

Abstract
Object
Cyclosporine neuroprotection has been reported in brain injury models but safety and dosing guidelines have not been determined in humans with severe traumatic brain injury (TBI). The purpose of this investigation was to establish the safety of cyclosporine using 4 clinically relevant dosing schemes.
Methods
The authors performed a prospective, blinded, placebo-controlled, randomized, dose-escalation trial of cyclosporine administration initiated within 8 hours of TBI (Glasgow Coma Scale score range 4–8; motor score range 2–5). Four dosing cohorts (8 patients treated with cyclosporine and 2 receiving placebo treatment per cohort) received cyclosporine (1.25–5 mg/kg/day) or placebo in 2 divided doses (Cohorts I–III) or continuous infusion (Cohort IV) over 72 hours. Adverse events and outcome were monitored for 6 months.
Results
Forty patients were enrolled over 3 years (cyclosporine cohorts, 24 male and 8 female patients; placebo group, 8 male patients). Systemic trough concentrations were below 250 ng/ml during intermittent doses. Higher blood concentrations were observed in Cohorts III and IV. There was no significant difference in immunological effects, adverse events, infection, renal dysfunction, or seizures. Mortality rate was not affected by cyclosporine administration, independent of dose, compared with placebo (6 of 32 patients receiving cyclosporine and 2 of 8 receiving placebo died, p > 0.05). At 6 months, a dose-related improvement in favorable outcome was observed in cyclosporine-treated patients (p < 0.05).
Conclusions
In patients with acute TBI who received cyclosporine at doses up to 5 mg/kg/day, administered intravenously, with treatment initiated within 8 hours of injury, the rate of mortality or other adverse events was not significantly different from that of the placebo group.
Keywords: cyclosporine, neuroprotection, traumatic brain injury
Traumatic brain injury continues to be a leading cause of death and disability in children and young adults, with an estimated 1.5 million Americans affected annually.63 The outcomes from this injury vary significantly depending on severity, with estimates of 230,000 hospitalized survivors and up to 90,000 experiencing long-term disability.5,24,39 Mortality rates from TBI have declined by 20% since 1980. The decline is primarily due to earlier transportation to the hospital and improved resuscitative measures. No pharmacological agent has been shown to significantly improve outcome from severe brain injury. The long-term disability associated with these injuries remains a significant health issue. It is estimated that 5.3 million individuals in the US are currently living with a permanent disability related to TBI. In persons between 15 and 24 years of age, TBI is the second most common cause of hospitalization, with males affected twice as often as females. The US Centers for Disease Control identified TBI as an “invisible epidemic” because of the magnitude of indolent morbidity accompanying these injuries.63 Neurological and systemic metabolic sequelae accompany acute brain injury and contribute to this poor outcome.46,49,50
Brain injury occurs in 2 phases—the primary structural deformity followed by secondary damage to the surrounding brain.9,46,49 The primary insult to the brain tissue at the time of injury causes initial irreversible cellular damage. Secondary injury ensues when the surviving tissue suffers a cascade of neurochemical events that jeopardize both white and gray matter.3,10,27,33,45 The importance of the induction of apoptotic pathways, excessive release of excitotoxic neurotransmitters and inflammatory chemokines, production of oxidative reactive species, calpain proteolysis, and axonal stretch are well established.16,25,26,43,47,50,56,62 Higher intracellular calcium concentrations trigger the processes activating secondary cell death. Mitochondria play an important role in the maintenance of intracellular Ca homeostasis by sequestering Ca++.23,29,58,59 A therapeutic strategy modulating Ca signaling activation targets a pivotal mechanism associated with secondary sequelae following TBI.2,17,22,30,37,49,55
Cyclosporine, a widely used immunosuppressive drug, has demonstrated neuroprotective properties after neural trauma in animal models by alleviating mitochondrial dysfunction and attenuating axonal disruption.1,2,8,11,14,15,28,43, 44,48,51–53,57,60,61 In humans, the pharmacokinetic profile of cyclosporine is variable and population dependent. Brain injury can lead to alterations in drug metabolism, protein binding, and clearance for many therapeutic agents.21,64 In patients with acute TBI, cyclosporine is cleared more rapidly and has a larger distribution volume than in non-TBI populations.12 The physiochemical properties of cyclosporine limit penetration into the CNS under normal physiological conditions, but the BBB is disrupted following TBI.4 The biphasic opening of the BBB following TBI affords a window of opportunity for cyclosporine to gain access to the injured brain. Defining the minimal effective systemic or central concentration is a key factor in optimizing this treatment strategy following TBI.
The complex metabolic changes following TBI combined with the variability of cyclosporine pharmacodynamics require prospective studies defining the dose-concentration relationship in this population prior to advancing this treatment to larger numbers of patients. In this clinical trial a dose-escalation design was used to systematically determine cyclosporine safety in a homogeneous population of patients with severe TBI. Findings from this investigation will be used to determine the optimal dosing strategy for future evaluations of cyclosporine safety and efficacy in this population. The study hypothesis was that clinically approved doses of cyclosporine would achieve measurable central and systemic concentrations and be safe when administered to patients with acute TBI.
Methods
Study Design and Population
This study was a prospective randomized, double-blind, placebo-controlled, dose-escalation trial of cyclosporine administration to 40 patients with acute severe nonpenetrating TBI who were admitted to the University of Kentucky Chandler Medical Center. Patients between 16 and 65 years of age with a GCS score between 4 and 8 and a motor score between 2 and 5 within 8 hours of injury were screened for eligibility. Eligibility criteria included the presence of 1 reactive pupil, positive CT findings, hemodynamic stability, and placement of an intraventricular catheter. The exclusion criteria included significant concomitant diseases, history of neurological disorder, renal dysfunction, immunosuppressive therapy, and participation in other investigational trials. The study was approved by the institutional review board, and informed consent was obtained prior to randomization.
Clinical Care
Patients were treated according to the American Association of Neurological Surgeons' Guidelines for the Management of Severe Traumatic Brain Injury.7 Intracranial pressure was monitored by means of an intraventricular catheter in all patients when clinically indicated. Steroids were not administered. All patients received nutritional support. Prophylactic anticonvulsants were not routinely administered. Cerebral hemodynamic goals included maintaining CPP > 60 mm Hg. Vasoactive drugs such as dopamine hydrochloride, dobutamine hydrochloride, phenylephrine, and norepinephrine bitartrate were used along with intraventricular drainage of CSF when ICP was > 20 mm Hg. Mannitol was used when CSF drainage failed to maintain CSF pressures < 20 mm Hg. Hypothermia and chronic hyperventilation to maintain PCO2 < 30 mm Hg were not used. Patients were monitored for adverse events reported in FDA labeling from other patient populations requiring chronic cyclosporine therapy. Definitions of adverse events were established prior to initiation of the study. These included CNS events, such as headache, tremor, seizures, and hallucinations, and systemic events, such as infection, hypertension, cardiovascular events, altered liver enzyme concentrations, ophthalmic changes, and kidney dysfunction. It was known that up to 25% of patients receiving cyclosporine for prevention of organ rejection experience CNS adverse events.
Study Procedures
Cyclosporine was prepared by the University of Kentucky's investigational drug pharmacy. Normal saline was the placebo used for this investigation. Within 8 hours of injury, each patient (10 patients/cohort) was randomly assigned to a treatment regimen and began to receive either placebo (2 patients/cohort) or cyclosporine (8 patients/cohort) intravenously based on the dosing scheme outlined in Table 1. Intermittent doses were infused intravenously over 2 hours (Cohorts I–III). Doses were selected based on FDA-approved labeling for intermittent administration of cyclosporine to transplant patient populations. Whole-blood cyclosporine concentrations were determined by means of HPLC or, for Cohort IV, by the University of Kentucky Hospital clinical laboratory using a validated HPLC-MS procedure. The continuous infusion dosing strategy was determined by modeling the pharmacokinetic profiles of Cohorts I–III.12 The Cohort IV loading dose/continuous infusion dosing strategy was determined by modeling the pharmacokinetic profiles of Cohorts I– III. The doses tested are shown in Table 1.
TABLE 1. Cyclosporine doses tested.
| Cohort | Dosing Schedule |
|---|---|
| I | 0.625 mg/kg/dose every 12 hrs for 72 hrs (6 doses) |
| II | 1.25 mg/kg/dose every 12 hrs for 72 hrs (6 doses) |
| III | 2.5 mg/kg/dose every 12 hrs for 72 hrs (6 doses) |
| IV | 2.5 mg/kg loading dose, then 5 mg/kg/day continuous infusion for 72 hrs |
Prior to the study, a safety response algorithm was generated. This algorithm was initiated whenever a cyclosporine concentration reached the “alert” threshold. Daily cyclosporine blood concentrations were obtained each morning for the first 7 days. Serial concentrations were also collected throughout the dosing period. Daily trough concentration values were reported to the study data manager and assessed for activation of the safety algorithm. In Cohort III, CSF samples were analyzed using HPLC to determine if cyclosporine could be detected in this matrix.
To maintain the blinding procedure, the safety monitoring for patient protection included “alert” status phone calls for both placebo- and cyclosporine-treated patients. Prior to the study, a computer-generated table of “false” cyclosporine concentrations was created for the 40 patients. Upon notification of a patient's cyclosporine concentration, the data manager reviewed the concentration table and compared the generated “false” value with the actual laboratory value to determine if the safety algorithm would be activated. The highest cyclosporine value, either generated or actual measured value, was the trigger for activation. The data manager communicated the concentration to an unblinded physician. When a qualifying concentration was met, other laboratory and clinical parameters were evaluated by this physician using the algorithm. A rise in serum creatinine > 2 mg/dl and seizure onset were each considered thresholds for intervention for patients participating in this trial.20
Safety Monitoring Procedures
Any serum cyclosporine value > 300 ng/ml in Cohorts I–III or 750 ng/ml in Cohort IV accompanied by a 50% increase in serum creatinine concentration resulted in the second dose for that day being withheld and a 50% reduction in dose for the next dosing day. Serum cyclosporine concentration was evaluated prior to the next dose, and if serum concentrations remained elevated, the subsequent dose was withheld until serum concentrations fell to the target concentration range. If > 72 hours was required for this decline, no additional doses were given. In the event of seizure, a serum cyclosporine level was obtained. If the value remained elevated, the next dose of study drug was withheld. Therapy was reinstituted if seizures resolved and serum cyclosporine concentrations were below the alert thresholds.
In addition to the daily cyclosporine concentrations, clinical safety parameters were followed. Vital signs, GCS score, and systemic and cerebrovascular hemodynamics were recorded hourly during the dosing phase. Daily laboratory monitoring included assessment of serum chemistry, triglyceride concentrations, and hematological and hepatic function parameters. Anergy panels were placed prior to the first dose and again in the 2nd week following the injury. Eye examinations were obtained during the dosing phase and were repeated prior to discharge and at 3 and 6 months after TBI. Adverse events were recorded up to the 6-month examination.
Safety Monitoring Board
An independent safety monitoring board was established by the National Institute of Neurological Disorders and Stroke. A report for each patient was generated and submitted to members of this board upon patient discharge from the hospital. Serious adverse events were defined as any life-threatening event, including refractory sustained increase in ICP to > 30 mm Hg, CPP < 50 mm Hg, sepsis, or death. Major organ dysfunction—as defined by a ≥ 50% decrease in creatinine clearance within the 72-hour treatment period, new-onset seizures refractory to anticonvulsant therapy, elevation of liver function test values at least 3 times above baseline, or refractory hypertension in the absence of vasopressor therapy—was also considered a serious adverse event
Each serious adverse event was evaluated for cause by the principal investigator, who remained blinded to treatment randomization. Causality was assigned as due to brain trauma, infection, systemic organ failure, or other or indeterminate. All reports to the Data Safety Monitoring Board (DSMB) were unblinded so adverse events could be evaluated for relationship to cyclosporine treatment. For each cohort, a comprehensive report summarizing all 10 patients in the cohort was provided to the DSMB at the completion of the 10th patient's discharge from the hospital.
Criteria for stopping treatment and criteria for dose escalation were developed. Unacceptable drug toxicity, maximum tolerated dose, death rate, and cause-of-death criteria were defined, along with the procedures to be followed if any of the thresholds were met. Assigning 10 patients per cohort enabled identification of toxicity levels of ≥ 33% and ≤ 5%. Statistical evaluation was provided in the cohort summary reports to the board. Advancing to the next dosing cohort was only initiated upon the board's review and approval of the previous cohort summary report.
Data Analysis
Descriptive statistics were used to summarize the responses and adverse events. Serum creatinine, anergy responses, and infection rates were compared between groups. Once the dose-finding studies were completed, mean responses for all dose levels that were considered safe to use were compared with the pooled data from the placebo patients using a linear mixed model. In this model the between-groups factor corresponded to dose while the within-groups factor corresponded to treatment Days 1–5.
At each of the follow-up periods (3 and 6 months) the proportion of patients with favorable outcomes (good and moderate disability on the GOS scale) were compared between placebo and cyclosporine by using a chi-square statistic. Adverse event rates were compared between doses and placebo using chi-square statistics or the Fisher exact test. Probability values of ≤ 0.05 were considered statistically significant throughout.
Results
Forty patients were enrolled in this dose escalation trial. Ten patients were enrolled into each dosing cohort with 8 patients randomly assigned to the cyclosporine study drug and 2 assigned to the placebo (Table 2). The cohorts were well matched. Placebo patients did not differ demographically from the cyclosporine-assigned patients.
TABLE 2. Summary of demographic data*.
| Cyclosporine Groups | |||||
|---|---|---|---|---|---|
| Variable | I | II | III | IV | Placebo Group |
| no. of patients | 8 | 8 | 8 | 8 | 8 |
| M | 7 | 7 | 7 | 3 | 8 |
| F | 1 | 1 | 1 | 5 | 0 |
| GCS score (± SD) | 6.5 ± 0.93 | 5.5 ± 1.5 | 5.9 ± 0.99 | 6.3 ± 1.4 | 6.0 ± 0.76 |
| 4 | 1 | 1 | 1 | 1 | 1 |
| 5–7 | 7 | 6 | 7 | 4 | 7 |
| 8 | 0 | 1 | 0 | 3 | 0 |
| age (± SD) | 29 ± 6.0 | 32 ± 14.6 | 23 ± 8.2 | 34 ± 14.8 | 6 ± 6.6 |
| Marshall CT classification† | |||||
| I | 0 | 0 | 0 | 0 | 0 |
| II | 1 | 2 | 1 | 0 | 1 |
| II + SAH | 1 | 3 | 3 | 4 | 2 |
| III | 0 | 0 | 0 | 1 | 0 |
| III + SAH | 2 | 0 | 1 | 0 | 0 |
| V | 2 | 2 | 0 | 1 | 4 |
| IV + SAH | 1 | 0 | 0 | 1 | 0 |
| SAH only | 0 | 1 | 1 | 0 | 0 |
| evacuated mass lesion | 1 | 0 | 1 | 0 | 0 |
| nonevcuated mass lesion + SAH | 0 | 0 | 1 | 1 | 1 |
Values represent numbers of patients unless otherwise indicated. Abbreviations: SAH = subarachnoid hemorrhage; SD = standard deviation.
As defined in Marshall et al., 1991.
The incidence of serious adverse events did not differ significantly between the cyclosporine and placebo groups (Fig. 1). One patient did not complete the dosing protocol due to death; elevated cyclosporine concentrations accompanied increases in hepatic enzymes and creatinine. The nonblinded physician investigator monitored this patient and applied the safety algorithm. None of the events were attributed to the study drug. There was no statistically significant difference in the mortality rate between cyclosporine-treated patients (6 [18.8%] of 32 patients) and placebo-treated patients (2 [25%] of 8 patients, p = 0.65). No effect of cyclosporine dose on mortality rate was detected (Fig. 2). The other serious adverse events were persistent or significant disability (2 patients), hospital readmissions or prolonged hospital stay (11 patients), and a diagnosis of testicular cancer reported by 1 patient during the 6 month follow-up period.
Fig. 1.
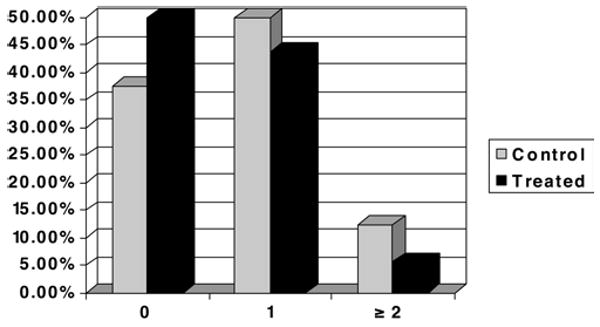
Graph showing the percentage of patients who experienced serious adverse events. The majority of the 32 cyclosporine-treated patients had no serious adverse event, and there was no significant difference in the incidence between the placebo controls and treated patients.
Fig. 2.
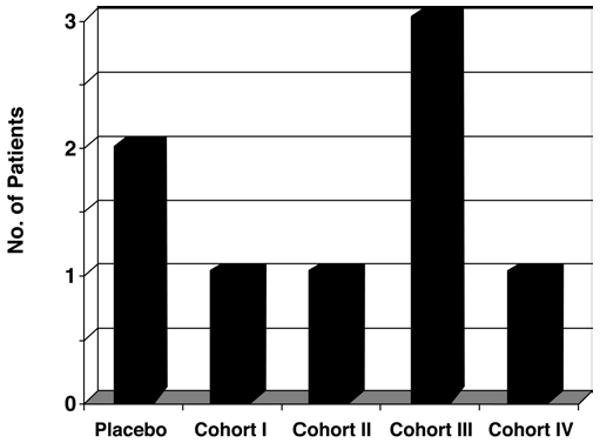
Graph showing the number of patient deaths in each group. There was no effect of cyclosporine dose on mortality end points. There were 6 deaths in the cyclosporine-treated group and 2 deaths in the placebo group.
All observed adverse events were recorded. No clinical safety concerns were identified in patients receiving cyclosporine at any of the doses tested. All patients had negative anergy panel reactions at baseline and upon repeat challenge at Day 14. No significant increase in risk of infection, renal dysfunction, or seizures was observed between cyclosporine dose levels or placebo-assigned patients. The most common infections were pneumonia, urinary tract infections, meningitis, and bacteremia (Fig. 3). Bilirubin levels increased during the first 14 days of hospitalization, but when they were evaluated during the dosing week and compared with baseline, no statistical differences were observed between the cyclosporine-treated patients and those who received the placebo treatment (Fig. 4). None of the patients experienced conjunctivitis, corneal deposits, or cortical blindness during the trial. Visual disturbances were present at both 3- and 6-month follow-ups, but the incidence was not different between patients who received the placebo and those who received cyclosporine.
Fig. 3.
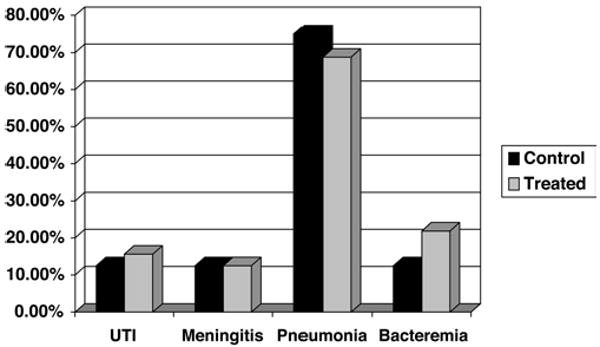
Graph showing the incidence of infections. No clinical safety concerns were observed in the cyclosporine-treated patients. The incidence of infectious complications did not differ between dosing groups or between cyclosporine-treated patients and placebo controls. UTI = urinary tract infection.
Fig. 4.
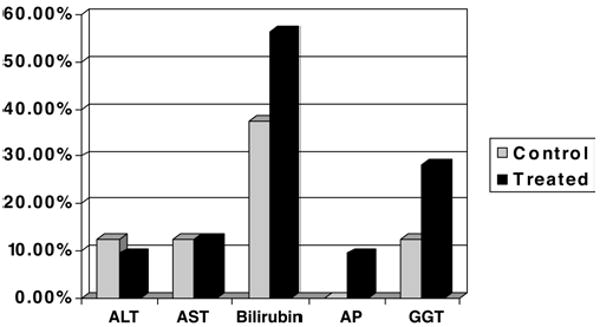
Graph showing the results of liver function tests. No significant effect of cyclosporine treatment on liver function was observed. During the 1st week after injury, rises in bilirubin were observed in both placebo controls and cyclosporine-treated patients. ALT = alanine transaminase; AP = alkaline phosphatase; AST = aspartate transaminase; GGT = gamma glutamyl transferase.
Although cyclosporine-treated patients did not experience statistically significant differences in safety end points compared with patients assigned to placebo treatment, there were some differences among the cyclosporine-treated patients in the different cohorts. The most frequent events in the lowest dosing cohorts were changes in serum chemistry parameters. In the patients who received 5 mg/kg/day (Cohorts III and IV), cerebral hemodynamic changes were more frequent than in patients who received the lower doses. These changes included a rise in ICP of ≥ 25% baseline (p = 0.04), CPP < 60 mm Hg (p = 0.02), and the combination of these events (p = 0.0003). Other events reaching statistical significance were bacteremia, decerebrate posturing, rash, decreased numbers of eosinophils, and urinary tract infections. There was, however, no statistically significant difference between the placebo and cyclosporine groups with respect to these parameters or other CNS safety end points (Table 3).
TABLE 3. Central nervous system adverse events during the dosing period in 32 patients receiving cyclosporine and 8 receiving placebo*.
| Event | Cyclosporine Groups | Placebo Group | p Value |
|---|---|---|---|
| cerebral edema | 0 | 1 | 0.05 |
| intracranial hypertension | 21 | 6 | 0.45 |
| decerebrate posturing | 3 | 1 | 0.79 |
| decreased CPP | 28 | 7 | 0.89 |
| seizures | 2 | 0 | 0.47 |
Values represent numbers of patients unless otherwise indicated. Definitions for all adverse events were agreed upon prior to initiation of the protocol in any patient. The events recorded in this table met the definition at any time point following the first dose of the study drug. Cerebral edema was defined as a new onset of fluid accumulation in the brain tissue; intracranial hypertension was defined as a clinically significant elevation of ICP that disrupts autoregulation (usually > 20 mm Hg; > 40 mm Hg sustained was defined as severe elevation); decerebrate posturing was defined as decerebrate rigidity with the extremities stiff and extended; decreased CPP was defined as any recorded value < 60 mm Hg; seizures were defined as sudden, involuntary contractions accompanied by electroencephalography changes.
Daily trough cyclosporine values were monitored for Cohorts I–III (Table 4). Trough values were < 250 ng/ml in all patients who received cyclosporine in each of these cohorts. In Cohort III, cyclosporine troughs were still within the therapeutic range (100–200 ng/ml) 12 hours following the sixth dose of 2.5 mg/kg but fell to < 40 ng/ml within 24 hours. The presence of cyclosporine in the CSF matrix was determined using HPLC. This assay has not been validated using CSF, so pharmacokinetic modeling was not possible. Despite these limitations, cyclosporine was detectable in CSF samples obtained from Cohort III during the 72-hour dosing interval and up to 6 days postinjury. Maximum blood concentrations achieved during cyclosporine treatment were not routinely monitored; they were, however, estimated using a computer-generated model (Fig. 5). Predicted maximum concentrations in the intermittent dosing groups occurred just following the sixth dose and were 398 ± 159 ng/ml, 645 ± 228 ng/ml, and 949 ± 640 ng/ml for Cohorts I, II, and III, respectively.
TABLE 4. Trough concentrations of cyclosporine (ng/ml) in intermittent dosing cohorts*.
| Time Point (hrs) | ||||
|---|---|---|---|---|
| Cohort | 12 | 24 | 48 | 72 |
| I | 38 ± 14.7 | 34 ± 5.8 | 54 ± 13.4 | 82 ± 49 |
| II | 44 ± 18.4 | 66 ± 23 | 94 ± 27.2 | 116 ± 39.9 |
| III | 119 ± 61.0 | 154 ± 48.2 | 169 ± 30.6 | 193 ± 28.6 |
Values represent means ± SDs.
Fig. 5.
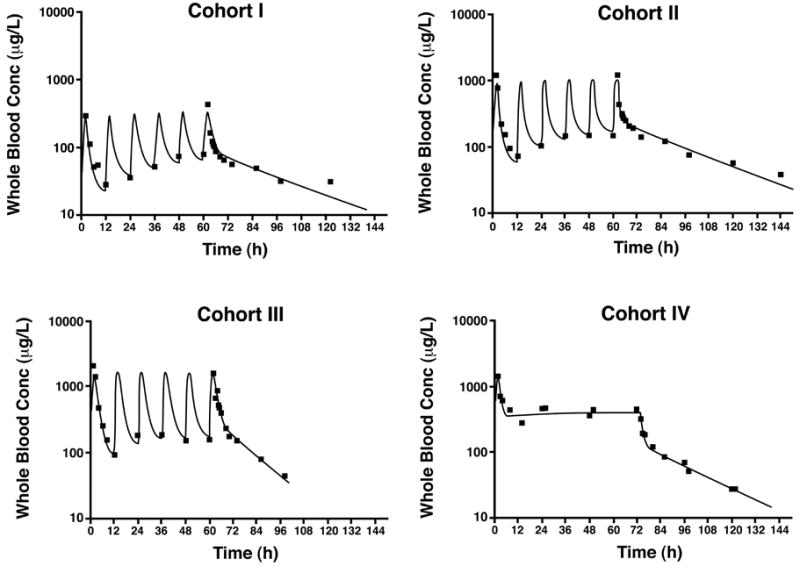
Graphs of pharmacokinetic modeling. Observed (squares) and predicted (lines) cyclosporine whole blood concentrations in a representative patient at each dosing level. (See Table 1 for the cyclosporine doses.) Conc = Concentration.
In Cohort IV, the maximum cyclosporine concentration was predicted to occur just following the loading dose; the predicted maximum was 1636 ± 569 ng/mL. The mean predicted concentration at the end of the 72-hour infusion was 461 ± 118 ng/mL in this cohort, and the mean concentration was predicted to remain within the therapeutic range at least 4 hours following cessation of the infusion.
Outcome
Functional outcome was not a primary objective of this study; nevertheless, 3- and 6-month examinations were conducted for long-term safety observations in surviving patients. No statistical difference in GOS or GOSE was observed between the placebo- and cyclosporine-treated patients (Fig. 6). Outcome scores in 7 (35%) of 20 cyclosporine-treated patients improved from poor to good at the 6-month assessment compared with no improvement in the placebo-treated patients (improvement in 0 of 6 patients, p = 0.15). While these early results are encouraging in this small sample, they did not meet the conventional definition of a statistical “trend” which is customarily defined as p < 0.10. The probability of a favorable outcome varied by cyclosporine dose with the continuous infusion protocol generating the most improved scores (p < 0.05) (Fig. 7).
Fig. 6.
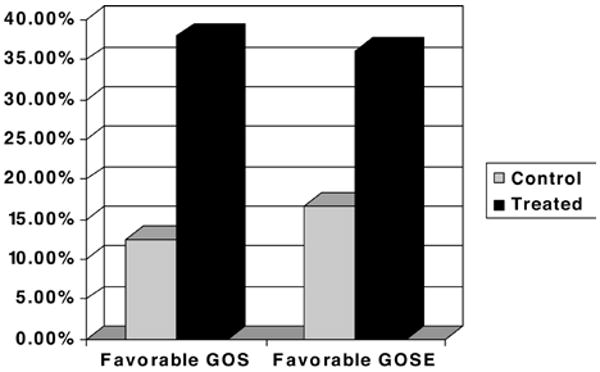
Graph showing the percentages of cyclosporine-treated and control patients with favorable GOS and GOSE scores. Data were examined for trends associated with cyclosporine treatment effect on functional outcome at 3 and 6 months following injury. There was no statistically significant difference in GOS scores between the surviving patients treated with cyclosporine and placebo controls.
Fig. 7.
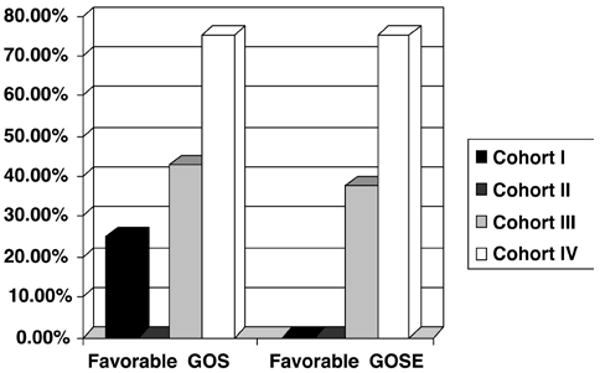
Graph showing percentages of patients with favorable GOS and GOSE scores stratified by cohort. Data were examined for functional outcome trends in response to differing dosing strata for cyclosporine. Increasing doses showed a probability of favorable outcome. The patients in Cohort IV, who were treated with the continuous infusion dosing paradigm, had the most improved scores (p < 0.05).
Discussion
There was no difference in mortality or adverse events when cyclosporine was administered to patients with acute severe TBI beginning within 8 hours of injury. The effects of cyclosporine on immune response in the acute TBI patient were not clinically significant in this trial. Of the doses investigated, the optimal cyclosporine dosing regimen for future evaluations of neuroprotective potential was a 2.5 mg/kg loading dose followed by a continuous infusion of 5 mg/kg/day. The 72-hour treatment duration did not generate safety concerns and is a reasonable starting point for examination of CNS penetration and efficacy outcomes. Recognizing the safety profile and challenging dosing paradigms associated with cyclosporine, we attempted to follow conservative strategies and extensive follow-up in our trial. The small number of patients prevents full extrapolation of these preliminary safety and efficacy findings until larger Phase III investigations can be completed. Further development and validation of an analytical method for quantifying cyclosporine in the CSF matrix is essential for assessment of pharmacokinetic parameters within the CNS.
The amount of drug reaching the injured brain is an important consideration in clinical trial design.30,32,34,37,42 Traumatic brain injury may alter BBB permeability, providing a window of dosing opportunity.4,55 Cyclosporine penetration of the BBB is critical for neuroprotective effects,6,8,11,14,28,44,48,51–53,57,60,61,65,66,68 and the drug was detectable in CSF from patients in whom treatment with a 5 mg/kg/day intermittent dose was initiated within 8 hours of injury. Animal models indicate that the window for therapeutic intervention is at least 1 hour and may even be as long as 24 hours.57 Systemic cyclosporine administration before or after cerebral contusion in animals with TBI significantly reduces lesion size. Postinjury administration of cyclosporine resulted in a 40% reduction in lesion volume.11,61 In animal models, a 74% reduction in lesion volume was observed with the higher continuous infusion dose.60 These findings suggest that continuous exposure to cyclosporine during a dosing period increases neuroprotection, overcoming time limitations for BBB penetration. The continuous infusion regimen used in our protocol was well tolerated with encouraging trends in outcome. Findings in both animal and human TBI suggest that this is the optimal dosing strategy for future clinical trials of cyclosporine.
The cellular mechanisms of secondary injury associated with TBI involve a number of endogenous mediators. Many agents have been evaluated in clinical trials over the past decade, but they failed to demonstrate significant benefit in overall morbidity or mortality rates.17–19,30,32,42 Neuroprotection by cyclosporine has been demonstrated in a variety of models, and a number of potential mechanisms have been described.43,44,46,53 Effects of cyclosporine on mitochondrial function and axonal disruption continue to be explored. The drug may protect against secondary neuronal death by preventing Ca efflux via inhibition of mitochondrial permeability transition pores.58,59,61
Mitochondria play an important role in the maintenance of intracellular calcium homeostasis by sequestering Ca++. The mitochondria function to buffer intracellular Ca and protect against a high level of cytosolic Ca++. Calcium enters the mitochondria by a low-capacitor antiporter or an electronic uniporter. Mitochondria pump Ca out when the cytosolic levels of Ca++ are high. The massive influx of Ca following glutamate activation of the N-methyl-d-aspartate receptor causes secondary neuronal injury. Mitochondria protect against this excitotoxic injury by accumulating Ca when exposed to glutamate. Excessive accumulation of Ca, however, causes hyperpolarization and opening of the mitochondria permeability transition pore to Ca. This efflux of Ca may then potentiate the secondary biochemical cascade leading to neuronal death. Although cyclosporine may protect by blocking Ca efflux, it has also shown other effects on mitochondria. Signoretti et al.53 described a significant restoration of adenosine triphosphate along with diminution of N-acetylaspartate reduction with cyclosporine therapy, illustrating attenuation of mitochondrial dysfunction. The preservation of mitochondrial function by cyclosporine is perhaps only 1 of several mechanisms underlying cyclosporine's neuroprotective effect.1,2,8,46,67,68
The complex central and metabolic events accompanying TBI make identification of a pharmacodynamic end point, a surrogate marker, a rationale consideration for evaluating drug response.35,36,54 The severity of brain injury appears to affect endogenous protein concentrations following injury.62 Identifying surrogate markers of cyclosporine effects remains critical for defining the optimal dose response profile in TBI patients. Several potential surrogate markers were explored during the course of this trial and remain under investigation. Clinical trials of possible treatment strategies continue, and alternative study designs are under consideration, including recommendations for the use of surrogate markers for drug response.13,31,37,38,41
Conclusions
Identifying a therapeutic strategy to mitigate the cognitive and physical impairments associated with TBI is essential to address the personal and societal impact of this condition. Our findings show that clinically approved doses of cyclosporine can be safely administered to patients with TBI. The optimal dose in this investigation was 2.5 mg/kg administered over 2 hours followed by a continuous infusion of 5 mg/kg/day for 72 hours. Biomarkers collected from patients with TBI will help define the mechanisms responsible for neuroprotective actions of cyclosporine and may be useful surrogates for predicting drug response. Although significant adverse events were not observed in this Phase II trial, future Phase III investigations with larger numbers of patients will be needed to fully define the role of cyclosporine as a potential treatment for acute TBI.
Acknowledgments
We thank George Davis, Pharm.D., Max Yau, M.S., Loran Karslosky, Sylvia Nicholson, Terri Cheak, R.N., Tina Brooks, R.N., Thomas Wade, M.D., Milford Geralds, Ph.D., Brenda Fahey, M.D., Aaron Cook, Pharm.D., Nurdan Shafaghi, R.N., and others from the neurosurgery clinical research staff, the Neurosurgery Intensive Care Unit nurses, neurosurgery house staff officers, and attending physicians.
Disclosure: This work was supported by National Institutes of Health Grant No. R01 NS41239-02, General Clinical Research Center US Public Health Service Grant No. M01RR02602, and Kentucky Spinal Cord and Head Injury Research Trust Grant No. 1R01NS 41239-01 (all to Drs. Young and Hatton).
Abbreviations used in this paper
- BBB
blood–brain barrier
- CNS
central nervous system
- CPP
cerebral perfusion pressure
- CSF
cerebrospinal fluid
- FDA
US Food and Drug Administration
- GCS
Glasgow Coma Scale
- GOS
Glasgow Outcome Scale
- GOSE
Extended Glasgow Outcome Scale
- HPLC
high-performance liquid chromatography
- ICP
intracranial pressure
- MS
mass spectrometry
- TBI
traumatic brain injury
Footnotes
Disclaimer: The authors do not report any conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
References
- 1.Albensi BC, Sullivan PG, Thompson MB, Scheff SW, Mattson MP. Cyclosporin ameliorates traumatic brain-injury induced alterations of hippocampal synaptic plasticity. Exp Neurol. 2000;162:385–389. doi: 10.1006/exnr.1999.7338. [DOI] [PubMed] [Google Scholar]
- 2.Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- 3.Avramut M, Achim CL. Immunophilins in nervous system degeneration and regeneration. Curr Top Med Chem. 2003;3:1376–1382. doi: 10.2174/1568026033451871. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin SA, Fugaccia I, Brown DR, Brown LV, Scheff SW. Blood-brain barrier breach following cortical contusion in the rat. J Neurosurg. 1996;85:476–481. doi: 10.3171/jns.1996.85.3.0476. [DOI] [PubMed] [Google Scholar]
- 5.Binder S, Corrigan JD, Langlois JA. The public health approach to traumatic brain injury: an overview of CDC's research and programs. J Head Trauma Rehabil. 2005;20:189–195. doi: 10.1097/00001199-200505000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Borlongan CV, Emerich DF, Hoffer BJ, Bartus RT. Bradykinin receptor agonist facilitates low-dose cyclosporin-A protection against 6-hydroxydopamine neurotoxicity. Brain Res. 2002;956:211–220. doi: 10.1016/s0006-8993(02)03474-1. [DOI] [PubMed] [Google Scholar]
- 7.Brain Trauma Foundation. The American Association of Neurological Surgeons, Congress of Neurological Surgeons, AANS/CNS Joint Section on Neurotrauma and Critical Care: Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl):S1–S95. [Google Scholar]
- 8.Büki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- 9.Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: lessons from animal models and clinical studies. Neurosurgery. 1999;45:207–219. doi: 10.1097/00006123-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domañska-Janik K, Buzañska L, Dluzniewska J, Kozlowska H, Sarnowska A, Zablocka B. Neuroprotection by cyclosporin A following transient brain ischemia correlates with the inhibition of the early efflux of cytochrome C to cytoplasm. Brain Res Mol Brain Res. 2004;121:50–59. doi: 10.1016/j.molbrainres.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Empey PE, McNamara PJ, Young B, Rosbolt MB, Hatton J. Cyclosporin A disposition following acute traumatic brain injury. J Neurotrauma. 2006;23:109–116. doi: 10.1089/neu.2006.23.109. [DOI] [PubMed] [Google Scholar]
- 13.Farin A, Marshall LF. Lessons from epidemiologic studies in clinical trials of traumatic brain injury. Acta Neurochir (Wien) 2004;89:101–107. doi: 10.1007/978-3-7091-0603-7_14. [DOI] [PubMed] [Google Scholar]
- 14.Folbergrova J, Li PA, Uchino H, Smith ML, Siesjö BK. Changes in the bioenergetic state of rat hippocampus during 2.5 min of ischemia, and prevention of cell damage by cyclosporin A in hyperglycemic subjects. Exp Brain Res. 1997;114:44–50. doi: 10.1007/pl00005622. [DOI] [PubMed] [Google Scholar]
- 15.Gogarten W, Van Aken H, Moskopp D, Roos N, Schleef M, Marcus M, et al. A case of severe cerebral trauma in a patient under chronic treatment with cyclosporine A. J Neurosurg Anesthesiol. 1998;10:101–105. doi: 10.1097/00008506-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- 17.Hatton J. Pharmacological treatment of traumatic brain injury: a review of agents in development. CNS Drugs. 2001;15:553–581. doi: 10.2165/00023210-200115070-00005. [DOI] [PubMed] [Google Scholar]
- 18.Hatton J, Kryscio R, Ryan M, Ott L, Young B. Systemic metabolic effects of combined insulin-like growth factor-I and growth hormone therapy in patients who have sustained acute traumatic brain injury. J Neurosurg. 2006;105:843–852. doi: 10.3171/jns.2006.105.6.843. [DOI] [PubMed] [Google Scholar]
- 19.Hatton J, Rapp RP, Kudsk K, Brown RO, Luer MS, Bukar JG, et al. Intravenous insulin-like growth factor I (IGF-I) in moderate to severe head injury: a phase II safety and efficacy trial. J Neurosurg. 1997;86:779–786. doi: 10.3171/jns.1997.86.5.0779. [DOI] [PubMed] [Google Scholar]
- 20.Kagawa Y, Sawada J, Yamada S, Matsuda H, Kageyama S, Masuya M, et al. Relationship between development of nephrotoxicity and blood concentration of cyclosporine A in bone-marrow transplanted recipients who received the continuous intravenous infusion. Biol Pharm Bull. 2003;26:1115–1119. doi: 10.1248/bpb.26.1115. [DOI] [PubMed] [Google Scholar]
- 21.Kalsotra A, Turman CM, Dash PK, Strobel HW. Differential effects of traumatic brain injury on the cytochrome p450 system: a perspective into hepatic and renal drug metabolism. J Neurotrauma. 2003;20:1339–1350. doi: 10.1089/089771503322686139. [DOI] [PubMed] [Google Scholar]
- 22.Kaminska B, Gaweda-Walerych K, Zawadzka M. Molecular mechanisms of neuroprotective action of immunosuppressants–facts and hypotheses. J Cell Mol Med. 2004;8:45–58. doi: 10.1111/j.1582-4934.2004.tb00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kristal BS, Stavrovskaya IG, Narayanan MV, Krasnikov BF, Brown AM, Beal MF, et al. The mitochondrial permeability transition as a target for neuroprotection. J Bioenerg Biomembr. 2004;36:309–312. doi: 10.1023/B:JOBB.0000041759.35731.70. [DOI] [PubMed] [Google Scholar]
- 24.Langlois JA, Rutland-Brown W, Thomas KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Atlanta: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2006. [Google Scholar]
- 25.Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. 2001;24:169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- 26.Lenzlinger PM, Saatman KE, Hoover RC, Cheney JA, Bareyre FM, Raghupathi R, et al. Inhibition of vascular endothelial growth factor receptor (VEGFR) signalizing by BSF476921 attenuates regional cerebral edema following traumatic brain injury in rats. Restor Neurol Neurosci. 2004;22:73–79. [PubMed] [Google Scholar]
- 27.Lewén A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 28.Li PA, Uchino H, Elmer E, Siesjö BK. Amelioration by cyclosporin A of brain damage following 5 to 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res. 1997;753:133–140. doi: 10.1016/s0006-8993(97)00005-x. [DOI] [PubMed] [Google Scholar]
- 29.Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion. 2004;4:705–713. doi: 10.1016/j.mito.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 30.Maas AI, Marmarou A, Murray GD, Steyerberg EW. Clinical trials in traumatic brain injury: current problems and future solution. Acta Neurochir Suppl. 2004;89:113–118. doi: 10.1007/978-3-7091-0603-7_16. [DOI] [PubMed] [Google Scholar]
- 31.Maas AI, Murray G, Henney H, III, Kassem N, Legrand V, Mangelus M, et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006;5:38–45. doi: 10.1016/S1474-4422(05)70253-2. [DOI] [PubMed] [Google Scholar]
- 32.Maas AI, Steyerberg EW, Murray GD, Bullock R, Baethmann A, Marshall LF, et al. Why have recent trials of neuroprotective agents in head injury failed to show convincing efficacy? A pragmatic analysis and theoretical considerations. Neurosurgery. 1999;44:1286–1298. [PubMed] [Google Scholar]
- 33.MacGregor DG, Avshalumov MV, Rice ME. Brain edema induced by in vitro ischemia: causal factors and neuroprotection. J Neurochem. 2003;85:1402–1411. doi: 10.1046/j.1471-4159.2003.01772.x. [DOI] [PubMed] [Google Scholar]
- 34.Machado SG, Murray GD, Teasdale GM. Evaluation of designs for clinical trials of neuroprotective agents in head injury. J Neurotrauma. 1999;16:1131–1138. doi: 10.1089/neu.1999.16.1131. [DOI] [PubMed] [Google Scholar]
- 35.Marciano P, Eberwine JH, Raghupathi R, McIntosh TK. The assessment of genomic alterations using DNA arrays following traumatic brain injury: a review. Restor Neurol Neurosci. 2001;18:105–113. [PubMed] [Google Scholar]
- 36.Marciano PG, Eberwine JH, Ragupathi R, Saatman KE, Meaney DF, McIntosh TK. Expression profiling following traumatic brain injury: a review. Neurochem Res. 2002;27:1147–1155. doi: 10.1023/a:1020973308941. [DOI] [PubMed] [Google Scholar]
- 37.Marklund N, Bakshi A, Castelbuono DJ, Conte V, McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12:1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 38.Marmarou A, Guy M, Murphey L, Roy F, Layani L, Combal JP, et al. A single dose, three-arm, placebo-controlled, phase I study of the bradykinin B2 receptor antagonist Anatibant (LF16-0687Ms) in patients with severe traumatic brain injury. J Neurotrauma. 2005;22:1444–1455. doi: 10.1089/neu.2005.22.1444. [DOI] [PubMed] [Google Scholar]
- 39.Marr A, Coronado V. Central Nervous System Injury Surveillance: Annual Data Submission Standards for the Year 2002. Atlanta: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2001. [Google Scholar]
- 40.Marshall LF, Marshall SB, Klauber MR, van Berkum Clark M, Eisenberg HM, Jane JA, et al. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75(Suppl):S14–S20. [Google Scholar]
- 41.Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, et al. Neuroscience experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 42.Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okonkwo DO, Melon DE, Pellicane AJ, Mutlu LK, Rubin DG, Stone JR, et al. Dose-response of cyclosporin A in attenuating traumatic axonal injury in rat. Neuroreport. 2003;14:463–466. doi: 10.1097/00001756-200303030-00033. [DOI] [PubMed] [Google Scholar]
- 44.Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporine A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Pratico D, Reiss P, Tang LX, Sung S, Rokach J, McIntosh TK. Local and systemic increase in lipid peroxidation after moderate experimental traumatic brain injury. J Neurochem. 2002;80:894–898. doi: 10.1046/j.0022-3042.2002.00777.x. [DOI] [PubMed] [Google Scholar]
- 46.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17:927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- 48.Riess P, Bareyre FM, Saatman KE, Cheney JA, Lifshitz J, Raghupathi R, et al. Effects of chronic, post-injury cyclosporin A administration on motor and sensorimotor function following severe, experimental traumatic brain injury. Restor Neurol Neurosci. 2001;18:1–8. [PubMed] [Google Scholar]
- 49.Royo NC, Shimizu S, Schouiten JW, Stover JF, McIntosh TK. Pharmacology of traumatic brain injury. Curr Opin Pharmacol. 2003;3:27–32. doi: 10.1016/s1471-4892(02)00006-1. [DOI] [PubMed] [Google Scholar]
- 50.Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Curr Pharm Des. 2001;7:1475–1503. doi: 10.2174/1381612013397311. [DOI] [PubMed] [Google Scholar]
- 51.Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- 52.Shiga Y, Onodera H, Matsuo Y, Kogure K. Cyclosporin A protects against ischemia-reperfusion injury in the brain. Brain Res. 1992;595:145–148. doi: 10.1016/0006-8993(92)91465-q. [DOI] [PubMed] [Google Scholar]
- 53.Signoretti S, Marmarou A, Tavazzi B, Dunbar J, Amorini AM, Lazzarino G, et al. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J Neurotrauma. 2004;21:1154–1167. doi: 10.1089/neu.2004.21.1154. [DOI] [PubMed] [Google Scholar]
- 54.Siman R, McIntosh TK, Soltesz FM, Chen Z, Neumar RW, Roberts VL. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol Dis. 2004;16:311–320. doi: 10.1016/j.nbd.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 55.Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- 56.Sullivan PG, Keller JN, Bussen WL, Scheff SW. Cytochrome C release and caspase activation after traumatic brain injury. Brain Res. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- 57.Sullivan PG, Rabchevsky AG, Hicks RR, Gibson TR, Fletcher-Turner A, Scheff SW. Dose-response curve and optimal dosing regimen of cyclosporine A after traumatic brain injury in rats. Neuroscience. 2000;101:289–295. doi: 10.1016/s0306-4522(00)00380-8. [DOI] [PubMed] [Google Scholar]
- 58.Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transitions in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan PG, Springer JE, Hall ED, Scheff SW. Mitochondrial uncoupling as a therapeutic target following neuronal injury. J Bioenerg Biomembr. 2004;36:353–356. doi: 10.1023/B:JOBB.0000041767.30992.19. [DOI] [PubMed] [Google Scholar]
- 60.Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol. 2000;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- 61.Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- 62.Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 63.Thurman DJ, Alverson C, Browne D, Dunn KA, Guerrero J, Johnson R, et al. Traumatic Brain Injury in the United States: A Report to Congress. December 1999. Atlanta: Centers for Disease Control and Prevention, National Center for In jury Prevention and Control; 1999. [Google Scholar]
- 64.Toler SM, Young AB, McClain CJ, Shedlofsky SI, Bandyopadhyay AM, Blouin RA. Head injury and cytochrome P-450 enzymes. Differential effect on mRNA and protein expression in the Fischer-344 rat. Drug Metab Dispos. 1993;21:1064–1069. [PubMed] [Google Scholar]
- 65.Uchino H, Ishii N, Shibasaki F. Calcineurin and cyclophilin D are differential targets of neuroprotection by immunosuppressants CsA and FK506 in ischemic brain damage. Acta Neurochir Suppl. 2003;86:105–111. doi: 10.1007/978-3-7091-0651-8_24. [DOI] [PubMed] [Google Scholar]
- 66.Uchino H, Minamikawa-Tachino R, Kristián T, Perkins G, Narazaki M, Siesjö BK, et al. Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol Dis. 2002;10:219–233. doi: 10.1006/nbdi.2002.0514. [DOI] [PubMed] [Google Scholar]
- 67.Van Den Heuvel C, Donkin JJ, Finnie JW, Blumbergs PC, Kuchel T, Koszyca B, et al. Downregulation of amyloid precursor protein (APP) expression following post-traumatic cyclosporine-A administration. J Neurotrauma. 2004;21:1562–1572. doi: 10.1089/neu.2004.21.1562. [DOI] [PubMed] [Google Scholar]
- 68.Zieminska E, Matyja E, Nalecz M, Salinska E, Ziembowicz A, Lazarewicz JW. In vivo brain microdialysis as a tool in studies of neuroprotective effects of cyclosporin A in acute excitotoxicity. Acta Pol Pharm. 2000;57(Suppl):129–133. [PubMed] [Google Scholar]