C irradiation (Figure 2C).
Figure 2.

(A) Schematic showing the SSA assay reporter plasmid [modified from (17)]. The reporter plasmid, pJF6 can be repaired by either SSA or GC. Triple digestion of the plasmid by PstI, SmaI and HindIII gives 5.6 and 4.4 kb bands, respectively, that hybridize with the 800 bp lacZ probe. Following cleavage by the HO endonuclease, the HO cut site can be repaired by SSA, which eliminates the HindIII restriction site to give a 5.8 kb product. Alternatively, repair by GC retains the HindIII site but generates a 4.3 kb fragment. The dark gray lines show the position of probe hybridization. (B) Rad10-YFP SSA efficiency is indistinguishable from that of the wild-type strain. Lanes 1 and 2 show sizing markers of 4.4 and 5.6 kb, respectively. Lanes 3, 5 and 7 show uninduced controls. Lanes 4, 6 and 8 show samples from cultures in which expression of the HO endonuclease was induced. Lanes 3 and 4 show the wild-type strain (PF044-24A); lanes 5 and 6 show the isogenic rad10 deletion mutant (WPF033-6C); lanes 7 and 8 show the Rad10-YFP strain (WPF006-4C). (C) UV survival experiments were conducted on the Rad10-YFP strain as well as isogenic wild-type and rad10 deletion mutant strains. The strains shown are the unlabeled wild-type strain W1588-4C (closed diamond), the Rad10-YFP strain, WPF006-4C (closed circle) and the rad10 deletion mutant strain, rad10Δ, PF033-6C (closed square). Percentage survival is reported as a function of UV-C dosage at 254 nm (J/m2). Error bars show the standard error of three independent experiments as a percentage of plated cells.
To determine whether Rad10 protein is recruited to DSBs, such lesions were generated in vivo in a strain containing both Rad10-YFP and a unique DSB labeling system previously reported (Figure 3A) (4). Specifically, a yeast strain was prepared that expresses the coding sequence for the TetR fused to monomeric red fluorescent protein (TetR-RFP). The strain additionally contains a DNA cassette with 224 copies of the TetR binding sequence abutted with one copy of the I-SceI restriction endonuclease site. I-SceI enzyme was introduced into the strain via a selectable extrachromosomal plasmid under the control of the GAL1 promoter (pWJ1320) (4). Subsequent induction of I-SceI generates a single DSB in close physical proximity to many copies of TetR-RFP protein bound to DNA, thereby enabling visualization of the DSB site as a bright red focus (DSB-RFP) within the nucleus (Figure 3B). Such induction of DSBs results in increased punctate yellow foci (Figure 3B, Rad10-YFP panel), indicating the presence of a high local concentration of Rad10-YFP. A significant subset of Rad10-YFP foci colocalized with DSB-RFP foci in DSB-induced cells, indicating that Rad10-YFP protein is indeed recruited to DSB sites (Figures 3B and 4). Rad10-YFP foci were also observed spontaneously in uninduced cells, but in fewer numbers and they rarely colocalized with the DSB site (see below). Some of these spontaneous events likely reflect the recruitment of the tagged protein to spontaneous DSBs, as previously reported for proteins of the RAD52 epistasis group, suggesting that Rad10 is recruited to both endonuclease-induced and naturally occurring DSBs (4). Indeed, cells co-expressing Rad10-YFP and Rad52-CFP exhibited spontaneous Rad52-CFP foci that colocalized with Rad10-YFP (Figure 6). Since spontaneous Rad10 foci were consistently observed at a higher frequency (70–80%) than spontaneous Rad52 foci (5–10%), many of the Rad10 foci likely reflect the involvement of Rad10 in other DNA repair pathway(s), such as NER and MMEJ.
Figure 3.

(A) Scheme showing experimental design. The I-SceI gene is introduced to the cells on an adenine selectable (ADE) plasmid under the control of the galactose-inducible (GAL1) promoter. Upon induction with galactose, the I-SceI gene is switched on to produce I-SceI enzyme which then cuts the single I-SceI recognition sequence installed on chromosome V. The I-SceI restriction site is adjacent to 224 copies of the TetR-binding site. The TetR gene fused to monomeric Red Fluorescent Protein (TetR-RFP) is constitutively expressed from the intergenic region iYGL119W on chromosome VII. TetR-RFP binds to the array of 224 Tet repressor binding sites, thereby labeling the site of the induced DSBs to which other fluorescently labeled protein factors are recruited. (B) Examples of Rad10-YFP foci that are colocalized with TetR-RFP (DSB-RFP) foci. Fluorescence and DIC images are shown from cells of strain PF025-7A transformed with plasmid pWJ1320 and induced with galactose as described in ‘Materials and Methods’ section. Arrows point to colocalizing YFP and RFP foci.
Figure 4.

Rad10-YFP foci are induced in response to DNA DSBs and colocalize with the DSBs. Yeast strains (PF025-7A, WPF021-4A, PF030-49A and PF023-15A) containing the Rad10-YFP/DSB-RFP labeling system were transformed with plasmid pWJ1320, and cultures of transformants induced with galactose as described in ‘Materials and Methods’ section. YFP and RFP foci in fluorescent images from the resulting cells were counted. Black bars show results corresponding to cells following induction with galactose. White bars show results corresponding to uninduced cells. Error bars indicate the standard error from three independent experiments in each of which at least 100 cells were counted and classified. Data corresponding to Rad10-YFP/DSB-RFP colocalized foci are shown for wild-type experiments alongside those conducted in isogenic strains deleted of either the MRE11 (mre11Δ), RAD52 (rad52Δ) or RAD51 (rad51Δ) genes.
To determine the cell cycle dependence of Rad10 foci, cells were scored not only for Rad10-YFP and DSB-RFP foci, but also for cell cycle phase (S, G2/M or G1, see ‘Materials and Methods’ section). S phase cells manifested the greatest magnitude of DSB-colocalized Rad10 foci, exhibiting approximately a 12-fold increase in DSB-induced cells over that in uninduced controls (Figure 4, wild-type). Colocalized foci were also induced in G2/M and G1 cells, but to a much lesser extent (Figure 4), suggesting that the repair of DSBs by HR transpires primarily during S phase of the cell cycle, presumably due to ongoing functions of Rad10 such as repair of spontaneous DSBs and NER. The great majority (70–80%) of uninduced cells contained at least one Rad10-YFP focus. However, upon DSB induction the increase in the numbers of Rad10-YFP foci matched the increase in the numbers of Rad10-YFP foci colocalized to DSB sites.
We next examined the genetic requirements of recruitment of Rad10-YFP to DSBs. Isogenic strains deleted for the MRE11, RAD52 or RAD51 genes (mre11Δ, rad52Δ or rad51Δ) were transformed with the I-SceI expression plasmid, pWJ1320. DSBs were induced and the formation of DSB-colocalized Rad10-YFP foci was monitored in parallel with wild-type controls. The frequency of DSB-colocalized Rad10-YFP foci observed in the mre11Δ strain was not appreciably altered relative to the wild-type, (P-values > 0.19 in all phases of the cell cycle) indicating that Mre11 protein is not required to recruit Rad10-YFP to DSBs (Figure 4, compare black wild-type and mre11Δ bars). Hence, while visual examination of Figure 4 suggests a small reduction in the number of Rad10-YFP foci colocalized with DSB-RFP foci in the mre11Δ background, this difference is not statistically significant under the experimental conditions employed. In contrast, rad52 and rad51 deletion mutants exhibited very few Rad10-YFP foci localized to sites of DSBs upon I-SceI induction (Figure 4, compare wild-type, rad52Δ and rad51Δ bars).
To determine whether Rad10 colocalizes with Rad52 or Rad51 at DSB sites, DSB induction experiments were conducted using triple-labeled Rad10-YFP/DSB-RFP/Rad52-CFP or Rad10-YFP/DSB-RFP/Rad51-CFP strains (Figure 5A). Triple-labeled experiments were imaged as single focal planes owing to photobleaching of the Rad10-YFP chromophore which precluded the formation of Z-stack images (see ‘Materials and Methods’ section). Accordingly, the overall percentage of cells containing Rad10-YFP is reduced in single-plane images (Figure 5B) versus Z-stack images (Figure 4), since a single focal plane reveals only a portion of the total thickness of the average cell.
Figure 5.

(A) A strain containing Rad10-YFP, the DSB-RFP labeling system and Rad52-CFP (WPF019-26C) was transformed with plasmid pWJ1320 and cultures of transformants induced with galactose as described in ‘Materials and Methods’ section. Single-focal plane images were acquired for all three fluorescent chromophores (YFP, CFP, RFP). The YFP, CFP and RFP images are shown as the top three panels of images, respectively; a merged image of the top three panels is shown as the fourth panel and the corresponding DIC image is shown as the bottom panel. (B) Quantitation of Rad10–Rad52 colocalization. Single-focal plane images for experiments described for Panel A were analyzed for the percentages of cells containing at least one focus of the type indicated for each bar of the graph. ‘YFP’ indicates total percentages of Rad10-YFP foci, ‘CFP’ indicates total percentages of Rad52-CFP foci, ‘RFP’ indicates total percentages of DSB-RFP foci. ‘YFP/RFP’, ‘CFP/RFP’ or ‘YFP/CFP/RFP’ indicate the percentages of cells containing the corresponding double or triple colocalized foci, respectively.
In both strains, colocalization of all three labels (YFP, RFP and CFP) was observed, indicating that Rad10-YFP is present at DSB sites at the same time as Rad52-CFP (Figure 5A) or Rad51-CFP (data not shown). Furthermore, ∼75% of the Rad10-YFP foci that colocalized with the DSB-RFP marker were also colocalized with Rad52-CFP, both in S and G2/M phase cells, while essentially no G1 cells exhibit similar triple colocalization (Figure 5B). Hence, most of the Rad10-YFP foci localized to the induced DSBs also had Rad52-CFP present indicating considerable temporal overlap of the functions of Rad10 with Rad52. Altogether, we conclude that Rad10 protein colocalizes with Rad51 and Rad52 proteins at DSBs during S and G2/M phases of the cell cycle.
Since the repair of DSBs induced by a restriction enzyme can differ from the repair of breaks induced by IR (20,21), a series of experiments were done using a gamma source to induce DSBs. Experiments were conducted in both RAD51 and rad51Δ strains containing Rad10-YFP and Rad52-CFP. Two hours post-irradiation, individual and colocalized foci were counted and compared with results from non-irradiated controls. Following IR, the numbers of cells containing at least one Rad10-YFP focus increased similarly in both the RAD51 and the rad51Δ strains relative to non-irradiated controls (1.3- and 1.7-fold, respectively; data not shown). The numbers of cells containing at least one Rad10-YFP/Rad52-CFP colocalized focus also increased in both strains relative to non-irradiated controls (6.2-fold for RAD51 and 4.2-fold for rad51Δ, respectively; data not shown). As with the I-SceI experiments, most of the foci were observed in the S, G2 or M phases of cell cycle. Upon IR induction, the number of S, G2 or M phase cells containing at least one colocalized Rad10-YFP/Rad52-CFP focus increased 4.8-fold for RAD51 cells and 2.5-fold for rad51Δ cells (Figure 6). Thus, IR induction of colocalizing Rad10 and Rad52 foci is RAD51-independent unlike that observed following I-SceI induction. Interestingly, in the absence of IR induction, the number of S, G2 and M phase cells containing at least one spontaneous colocalized Rad10-YFP/Rad52-CFP focus was 2.5-fold higher in rad51Δ cells than in RAD51 cells (Figure 6).
DISCUSSION
Here we monitored in real time, in living yeast cells, several proteins involved in DNA repair, which are expressed from their native chromosomal locations and endogenous promoters. This approach provides a more physiologically relevant aspect to in vitro biochemical experiments involving cell free extracts or overexpression systems. Our experiments demonstrate that Rad10-YFP foci are induced in response to the formation of DNA DSBs, and that such foci are localized specifically to such sites in S. cerevisiae. The total number of cells that contained at least one Rad10-YFP focus was high (∼70–80%) even without induction of DSBs and many of the foci observed are not localized to the induced DSBs. These foci presumably reflect other roles of the Rad1–Rad10 complex in yeast, notably NER, MMEJ and spontaneous mitotic recombination, and indicate that Rad1–Rad10 is active in a majority of cells at all phases of cell cycle. The observation that the number of spontaneous Rad10-YFP foci is reduced in G1 cells when compared with S and G2/M phase cells suggests that a significant fraction of foci observed is associated with DNA replicative activity, such as spontaneous mitotic recombination events.
Although the Rad10-YFP protein is regulated by its natural promoter and from its native site, it appears slightly compromised in SSA function (86% of the wild-type level) and GC function (54% of the wild-type level), the two DSB repair processes in which Rad10 plays a role (Figure 2B and data not shown). We suspect that this attenuated functionality is the result of a kinetic lag in the recruitment of Rad10-YFP to the repair site, the YFP tag occluding the binding of other accessory factors, or the YFP tag causing faster dissociation from DSB sites, thus limiting the time available to execute Rad1–Rad10 function. However, the fact that the YFP tag does not compromise Rad10 function in NER as measured by UV resistance (Figure 2C) suggests that the Rad1–Rad10 endonuclease activity itself is fully functional. Indeed, the functionality of the Rad10-YFP fusion protein is comparable to other YFP-tagged DNA repair and checkpoint proteins (4).
Previous genetic studies have implicated RAD1 and RAD10 with RAD51 and RAD52 in DSB repair (6). However, this study constitutes the first direct experimental evidence for the involvement of either RAD51 or RAD52 in recruiting Rad10 to DSBs. It is well established that RAD1 and RAD10 are required for several modes of HR in yeast, including SSA, MMEJ and synthesis-dependent strand annealing (SDSA) (6). Here we show that two ways of creating of DSBs (I-SceI and IR) give rise to the formation of Rad10-YFP foci and recruitment to sites of DSBs. However, the genetic requirements observed for Rad10 recruitment are dependent on how and, hence, where we create the break in the genome and likely reflect which modes of repair are being used.
Following IR, Rad10-YFP/Rad52-CFP colocalized foci are induced (Figure 6), suggesting that Rad10 is recruited to IR-induced DSBs. The extent of induction is approximately the same whether or not the RAD51 gene is present (Figure 6). These observations auger well with existing genetic and biochemical evidence for the several modes of DSB repair requiring RAD10, not all of which require RAD51 (6). For example, ribosomal DNA, which makes up almost 10% of the genome, is a multiple tandem array containing 150–200 repeats. When a DSB occurs in one of these repeats, it is largely repaired by SSA (6,17,22). Similarly, events initiated in other repetitive sequences such as Ty elements or delta sequences can also be repaired by SSA (23). SSA requires RAD52 but does not strictly require RAD51 (22,24,25). Since DSBs induced by IR occur in a broad range of DNA sequence contexts throughout the genome, many sequences flanked by DNA repeats are likely repaired by SSA (Figure 7). Therefore, we explain the 2.5-fold increase in the percentage of rad51Δ cells containing spontaneous Rad10-YFP/Rad52-CFP colocalized foci relative to that observed in RAD51 cells (Figure 6) by a shift in repair of spontaneous damage from RAD51-dependent GC to RAD51-independent SSA.
Figure 7.

Model for Rad1–Rad10 recruitment to sites of SDSA and SSA in response to either I-SceI- or IR-induced strand breaks. In response to I-SceI-induced strand breaks, Rad1–Rad10 is recruited to DSB repair locations following strand invasion and homology searching requiring RAD52 and RAD51. During SDSA, the invading strand copies the homology on the sister chromatid (shown in red) and, after it is displaced, it anneals with the homologous region on the originally cut chromosome (shown in black). Any overlap between the newly annealed strands is removed by the Rad1–Rad10 endonuclease. Alternatively, Rad1–Rad10 might be recruited to any of the branched structures present during the strand-invasion/DNA synthesis stage of repair without endonuclease function ultimately being utilized. Following IR, however, a broader range of DSB sequence contexts gives rise to Rad10 participation in other DSB repair modes in addition to SDSA. After IR, SSA would presumably represent a fraction of repair events involving Rad10 and would be recruited in a RAD51-independent fashion. Following annealing of repeat regions (shown in gray), Rad1–Rad10 trims overlap regions, which are then filled, if necessary, and ligated.
In contrast, the I-SceI-induced DSBs in this study (Figures 4 and 5) are not flanked by DNA repeats and therefore not repaired by SSA. We find that RAD51 is required for recruitment of Rad10 to the I-SceI sites, consistent with SSA-independent repair. It is unlikely these breaks are repaired via MMEJ, another mode of DSB repair known to require both RAD1 and RAD10 since MMEJ has an absolute requirement for MRE11 (8,10) and we show that Rad10 foci form independently of MRE11. The break-induced replication (BIR) pathway, which requires RAD52 and has both RAD51-dependent and RAD51-independent modes could, in principle, be consistent with the I-SceI-induced repair observed in this study (26,27). However, we do not think it is the preferred pathway since a strict requirement for RAD1 or RAD10 in BIR has not been established (28–30).
To explain the RAD52 and RAD51 dependence we observe for Rad10 focus formation after I-SceI-induced DSBs, we suggest that the breaks are repaired by SDSA (Figure 7). In a dividing cell where one sister chromatid has been cleaved by I-SceI and the other is still intact, SDSA would be initiated by RAD52 and RAD51-dependent strand invasion. The branched structure may be recognized by Rad1–Rad10 (Figure 7). Indeed, genetic evidence for the requirements of RAD52, RAD51 and RAD10 in SDSA is consistent with this notion (6,31,32).
Results from experiments in which triply labeled strains containing Rad10-YFP/Rad51-CFP/DSB-RFP or Rad10-YFP/Rad52-CFP/DSB-RFP were induced with I-SceI show that a significant number of Rad10-YFP/DSB-RFP foci also contained either Rad51-CFP or Rad52-CFP suggesting that significant temporal overlap occurs in the functions of Rad10 with either Rad51 or Rad52 (Figure 5B). The observation that I-SceI-induced DSBs require Rad51 to recruit Rad10 leads us to speculate that Rad51 strand-exchange activity may be required for efficient recruitment of Rad10 to I-SceI-induced DSBs. Conceivably, the strand-exchange reaction generates a DNA structure in which a region of duplex DNA is abutted next to a region lacking homology in the 3′ direction (i.e. a 3′ nonhomologous end). It is possible that Rad51 or Rad52 recruits the Rad1–Rad10 complex at the point-in-time when the junction between homologous and nonhomologous DNA is encountered. It will be interesting in the future to determine whether a rad51 mutant deficient in strand-exchange activity can still recruit Rad10 to I-SceI-induced DSBs and to determine the extent to which Rad51 and/or Rad52 precedes Rad10 temporally during a repair event. Nonetheless, our existing data support a model in which the Rad1–Rad10 complex functions after Rad51 and Rad52 helping to execute the final processing of DNA ends by trimming DNA ends prior to ligation.
In humans, Rad52 protein participates in a three-protein complex with XPF/ERCC1 endonuclease, the human counterpart of Rad1–Rad10 (33). However, it remains to be determined whether an analogous three-protein complex involving Rad52 and the Rad1–Rad10 endonuclease is generated in yeast, and, if so, whether Rad52 protein recruits Rad1–Rad10 to DSB sites via a direct physical interaction. The triple-labeled colocalization experiments presented herein (Figure 5) show that Rad10 and Rad52 are both present at DSB sites at the same time, but do not provide evidence for a direct protein–protein interaction since other proteins may ultimately ‘bridge’ direct physical interaction between Rad52 and the Rad1–Rad10 complex as has been shown for Saw1 in processing recombination intermediates (13). It will be of interest to see whether a direct Rad52–Rad1–Rad10 complex forms in vitro, and if so, whether this interaction is required for the recruitment of Rad10.
FUNDING
California State University Northridge’s Competition for Research, Scholarship and Creative Activity Awards [to P.L.F.], the National Institutes of Health [5-R01-ES011344 to E.C.F., SC2GM081155 to P.L.F., GM50237, GM67055 to R.R.], the Danish Agency for Science, Technology and Innovation [to M.L.], the Villum Kann Rasmussen Foundation [to M.L.] and Fundación Alfonso Martín Escudero [to S.G.-B.]. Funding for open access charge: National Institutes of Health (SC2GM081155).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Neal Sugawara and Dr Jim Haber for kindly providing plasmids and strains for the SSA assay. They thank Jeff Wagner and the Radiation Safety Department at UCLA for generously providing access to their 137Cs gamma irradiator in order to genotype rad51Δ and rad52Δ strains.
REFERENCES
- 1.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 2.Krogh BO, Symington LS. Recombination proteins in yeast. Annu. Rev. Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 3.San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, Sung P. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J. Biol. Chem. 2006;281:11649–11657. doi: 10.1074/jbc.M601249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Lisby M, Rothstein R. DNA repair: keeping it together. Curr. Biol. 2004;14:R994–996. doi: 10.1016/j.cub.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2nd edn. Washington, DC: ASM Press; 2005. [Google Scholar]
- 7.Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC. Specific cleavage of model recombination and repair intermediates by the yeast Rad1–Rad10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- 8.Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–2014. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daley JM, Wilson TE. Rejoining of DNA double-strand breaks as a function of overhang length. Mol. Cell Biol. 2005;25:896–906. doi: 10.1128/MCB.25.3.896-906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell Biol. 2003;23:8820–8828. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aboussekhra A, Al-Sharif IS. Homologous recombination is involved in transcription-coupled repair of UV damage in Saccharomyces cerevisiae. EMBO J. 2005;24:1999–2010. doi: 10.1038/sj.emboj.7600665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell Biol. 2008;28:5082–5092. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Dong J, Pan X, Oum JH, Boeke JD, Lee SE. Microarray-based genetic screen defines SAW1, a gene required for Rad1/Rad10-dependent processing of recombination intermediates. Mol. Cell. 2008;30:325–335. doi: 10.1016/j.molcel.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lisby M, Rothstein R. DNA damage checkpoint and repair centers. Curr. Opin. Cell Biol. 2004;16:328–334. doi: 10.1016/j.ceb.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Reid RJ, Lisby M, Rothstein R. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 2002;350:258–277. doi: 10.1016/s0076-6879(02)50968-x. [DOI] [PubMed] [Google Scholar]
- 16.Bankmann M, Prakash L, Prakash S. Yeast RAD14 and human xeroderma pigmentosum group A DNA-repair genes encode homologous proteins. Nature. 1992;355:555–558. doi: 10.1038/355555a0. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov EL, Haber JE. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 1995;15:2245–2251. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fishman-Lobell J, Rudin N, Haber JE. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell Biol. 1992;12:1292–1303. doi: 10.1128/mcb.12.3.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris DC. Quantitative Chemical Analysis. 7th edn. New York: W. H. Freeman and Co.; 2007. [Google Scholar]
- 20.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 21.Barlow JH, Lisby M, Rothstein R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell. 2008;30:73–85. doi: 10.1016/j.molcel.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mieczkowski PA, Lemoine FJ, Petes TD. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair (Amst) 2006;5:1010–1020. doi: 10.1016/j.dnarep.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Ira G, Haber JE. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishman-Lobell J, Haber JE. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–484. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]
- 26.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol. Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell. 1993;73:347–360. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- 28.Dong Z, Fasullo M. Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res. 2003;31:2576–2585. doi: 10.1093/nar/gkg352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith CE, Lam AF, Symington LS. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase {delta} complex. Mol. Cell Biol. 2009;29:1432–1441. doi: 10.1128/MCB.01469-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang LE, Symington LS. Aberrant double-strand break repair in rad51 mutants of Saccharomyces cerevisiae. Mol. Cell Biol. 2000;20:9162–9172. doi: 10.1128/mcb.20.24.9162-9172.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- 32.Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics. 2002;162:647–662. doi: 10.1093/genetics/162.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Motycka TA, Bessho T, Post SM, Sung P, Tomkinson AE. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004;279:13634–13639. doi: 10.1074/jbc.M313779200. [DOI] [PubMed] [Google Scholar]
- 34.Weiss WA, Friedberg EC. Molecular cloning and characterization of the yeast RAD10 gene and expression of RAD10 protein in E. coli. EMBO J. 1985;4:1575–1582. doi: 10.1002/j.1460-2075.1985.tb03819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]