

Abstract
Purpose
Age-related degradation of the elastic lamina in Bruch's membrane may have a permissive effect on the growth of choroidal neovascularization (CNV). This study investigated the influence of defective elastic fiber maintenance in the development of laser-induced CNV.
Methods
A mouse lacking lysyl oxidase-like (LOXL)-1, an enzyme essential for elastin polymerization, was studied. The morphologic characteristics of the elastic lamina within Bruch's membrane were examined in mutant and wild-type (WT) eyes. Laser-induced CNV was evaluated by fluorescein angiography and choroidal flat mounts. Immunohistochemistry for elastin was performed on the CNV lesions, and vascular endothelial growth factor (VEGF) levels were determined by ELISA. Soluble elastin and matrix metalloproteinase (MMPs) levels were also analyzed by immunoblotting.
Results
The elastic lamina of Bruch's membrane in the LOXL1-deficient mice was fragmented and less continuous than in the WT controls. The mutant mice showed increased levels of soluble elastin peptides and reduced elastin polymer deposition in neovascular membranes. Significantly larger CNV with greater leakage on fluorescein angiography developed in mutant mice. VEGF levels in the RPE/choroid were higher in the knockout mice on days 7 and 14 after laser (P < 0.05). MT1-MMP (MMP14) was also elevated after laser in the LOXL1 mutant eyes compared to the WT controls.
Conclusions
These results show that a systemic defect in elastic fiber deposition affects Bruch's membrane integrity and leads to more aggressive CNV growth. The latter may be partially mediated by abnormal signaling from the accumulation of soluble elastin peptides.
Choroidal neovascularization (CNV) is the predominant cause of severe visual loss in age-related macular degeneration (AMD) and other macular diseases. Bruch's membrane integrity is known to be compromised in many conditions characterized by choroidal neovascularization, such as traumatic choroidal rupture, angioid streaks, and myopia. Additionally, laser rupture of Bruch's membrane is a well-established means of inducing experimental choroidal neovascularization. Such evidence indicates that Bruch's membrane serves as an important barrier to CNV formation, but the structural characteristics important in this barrier function are still under investigation.
Bruch's membrane lies between the neural retina and the choriocapillaris. There are five layers to this structure: the basement membrane of the retinal pigment epithelium, an inner collagenous layer, the central elastic lamina composed of elastic fibers, the outer collagenous layer, and the basement membrane of the choriocapillaris endothelium. Numerous morphologic changes in Bruch's membrane have been described with aging, particularly the development of drusen and basal laminar deposits, but also calcification, changes in thickness, and decreases in hydraulic conductivity.1 Loss of elastic fibers is a hallmark of connective tissue aging, and the central elastic lamina of Bruch's membrane has long been postulated to have a barrier function against CNV.2,3 Recently, Chong et al.4 observed decreases in thickness and integrity of the elastic layer of Bruch's membrane in the maculae of eyes with AMD compared to normal age-matched controls. Similar changes were also noted between macular and extramacular regions. The association of mutations in fibulin-5, a protein involved in elastin polymerization, with AMD also points to the importance of the elastin component of Bruch's as a barrier to choroidal neovascularization.5
Elastic fibers are amorphous polymers composed of the protein elastin, known as tropoelastin in its monomeric form. Oxidative deamination of lysine residues, an initial step of elastin polymerization, requires lysyl oxidases.6 Lysyl oxidase-like (LOXL) protein 1 has been shown to guide the spatially defined deposition of elastin and is essential for the maintenance of elastic fibers.7 LOXL1-deficient mice develop multiple systemic defects including pelvic organ prolapse, emphysematous changes in the lungs, and vascular abnormalities attributable to a failure in elastic fiber maintenance.7–9 To explore the relationship between elastic fiber maintenance and the development of CNV, we chose to study laser-induced CNV in this mutant model.
Methods
Animals
All animal experiments followed the guidelines of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Animal Care Committee of Massachusetts Eye and Ear Infirmary. Homozygous LOXL1-mutant mice were generated by gene targeting as previously described.7 Genotypes were confirmed by PCR of genomic DNA extracted from tail snips. Wild-type (WT) C57BL/6J mice, which served as controls, were purchased from Jackson Laboratories (Bar Harbor, ME). Mice of either sex between 3 and 6 months of age were used. For all procedures, anesthesia was achieved by intramuscular injection of 50 mg/kg ketamine hydrochloride (Phoenix Pharmaceutical, Inc., St. Joseph, MO) and 10 mg/kg xylazine (Phoenix Pharmaceutical, Inc.), and pupils were dilated with topical 0.5% tropicamide (Alcon, Humacao, Puerto Rico).
Induction of CNV
Laser photocoagulation (532 nm, 0.05 second, 50 μm; Oculight GLx, IRIDEX Corp., Mountain View, CA) was performed on both eyes of each animal (n = 10 animals per group) by an operator masked to its genetic identity. Power of 200 or 400 mW was randomly chosen between the right and left eyes. Four laser spots were applied in a peripapillary distribution in a standardized fashion, approximately one to two disc diameters from the optic nerve, using a slit lamp delivery system and a coverslip as a contact lens. The appearance of a cavitation bubble, a sign thought to correlate with the disruption of Bruch's membrane, was recorded. Those spots with hemorrhagic complications were excluded from further evaluation. For measurement of vascular endothelial growth factor (VEGF) and morphologic analysis, both eyes of each animal in another group were treated with a laser power of 400 mW.
Fluorescein Angiography
Fluorescein angiography was performed by an operator masked to the genetic identity of the animal, with a commercial camera and imaging system (TRC 50 VT camera and IMAGEnet 1.53 system; Topcon, Paramus, NJ), at 3 days and at 1 and 2 weeks after laser photocoagulation. Photographs were captured with a 20 D lens in contact with the fundus camera lens after intraperitoneal injection of 0.1 mL of 2% fluorescein sodium (Akorn, Decatur, IL). Two masked retina specialists not involved in laser photocoagulation or angiography evaluated the 2-week post-laser fluorescein angiograms at a single sitting. Lesions were graded using a previously established scheme as follows: 0 (not leaky), faint hyperfluorescence or mottled fluorescence without leakage; 1 (questionable leakage), hyperfluorescent lesion without progressive increase in size or intensity; 2A (leaky), hyperfluorescence increasing in intensity but not in size; 2B (pathologically significant leakage), hyperfluorescence increasing in intensity and in size.
FITC-Dextran Perfusion and CNV Size Measurement
The size and vascularity of CNV lesions were measured in choroidal flat mounts (n = 7 eyes per group). On day 14, mice were anesthetized and perfused with 5 mL phosphate-buffered saline containing 50 mg/mL fluorescein-labeled dextran (FITC-dextran; MW 2 × 106, Sigma-Aldrich, St. Louis, MO). The eyes were removed and fixed for 1 hour in 4% phosphate-buffered paraformaldehyde. The cornea and lens were removed and the entire retina was carefully dissected from the eyecup. Radial cuts (four to six) were made from the edge to the equator, and the eyecup was flat mounted in Vectashield (Vector Laboratories, Burlingame, CA) with the sclera facing down. Flat mounts were examined by fluorescence microscopy, and images were digitized using a CCD video camera. Modular imaging software (Openlab; Improvision Inc, Waltham, MA) was used, by an operator masked with respect to treatment group, to measure the total area of choroidal neovascularization associated with each burn. CNV lesions were identified as fluorescent blood vessels on the choroidal/retinal interface circumscribed by a region lacking fluorescence. In addition, the relative vascularity of CNV was measured by calculating the area of pixels brighter than background within the CNV lesion and dividing this area by the total CNV size.
Immunogold Labeling and Transmission Electron Microscopy
Mouse eyes (n = 3 per group) without laser treatment were fixed in 2% formaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer for 1 hour. Retinas were dissected out, soaked in 30% sucrose in PBS overnight, and frozen in liquid nitrogen. Thin (70-nm) sections were cut on a Leica (Wetzlar, Germany) cryo-ultramicrotome and collected on Formvar-coated nickel grids. Grids were incubated sequentially with 0.15 M glycine/PBS and Tween-Tris buffered saline (TTBS) supplemented with 100 mM 2-mercaptoethanol and were blocked in 5% goat serum and 1% fish gelatin in TTBS. Incubation with the primary antibody (Elastin PR385; Elastin Products Company, Owensville, MO) diluted at 1:1000 in the blocking solution proceeded overnight at room temperature. After washing in TTBS, grids were incubated with goat anti-rabbit secondary antibody conjugated to 5 nm gold and were washed again. To perform silver enhancement, a goat anti-rabbit secondary antibody conjugated to 1 nm gold particles was used instead. Sections were then treated with silver enhancement reagents (Aurion R-Gent; Electron Microscopy Sciences, Fort Washington, PA) for 8 minutes. Sections were poststained with 4% uranyl acetate, washed through drops of methyl cellulose, and air dried. Sections were viewed and photographed through a electron microscope (JEOL 100CX; JEOL, Tokyo, Japan).
For morphologic analysis, eyes (n = 3 per group) were removed 3 weeks after laser and then processed for transmission electron microscopy by conventional techniques. Tissue was placed in modified Karnovsky's fixative consisting of 2.5% glutaraldehyde and 2% formaldehyde in 0.1 M cacodylate buffer with 8 mM CaCl2 and was fixed for 12 to 24 hours at 4°C. The specimens were subsequently changed to 0.1 M cacodylate buffer for storage at 4°C. The tissue was trimmed to block size and was postfixed in 2% aqueous OsO4 for 2 hours at room temperature. After the tissue was dehydrated in ethanol, it was infiltrated with mixtures of propylene oxide and Epon (EMBed 812; Electron Microscopy Sciences), embedded in pure Epon, and polymerized at 60°C for 18 to 24 hours. One-micrometer sections and thin sections were cut on an ultramicrotome (Ultracut E; Leica, Deerfield, IL). The 1-μm sections were stained with 0.5% toluidine blue and the thin sections were stained with saturated aqueous uranyl acetate and Sato's lead stain and then examined under a transmission electron microscope (model CM-10; Philips, Eindhoven, Netherlands).
Immunofluorescence and Immunoblotting
For elastin staining, other enucleated eyes (n = 4 per group) were fixed with 4% paraformaldehyde for 2 hours. After cryoprotection, vertical sections of the eyes were cut on a cryostat (10 μm). Sections were rehydrated in PBS and incubated for 10 minutes in 8 M urea. Sections were washed in PBS and incubated in PBS containing 5% bovine serum albumin and 0.1% Triton (1 hour). Thereafter, sections were incubated overnight in PBS with a goat anti-mouse elastin antibody (PR387; Elastin Products Company). For the detection of endothelial cells, sections were also incubated with anti-mouse CD31. Next, the slides were washed with PBS (three times, 10 minutes each). After incubation with a secondary anti-goat immunoglobulin conjugated to Alexa 488 (Molecular Probes, Eugene, OR) for 30 minutes, the slides were washed again. Cell nuclei were stained with 4′,6′-diamino-1-phenylindole (DAPI; Molecular Probes). Slides were mounted with aqueous mounting medium and coverslipped.
To prepare samples for immunoblotting, posterior eye cups (anterior segment and lens removed) were homogenized in PBS. Supernatants containing the most soluble proteins were collected. Pellets were extracted again with RIPA buffer. Supernatants containing mostly membrane-bound proteins were collected. Proteins were separated on SDS-polyacrylamide gels and electroblotted to PVDF membranes according to standard protocols. Membranes were blocked and incubated with primary antibodies. The following primary antibodies were used: goat anti-mouse elastin antibody (PR387; Elastin Products Company), rabbit anti-VEGF (Chemicon, Temecula, CA), and rabbit anti-MT1-MMP (MMP14; Chemicon). After washing, membranes were incubated with peroxidase-conjugated secondary antibodies and then washed again, followed by detection with chemiluminescence.
VEGF ELISA
At days 0, 3, 7, and 14 after laser, each whole retina was dissected from the RPE-choroid complex and protein from these two tissue complexes were isolated separately (n = 3 animals from each group at each time point). Samples were sonicated in lysis buffer (20 mM imidazole HCl, 10 mM KCl, 1 mM MgCl2, 10 mM EGTA, 1% Triton X-100, 10 mM NaF, 1 mM Na molybdate, and 1 mM EDTA with protease inhibitor; Sigma-Aldrich) on ice. VEGF protein levels in the supernatant were determined with the use of an ELISA kit (threshold of detection 3 pg/mL; R&D Systems, Minneapolis, MN) that recognizes all splice variants. Readings were taken with the spectrophotometer at 450 to 570 nm (Emax; Molecular Devices, Sunnyvale, CA) and were normalized to total protein (Bio-Rad, Hercules, CA). Duplicate measurements were performed.
Statistical Analysis
Differences in incidence of grade 2B lesions and the appearance of a cavitation bubble between groups were analyzed by the χ2 test. Differences in CNV size and vascularity between groups were statistically compared using the Student's two-tailed t-test. Differences in the VEGF levels between WT and knockout (KO) mice were evaluated by the nonparametric Mann–Whitney U test at each time point. In addition, the Kruskal-Wallis test was used to identify differences between values obtained at different time points in the same group. P < 0.05 was considered significant.
Results
Constitutive Defect in the Elastic Layer of Bruch's Membrane in the Mutant
The elastic lamina in Bruch's membrane of WT mice appeared continuous and thick by transmission electron microscopy. In contrast, the elastic layer of the mutant mice appeared fragmented (Fig. 1A). Because elastin polymer is amorphous, these observations were difficult to quantify. Immunofluorescence showed a continuous line of elastin staining along the basal aspect of RPE in the WT mice; this signal was nearly absent in the mutant (Fig. 1B; arrowhead). Electron microscopic evaluation showed robust immunogold labeling for elastin in the WT but sparse staining in the mutant Bruch's membranes (Fig. 1C; arrowheads). Immunoblotting of retina/choroidal tissues showed detectable tropoelastin in the mutant but not in the WT, indicating an increased level of tropoelastin in the mutant (data not shown). An increase in soluble elastin is a common finding in many different tissues of the LOXL1-deficient mice and is consistent with a blockade of elastin polymer formation.7,9
Figure 1.
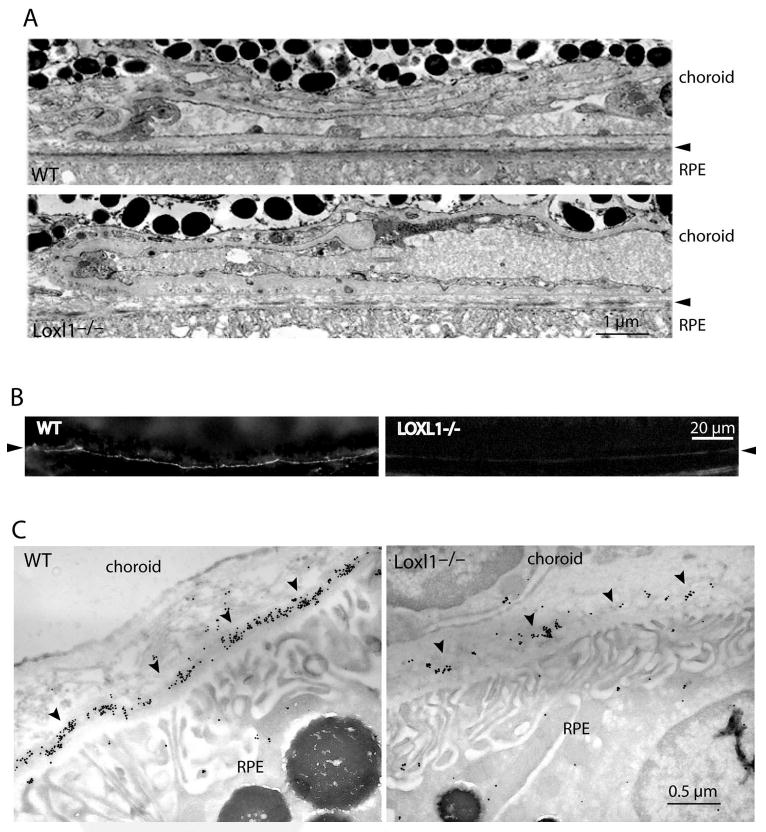
(A) Transmission electron micrographs showing the elastic lamina of Bruch's membrane in WT and LOXL1 mutant mice (at 3 months of age). Note the continuous elastic lamina in the WT in contrast to the many discontinuities (gaps) seen in the mutant. (B) Immunofluorescence staining for elastin in Bruch's membrane. WT (left) and LOXL1 mutant (right) mice show different amounts of elastin staining (arrowheads) in the elastic lamina of Bruch's membrane. The samples were lightly fixed and extracted with a denaturant (urea). In the WT, elastin immunostaining appears as a bright line that is continuous along Bruch's membrane. Elastin staining appears much weaker in the mutant. (C) Immunogold labeling of elastin in Bruch's membrane. Representative images of WT and LOXL1-deficient mice are shown. Elastin labeling (arrowheads) in the mutant mouse is scanty and fragmented, whereas that in the WT is more continuous and of a higher density.
Angiographic Leakage from CNV
The appearance of cavitation bubbles was not different between the WT and the mutant mice. They occurred in 83.5% and 91.5% with a laser power of 200 mW and in 93.5% and 94.5% with 400 mW in WT and LOXL1-deficient mice, respectively. These differences were not statistically significant. Hemorrhagic complications occurred only in four of all burns (two in each group). Comparisons of fluorescein angiograms revealed that LOXL1-deficient mice developed larger and leakier CNV (Fig. 2). At day 14, pathologically significant leakage (grade 2B lesions) was observed in 40.6% of the 200 mW lesions and in 45.5% of the 400 mW lesions in WT mice, whereas it developed in significantly higher proportions (71.9% and 83.8%) of lesions in LOXL1-deficient mice (P < 0.05; Fig. 3).
Figure 2.
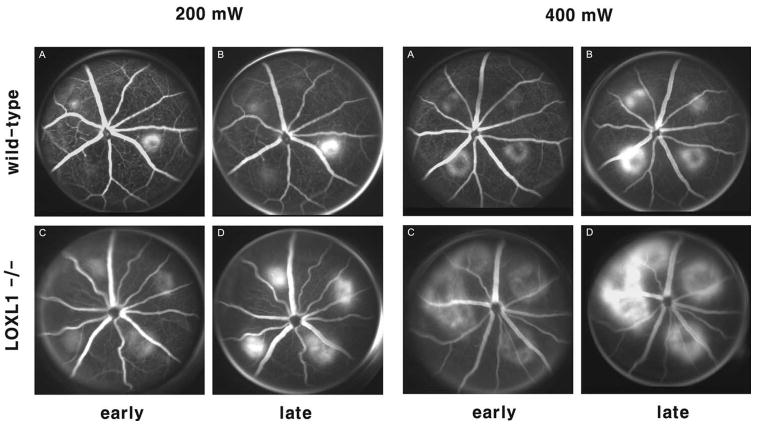
Representative fluorescein angiograms 2 weeks after laser treatment with power of 200 mW (A–D) and 400 mW (E–H) in WT (A, B, E, F) and LOXL1-deficient mice (C, D, G, H). Early-phase (1–2 minutes) and late-phase (6–8 minutes) frames are shown. LOXL1-deficient mice developed larger CNV lesions and showed increased angiographic leakage.
Figure 3.
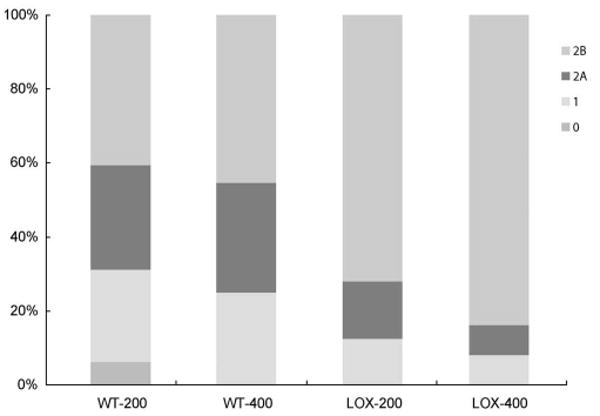
Histogram of angiographic leakage grades. Significantly greater numbers of grade 2B lesions occurred in LOXL1-deficient mice than in control animals 2 weeks after laser induction. WT-200, WT mice treated with 200 mW/spot power; WT-400, WT mice treated with 400 mW/spot power; LOX-200, LOXL1 mutant mice treated with 200 mW/spot power; LOX-400, LOXL1 mutant mice treated with 400 mW/spot power.
Incidence, Size, and Vascularity of CNV
We evaluated the incidence and size of CNV development using FITC-dextran perfusion. The incidence of CNV visualized by this method after laser using 200 mW was 69.2% and 89.5% in WT and LOXL1-deficient mice, respectively (P < 0.05), indicating more susceptibility to CNV development in the latter. In contrast, most 400-mW burns induced CNV growth in both WT and LOXL1−/− mice (91.5% vs. 97.2% in WT and LOXL1−/−; P > 0.05). The LOXL1-deficient mice also developed markedly larger CNV than WT mice at both laser powers (P < 0.05; Fig. 4E). On the other hand, the relative vascularity of CNV lesions was not significantly different between groups (Fig. 4F).
Figure 4.
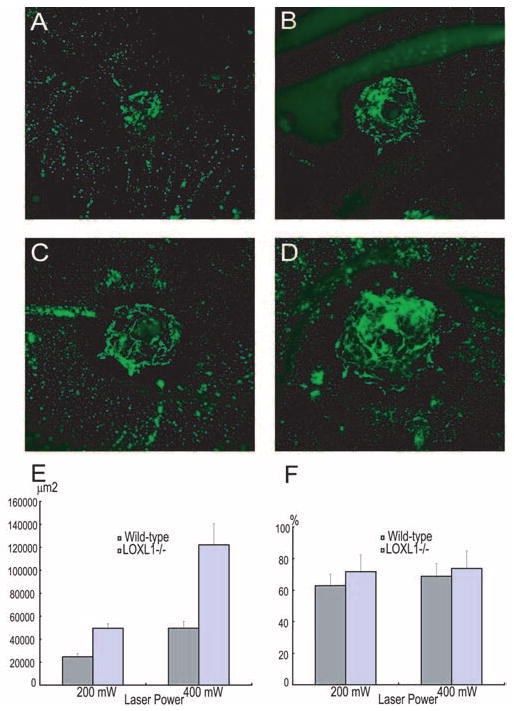
Quantitation of CNV size by choroidal flat mounts. CNV lesions were larger in LOXL1-defecient mice (C, D) than in WT mice (A, B) after laser induction at 200-mW (A, C) and 400-mW (B, D) power settings. (E) Differences in CNV size are shown graphically (P < 0.01 for both power settings). The relative vascularity of lesions (F) was also determined by computer-assisted image analysis and did not differ significantly between groups.
Expression of VEGF and Elastin in Experimental CNV
Light microscopy of laser-treated eyes showed more extensive CNV lesions in the mutant than in the WT mice (Figs. 5A, 5B), consistent with the fluorescein angiography and FITC-dextran perfusion analyses. When CNV lesions were examined for elastin expression, deposition of elastin fibers in the CNV lesions was seen in both WT and LOXL1 mutant mice, but the expression of elastin was much weaker in the LOXL1 mutant (Figs. 5C, 5D). In addition, this weak staining showed a diffuse pattern, in contrast to a fibrillar pattern in WT mice.
Figure 5.
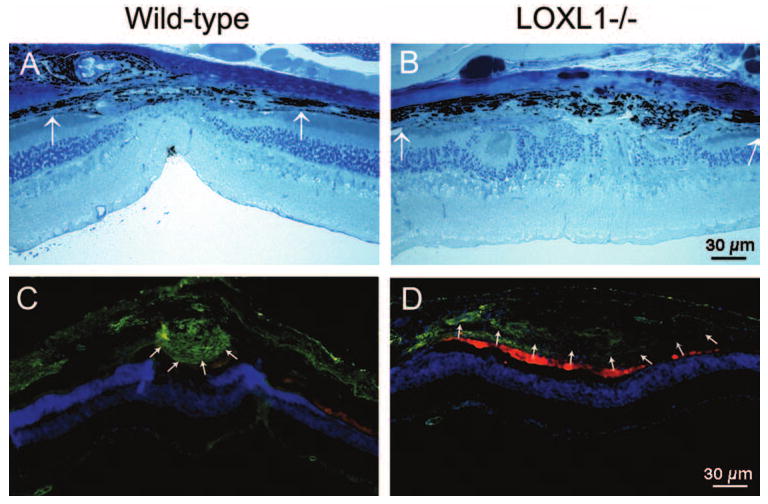
Photomicrographs of CNV lesions in WT and LOXL1-deficient mice. Toluidine blue stains of 1-μm plastic sections (A, B) and immunostaining for elastin on 10-μm cryosections (C, D). Both lesions were created with a laser power of 400 mW. The extent of the CNV lesion (between arrows) appears larger in the mutant mice, but the expression of elastin (green) is much weaker. Nuclei are stained blue. Autofluorescence from photoreceptor debris and pigmented epithelial cells around CNV lesions appears red.
As shown in Figure 6, VEGF protein levels were not different on days 0 and 3 after laser treatment between WT and LOXL1−/− mice. However, peak expression occurred on day 3 in WT mice and on day 7 in LOXL1 mutant mice, resulting in higher levels in LOXL1−/− mice on days 7 and 14 than in WT mice (P < 0.05).
Figure 6.
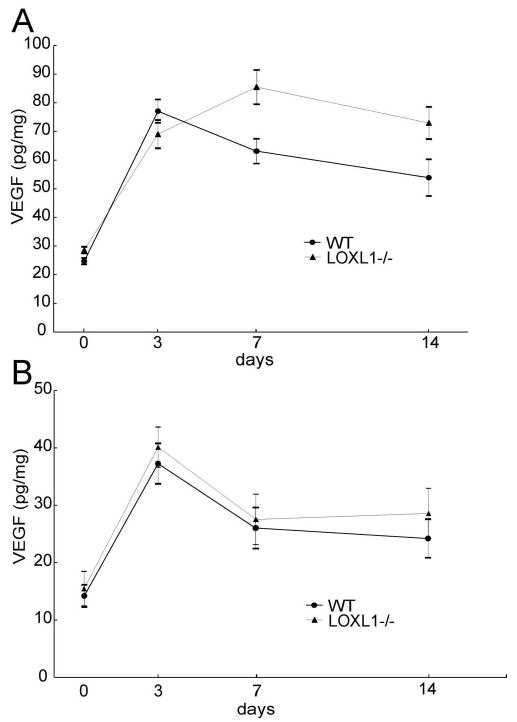
Quantitation of VEGF protein levels after laser-induction of CNV. (A) Time course of VEGF levels in the RPE/choroid complex of LOXL1-deficient and WT mice, as measured by ELISA. VEGF levels were significantly higher (P < 0.5) in the LOXL1-deficient mice at days 7 and 14 after laser treatment. (B) VEGF levels in the retinas of LOXL1-deficient and WT mice were not significantly different through 14 days after laser treatment.
We also analyzed the soluble elastin and VEGF contents in laser-treated eyes by immunoblotting. As shown in Figure 7A, soluble elastin and VEGF were readily detectable in mutant but not in WT tissues, confirming that both were present at higher levels in the mutant. Given that soluble elastin-derived peptides have been shown to have angiogenic properties mediated through the upregulation of MT1-MMP,10 immunoblotting for MT1-MMP was also performed. These studies demonstrated significantly elevated levels of MT1-MMP in the retina/choroid of LOXL1-deficient mice 7 days after laser induction of CNV (Fig. 7).
Figure 7.
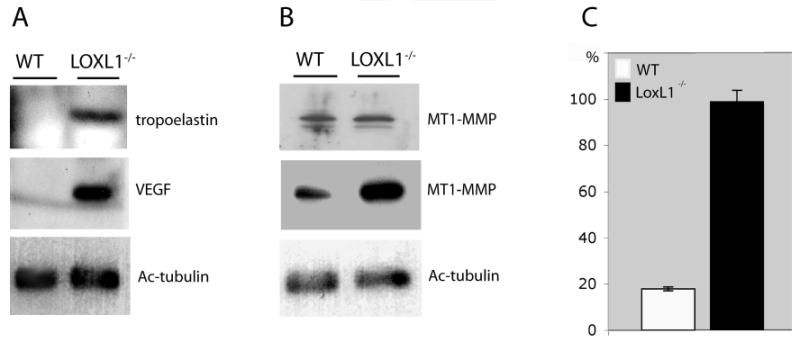
Immunoblotting analysis of WT and LOXL1 mutant retina/choroid tissues before and 7 days after treatment with laser. (A) Soluble elastin (tropoelastin) and VEGF were found at much higher levels in the mutant tissues after laser treatment. With this assay, tropoelastin was also elevated in the LOXL1 mutant before laser treatment, but VEGF was not detected (data not shown). (B) There is an apparent increase of MT1-MMP in the LOXL1 mutant after laser treatment. Only the detergent-soluble fractions are shown. Top: before laser treatment. Middle: after laser treatment. (A, B, bottom) Acetylated (Ac) tubulin as a loading control. (C) Quantitation of MT1-MMP immunoblots (n = 3) showing a significant increase in MT1-MMP levels in the mutant over WT eyes after laser treatment.
MMP-2 and MMP-9 have been detected in human Bruch's membrane11 and in surgically excised choroidal neovascular membranes.12 These MMPs have been shown to be upregulated in experimental CNV in rodents13,14; therefore, immunoblotting analysis for MMP-2 and MMP-9 was also performed in this study. However, no clear differences between WT and mutant mice were detected (data not shown).
Discussion
It has been suggested that the elastic lamina of Bruch's membrane may be a key barrier to CNV development.15 The integrity of the macular elastic lamina may be compromised in patients with early-stage AMD and active choroidal neovascularization compared with unaffected, age-matched controls.4 LOXL1-deficient mice demonstrate diminished and fragmented elastic fibers in systemic organs. Therefore, we hypothesized that these mice might also have similar defects of the elastic layer in Bruch's membrane and be more susceptible to the development of CNV. Electron microscopy revealed that the Bruch's membrane elastic fibers in the LOXL1−/− mice appeared fragmented and thinner than those of WT mice and contained less elastin polymer, as revealed by immunolabeling. LOXL1-deficient mice showed almost twofold larger CNV after laser treatment. Together these data suggest that a compromised elastic lamina in Bruch's membrane may be permissive for CNV growth.
This hypothesis is also supported by the observation that LOXL1−/− mice experienced higher rates of CNV at a lower laser power setting (90% vs. 69%). The sizes of CNV lesions created by the lower power in the LOXL1-deficient mice were similar to those of lesions created by the higher laser power in WT mice. Additionally, fluorescein angiography showed more significant leakage (grade 2B) in the CNV lesions of LOXL1−/− mice than of WT mice. CNV vessels in LOXL1-deficient mice appeared to demonstrate greater leakage regardless of the size of the CNV. On the other hand, the relative vascularity of the CNV lesions was not significantly different between groups. Other than the larger size of the lesions in the LOXL1−/− mice, CNV lesions appeared similar in composition on morphologic evaluation. However, elastin immunostaining did reveal a paucity of elastin deposition in lesions of the LOXL1-deficient mice.
The more aggressive CNV development in the LOXL1-deficient mice might have been influenced by two factors: the lack of physical containment of the CNV because of defective elastin polymerization within the lesion and a proangiogenic matrikine effect related to higher levels of elastin-derived peptides (EDPs) in the local ocular environment compared with that in WT mice. EDPs, which are degradation products of elastic fibers and include soluble tropoelastin, have been shown to have angiogenic properties through effects on endothelial cell migration and tube formation.10 Although ocular levels of EDPs have not been measured in patients with choroidal neovascularization, serum EDP levels, which reflect systemic elastin turnover, were recently reported to be significantly higher in AMD patients, especially those with the neovascular form, compared with age-matched controls.16 Here, we also demonstrated that LOXL1-deficient mice show increased levels of MT1-MMP, lending support to previous studies suggesting that the proangiogenic effects of EDPs may be mediated by the upregulation of MT1-MMP.10 MT1-MMP has been shown to mediate multiple steps in angiogenesis, including endothelial cell invasion and migration, tube formation, and recruitment of smooth muscle cells.17 Furthermore, studies in various tumor cell lines have demonstrated that MT1-MMP induces VEGF upregulation.18,19 Therefore, in the LOXL1−/− mouse, the presence of higher levels of soluble elastin derived peptides appears to increase levels of MT1-MMP, which may enhance CNV development directly and through its effect on VEGF expression.
Although MT1-MMP serves as a regulator of MMP-2 activation,20 the levels of MMP-2 after laser induction of CNV did not differ between the LOXL1 knockout and WT mice, suggesting that the angiogenic effects of MT1-MMP upregulation in this model are independent of MMP-2 activity. This observation is supported by evidence demonstrating that MMP-2 and MMP-9 are not required for angiogenesis in a collagen matrix, but MT1-MMP appears essential.21 However, in our study, because MMP-2 and MMP-9 levels were assayed by immunoblotting at a single time point after laser, subtle differences in expression level and timing of peak expression might not have been detected.
Clinical evidence also suggests that defective elastin polymerization may contribute to AMD pathogenesis. Mutations in fibulin-5 have been associated with age-related macular degeneration.5 LOXL1 and fibulin-5 interact in the process of elastogenesis, and the respective KO mice show similarities in phenotype. These results imply that defective elastic fiber homeostasis is permissive for the development of CNV and that the elastic lamina in Bruch's membrane is important in the prevention of CNV growth.
The increased CNV severity seen in LOXL1-deficient mice shows similarities to the increased CNV recently demonstrated in collagen XVIII-deficient mice.22 Both knockouts are deficient in a single Bruch's membrane component and display larger CNV with increased leakage after laser injury. CNV formation in the collagen XVIII-deficient mice can be decreased to WT levels by administration of recombinant endostatin at physiologic levels, suggesting that structural abnormalities in Bruch's membrane are not the only factor leading to increased CNV development. These two models suggest that proteolytic fragments derived from Bruch's membrane components may mediate both proangiogenic and antiangiogenic activity. The collagen XVIII knockout implicates the role of endogenous endostatin in inhibiting CNV, and the findings in the LOXL1 knockout presented here suggest a proangiogenic function of soluble elastin-derived peptides.
These studies provide more detailed characterization of the contribution of various Bruch's membrane components in providing barriers to CNV development. The LOXL1-deficient mouse provides a model with defects in Bruch's membrane similar to those seen in the aging human macula. Data generated with this model suggest that future pharmacologic therapies for CNV may target reinforcement of the elastic lamina in Bruch's membrane, and these therapies may have particular relevance not only for AMD but also for CNV arising in other conditions with Bruch's membrane pathology.
Acknowledgments
The authors thank Basil Pawlyk and Juliett Xu for helpful advice and technical assistance.
Supported by the Marion W. and Edward F. Knight AMD Fund and by National Institutes of Health Grant EY10309 (TL).
Footnotes
Disclosure: H.G. Yu, None; X. Liu, None; S. Kiss, None; E. Connolly, None; E.S. Gragoudas, Jerini (C, P); N.A. Michaud, None; O.V. Bulgakov, None; M. Adamian, None; M.M. DeAngelis, None; J.W. Miller, Bausch and Lomb (C, P), Genentech (C, P), Genzyme (C, P), AstraZeneca (C, P); T. Li, None; I.K. Kim, None
References
- 1.de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355:1474–1485. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- 2.Blumenkranz MS, Russell SR, Robey MG, Kott-Blumenkranz R, Penneys N. Risk factors in age-related maculopathy complicated by choroidal neovascularization. Ophthalmology. 1986;93:552–558. doi: 10.1016/s0161-6420(86)33702-3. [DOI] [PubMed] [Google Scholar]
- 3.Hogan MJ. Bruch's membrane and disease of the macula: role of elastic tissue and collagen. Trans Ophthalmol Soc U K. 1967;87:113–161. [PubMed] [Google Scholar]
- 4.Chong NH, Keonin J, Luthert PJ, et al. Decreased thickness and integrity of the macular elastic layer of Bruch's membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005;166:241–251. doi: 10.1016/S0002-9440(10)62248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 6.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Zhao Y, Gao J, et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 8.Liu G, Daneshgari F, Li M, et al. Bladder and urethral function in pelvic organ prolapsed lysyl oxidase like-1 knockout mice. BJU Int. 2007;100:414–418. doi: 10.1111/j.1464-410X.2007.06929.x. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Zhao Y, Pawlyk B, Damaser M, Li T. Failure of elastic fiber homeostasis leads to pelvic floor disorders. Am J Pathol. 2006;168:519–528. doi: 10.2353/ajpath.2006.050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinet A, Fahem A, Cauchard JH, et al. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J Cell Sci. 2005;118:343–356. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- 11.Guo L, Hussain AA, Limb GA, Marshall J. Age-dependent variation in metalloproteinase activity of isolated human Bruch's membrane and choroid. Invest Ophthalmol Vis Sci. 1999;40:2676–2682. [PubMed] [Google Scholar]
- 12.Steen B, Sejersen S, Berglin L, Seregard S, Kvanta A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1998;39:2194–2200. [PubMed] [Google Scholar]
- 13.Kvanta A, Shen WY, Sarman S, Seregard S, Steen B, Rakoczy E. Matrix metalloproteinase (MMP) expression in experimental choroidal neovascularization. Curr Eye Res. 2000;21:684–690. [PubMed] [Google Scholar]
- 14.Lambert V, Wielockx B, Munaut C, et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17:2290–2292. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- 15.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 16.Sivaprasad S, Chong NV, Bailey TA. Serum elastin-derived peptides in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:3046–3051. doi: 10.1167/iovs.04-1277. [DOI] [PubMed] [Google Scholar]
- 17.Genis L, Galvez BG, Gonzalo P, Arroyo AG. MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006;25:77–86. doi: 10.1007/s10555-006-7891-z. [DOI] [PubMed] [Google Scholar]
- 18.Deryugina EI, Soroceanu L, Strongin AY. Up-regulation of vascular endothelial growth factor by membrane-type 1 matrix metalloproteinase stimulates human glioma xenograft growth and angiogenesis. Cancer Res. 2002;62:580–588. [PubMed] [Google Scholar]
- 19.Sounni NE, Devy L, Hajitou A, et al. MT1-MMP expression promotes tumor growth and angiogenesis through an up-regulation of vascular endothelial growth factor expression. FASEB J. 2002;16:555–564. doi: 10.1096/fj.01-0790com. [DOI] [PubMed] [Google Scholar]
- 20.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206:1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 21.Chun TH, Sabeh F, Ota I, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757–767. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marneros AG, She H, Zambarakji H, et al. Endogenous endostatin inhibits choroidal neovascularization. FASEB J. 2007;21:3809–3818. doi: 10.1096/fj.07-8422com. [DOI] [PubMed] [Google Scholar]