

Abstract
Hemizygous interstitial deletions in human chromosome 22q11 are associated with velocardiofacial syndrome and DiGeorge syndrome and lead to multiple congenital abnormalities, including cardiovascular defects. The gene(s) responsible for these disorders is thought to reside in a 1.5-Mb region of 22q11 in which 27 genes have been identified. We have used Cre-mediated recombination of LoxP sites in embryonic stem cells and mice to generate a 550-kb deletion encompassing 16 of these genes in the corresponding region on mouse chromosome 16. Mice heterozygous for this deletion are normal and do not exhibit cardiovascular abnormalities. Because mice with a larger deletion on mouse chromosome 16 do have heart defects, the results allow us to exclude these 16 genes as being solely, or in combination among themselves, responsible for the cardiovascular abnormalities in velocardiofacial/DiGeorge syndrome. We also generated mice with a duplication of the 16 genes that may help dissect the genetic basis of “cat eye” and derivative 22 syndromes that are characterized by extra copies of portions of 22q11, including these 16 genes. We also describe a strategy for selecting cell lines with defined chromosomal rearrangements. The method is based on reconstitution of a dominant selection marker after Cre-mediated recombination of LoxP sites. Therefore it should be widely applicable to many cell lines.
Interstitial deletions of human chromosome 22q11 (HSA22q11) are frequent and are associated with velocardiofacial syndrome/DiGeorge syndrome [VCFS, Mendelian Inheritance in Man (MIM) 192430/DGS, MIM 188400]. VCFS/DGS is characterized by a series of anomalies that include facial dysmorphology, velopharyngeal insufficiency, cleft palate, cardiac abnormalities involving the outflow tract, and thymic and parathyroid gland hypoplasia or aplasia, resulting in immune deficiencies and hypocalcemia, respectively (1–3). Most VCFS/DGS patients are hemizygous for a large, 3-Mb, interstitial deletion with well-defined breakpoints (4). A small subset of patients have a nested deletion of 1.5 Mb (5). Other constitutional rearrangements such as tetrasomy and trisomy of HSA22q11 have been described in two other developmental disorders, “cat eye” syndrome (MIM 115470) and derivative 22 syndrome (6). The extra copies of the 22q11 region present in such individuals include the 1.5-Mb region that is deleted in some VCFS/DGS patients, suggesting the presence of genes in this interval causing dosage-sensitive effects on cardiac development and other aspects of fetal development (7).
Several of the tissues and organs affected in VCFS/DGS are derived from the pharyngeal arches and pouches, structures that are populated by migrating neural crest cells that have been shown to be essential for the proper development of the conotruncal region of the heart, the thymus, and the parathyroid glands (8, 9). Most of the 1.5-Mb VCFS/DGS region has been cloned and sequenced (10), and 27 genes have been identified within this region (Fig. 1a). Several approaches have been used to identify the gene(s) responsible for producing the VCFS/DGS phenotype, including mutation analysis of rare nondeleted patients, developmental gene expression analysis, and generation of gene-inactivated mice. However, these efforts have not led to a definitive identification of the responsible gene(s). Moreover, the fact that a small number of patients have nonoverlapping deletions raises the possibility that there is more than one gene in 22q11 that when deleted contributes to the VCFS/DGS phenotype (reviewed in ref. 12).
Figure 1.

(a) Comparison of the human VCFS/DGS region with the homologous region in mice. The relative order of genes on Homo sapiens chromosome 22q11 (HSA22q11) and Mus musculus chromosome 16 (MMU16) is shown (11, 18). The location of bacterial artificial chromosomes (BACs) 147J11 and 213A6 used for fluorescence in situ hybridization experiments and the region deleted in our experiments and those of Lindsay et al. (13) are shown on the right. Ym24do7, op53c05, me49f07, and vo59c07 correspond to independent genes identified by genscan analysis of GenBank sequences AC006082, AC003060, AC003066, AC008019, which are also represented in the 22q11 genomic sequence. (b) Scheme for selecting Cre/LoxP-mediated interstitial chromosomal deletion and duplication. Recombination between LoxP sites in cis leads to a deletion (upper two diagrams), and recombination between LoxP sites in trans leads to a deletion as well as a duplication (lower two diagrams). The locations of various diagnostic PCR products are represented by A–E. The Idd targeting vector contained a 4.3-kb XbaI genomic fragment including Idd exon 6, a 3.0-kb PvuII/SmaI genomic fragment including Idd exon 2, and part of exon 3 and a cassette containing a gene that confers resistance to neomycin (NeoR). The cassette also included a LoxP site and a solitary phosphoglycerate kinase (PGK) promoter (P). The Arvcf targeting vector contained a 0.7-kb EcoRI/MscI fragment containing a part of Arvcf exon 3 and an 8.0-kb XbaI fragment including Arvcf exons 5 and 6. A 3.1-kb cassette containing a hygromycin resistance (HygroR) gene, a LoxP site, and a promoterless puromycin resistance (PuroR) gene was inserted by blunt-end ligation into the MscI and XbaI sites.
Recently, mice with cardiovascular defects similar to those in VCFS/DGS patients were produced by deleting a region on Mus musculus chromosome 16 (MMU16) corresponding to most of the 1.5-Mb deletion seen in some VCFS/DGS patients (13). We sought to identify a subregion of MMU16 in which the gene(s) responsible for VCFS/DGS phenotypes resides. We describe here the use of a selectable system for producing a 550-kb deletion on MMU16, using the Cre–LoxP system. This deletion encompasses at least 16 genes, representing about 60% of the genes present in the 1.5-Mb VCFS/DGS region (Fig. 1). These mice do not exhibit cardiac abnormalities, allowing us to exclude many of the genes in this region as being solely responsible, when haploinsufficient, for such defects. Our results indicate either that the gene responsible for cardiovascular defects in VCFS/DGS patients lies outside of the region deleted in these mice or that haploinsufficiency of more than one gene is involved in the defects. To distinguish between these two possibilities, we also have generated mice with a duplication of the same region.
Materials and Methods
Primers.
Primers iddko2 (CACGTTGTCATTCTCAGACATG), iddko1 (CTGTTGTTGACACAGCACATG), 6x32t3 (AACTCTACCTGTTCCTACTG), PGK1 (GCTAAAGCGCATGCTCCAGAC), Neo5F (ACCGCTATCAGGACATAGCGT), HygKoF1 (GTGCTTTCAGCTTCGATGTAG), HygKoR1 (GTAGTGTATTGACCGATTCCTTG), PGKF2 (GCATTCTGCACGCTTCAA), 5′PuroR (CTTGTACTCGGTCATGGT), NeoR1 (CTGCGTGCAATCCATCTTGT), hygro2 (ACTCGCCGATAGTGGAAACC), arvf4 (TGGACAGGTCCCAGACATTC), 6R2R1 (AAATCGATGGAAGTCAGTGCAGATG), and 6R2R2 (AAATCGATCAGATGGCAAGCACACTG) were used for mouse genotyping and for identification of a homologous recombination event at the Idd and Arvcf loci. Primers (see Figs. 1C and 3) were combined as follows: iddko2 + PGK1 (A), iddko1 + neo5F (B), HygKOF1 + HygKOR1 (C), PGKF2 + 5′PuroR (D), HygKoF1 + NeoR1 (E).
Figure 3.

Genotype analysis of progeny of chimeric mice derived from ES cells with chromosomal rearrangements. Mouse tail DNA obtained from two litters of progeny of chimera Vc1 was analyzed by PCR, using primers designed to amplify a fragment of (from Top to Bottom) the PGK-Puro junction, the modified Idd allele on its 3′ end or its 5′ end, the Hyg–Neo junction, and the modified Arvcf allele on its 5′ end (indicated, respectively, as fragments D, A, B, E, and C in Fig. 1b). Mice positive for PCR fragments D and A contain a chromosome with an interstitial deletion, whereas mice positive for PCR fragments B, E, and C contain a chromosome with an interstitial duplication.
Generation of Embryonic Stem (ES) Cell Lines Containing Chromosomal Rearrangements.
WW6 ES cells were cultured according to the method of Ioffe et al. (14). To introduce a LoxP site at the Idd locus, ES cells were transfected with NotI-linearized Idd targeting vector and selected with G418 (150 μg/ml). DNA from pools of six G418-resistant ES cell colonies was analyzed by long-range PCR (Boehringer Mannheim) as recommended by the manufacturer, using primers 6R2R1 and Neo5F. Specificity of the PCR amplification was determined by hybridization with 20 ng of an internal primer, 6R2R2, that was labeled as described by Hazan et al. (15). To introduce a second LoxP site at the Arvcf locus, ES cell clone 14.9 targeted at the Idd locus was electroporated with 10 μg of NotI-linearized Arvcf targeting vector and selected in G418 (150 μg/ml) and hygromycin (200 μg/ml). DNA from HygR colonies was analyzed by long-range PCR, using primers hygro2 and arvf4. Doubly targeted cells were electroporated with either pCMV-Cre or pCAG-Cre (16). Cells were plated without selection at a density of 2 × 106 cells per plate for 36–48 h and then replated in medium containing puromycin (2.2 μg/ml). DNA from resistant colonies was analyzed by PCR for a DNA fragment indicative of joining the PGK promoter to the PuroR gene (Fig. 1b). Positive ES cell clones were injected into C57BL/6 recipient blastocysts, which were transferred to CD-1 pseudopregnant females (17).
Fluorescent in Situ Hybridization.
A high-resolution physical map of the HSA22q11/MMU16 region was generated as described by Morrow et al. (5). Mouse BACs were from the RPCI-22 (129S6/SvEvTac) mouse BAC library (http://bacpac.med.buffalo.edu/). The gene content of each BAC was identified as described by Puech et al. (18). Mouse BACs lying inside (RPCI22-213A6) and outside (RPCI22-174J11) of the targeted region (Fig. 1a) were used to generate DNA probes by degenerate oligonucleotide-primed (DOP)-PCR (19). PCR products were purified and labeled by nick translation with bio-16-dUTP (Roche Molecular Biochemicals) for BAC RPCI22-213A6 and with Cy3-dUTP (Amersham Pharmacia) for BAC RPCI22-174J11. In situ hybridization was performed as described by Cherif et al. (20). Slides were analyzed under a Leica fluorescence microscope (DMRXA) equipped with a cooled charged-coupled camera (C4880-06; Hamamatsu, Middlesex, NJ) and processed to create pseudocolor images with software developed by IMSTAR (Paris).
Pathologic Examination of Heterozygous Mutant Mice.
A complete gross and histological examination and weight measurements were conducted on all organs from 4- to 5-month-old heterozygotes and controls. Organs were fixed in 10% neutral buffered formalin and embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin. Sections of the following tissues were examined by light microscopy: brain, eye, nasal cavity, tongue, inner ear, middle ear, salivary glands, trachea, Harderian gland, esophagus, thyroid, parathyroid, thymus, pituitary, peripheral lymph nodes, heart, aorta, diaphragm, lung, spleen, liver, gall bladder, kidney, adrenal, stomach, small intestine, cecum, colon, pancreas, reproductive tract from each sex (ovaries, oviduct, uterus, vagina, or prostate, testis, penis, seminal vesicle), urinary bladder, skin, skeletal muscle, and bone marrow. For examination, the intact heart was first bisectioned in a basal-apical plane including both valves. The two halves were sectioned together to obtain approximately 25 serial sections. Different section levels (usually five per animal) were prepared using every fifth section and examined histologically. For visualization of the major blood vessels, embryonic day 18.5 (E18.5) embryos were perfused by intracardiac injection with red acrylic cast (Batson's no. 17 acrylic; Polysciences) and then fixed in 10% buffered formalin. The soft tissue in the anterior thorax was removed by dissection to examine the hardened vessels filled with corrosion cast, under a stereomicroscope.
Results
Engineering ES Cells with a Deletion and a Duplication of a Portion of MMU16: A Strategy for Selecting Cre-Mediated Chromosomal Rearrangements.
Some VCFS/DGS patients have an approximately 1.5-Mb deletion spanning the sequence from DGCR6 to ym24d07 on 22q11, causing them to be hemizygous for 27 known genes (10). We and others have examined the organization of this region in mice and found that it is located in a single region of mouse chromosome 16 (18, 21, 22). However, the order of genes in the two species is different, probably because of an inversion and exchange of other segments during evolution (Fig. 1a). Recently, mice hemizygous for a deletion spanning the sequence from Es2 to Ufd1l, encompassing 22 known genes, were generated using the Cre-LoxP system. About 25% of E18.5 embryos with this deletion exhibited various VCFS/DGS-associated cardiovascular abnormalities. Increased mortality during the first day of life was also noted in such mice, but mice surviving the first day were viable and grew normally (13). To examine the contribution of a subset of the genes in this region to these cardiovascular abnormalities, we first produced ES cells containing a deletion spanning the sequence from Idd to Arvcf, a region estimated to be 550 kb (Fig. 1a). The relative transcriptional orientation of the two genes on HSA22q11 was precisely determined by high-resolution physical mapping and complete sequencing of the HSA22qq11 region (10). Detailed comparative mapping allowed us to deduce the genes' relative transcriptional orientation in mice (18). This region encompasses 16 genes from among the 27 genes in the 1.5-Mb VCFS/DGS deleted region and includes 13 of the 22 genes hemizygous in the Es2 to Ufd1l deleted mice recently reported (13).
We used Cre-mediated recombination of LoxP sites to modify a portion of MMU16 in ES cells. Because the frequency of Cre-mediated recombination of LoxP sites separated by large distances in mammalian cells is quite low, a selection system was described in which recombination leads to reconstitution of a functional hypoxanthine phosphoribosyltransferase (HPRT) gene (23). However, this approach requires using HPRT-deficient ES cells, precluding its use in most ES cell lines. Therefore, we devised another strategy, using a dominant antibiotic resistance marker for selecting ES cells in which distant LoxP sites have recombined (Fig. 1b). The strategy involves inserting two cassettes containing LoxP sites into defined positions in ES cells by homologous recombination. One cassette contains a solitary PGK promoter proximal to a LoxP site, as well as a selectable marker (NeoR). The second cassette contains a promoterless PuroR gene distal to a LoxP site and a different selectable marker (hygromycin resistance, HygR). Recombination between the LoxP sites joins the PGK promoter to the promoterless PuroR gene, producing a functional gene that confers resistance to puromycin. Recombination in PuroR clones can be confirmed by PCR, using primers specific for the PGK promoter and the PuroR gene (PCR D, Fig. 1b). If the LoxP sites have been inserted in cis and in the same relative orientation, recombination leads to cells with a deletion on one chromosome and simultaneous loss of HygR and NeoR genes; the homologous chromosome is unaffected (Fig. 1b, upper two diagrams). On the other hand, if the LoxP sites have been inserted in trans on homologous chromosomes, recombination between LoxP sites results in cells containing two rearranged chromosomes, one with an interstitial deletion of the region between the LoxP sites and the other with a duplication of the same region (Fig. 1b, lower two diagrams). In this case, intact NeoR and HygR genes would be present on the chromosome containing the duplication, and the resulting cells should be resistant not only to puromycin but also to G418 and hygromycin. The two rearranged chromosomes should segregate in the progeny of chimeras obtained from these cells, generating animals with either a deleted chromosome or a chromosome containing an interstitial duplication.
To modify the region of MMU16 in ES cells, the Idd gene was targeted with a vector containing a solitary PGK promoter, a LoxP site, and a NeoR gene. Homologous recombination at the Idd locus would result in the deletion of a 2-kb segment including Idd exons 4 and 5. Genomic DNA from 384 NeoR clones was pooled (six colonies per pool) and tested for gene targeting events by PCR. Five pools gave the expected 4.6-kb PCR product, and upon subsequent analysis a total of five colonies exhibited the 4.6-kb product (data not shown). The overall targeting frequency was 1.3% among NeoR clones. To introduce a second LoxP site in MMU16, a line targeted at the Idd locus (clone 14.9) was transfected with a targeting vector containing two regions of the Arvcf gene flanking a HygR gene, a LoxP site, and a promoterless PuroR gene. Homologous recombination should result in the deletion of a segment of Arvcf corresponding to amino acids 83–276. Ninety-six HygR clones were selected and tested for gene targeting events. A total of 10 colonies exhibited the 1.4-kb product (not shown), corresponding to an overall targeting frequency of 10.4% among the HygR clones.
To generate ES cells with interstitial deletions and duplications, eight doubly targeted ES cell clones were transfected with one of two Cre expression plasmids (pCMV-Cre or pCAG-Cre), and PuroR clones were selected. Six cell lines gave rise to no (1c1, 3c4, 3b3) or very few (4b1, 4b4, 3c1) PuroR clones, whereas two lines (4c1, 4d1) yielded many more PuroR clones (Table 1). All PuroR clones except one were positive for a PCR fragment spanning the sequence from the PGK promoter to the PuroR gene (PCR D, Fig. 1b), indicating joining of these two segments of DNA. Clones obtained from cell lines yielding few PuroR clones were HygR and NeoR and were positive for PCR E (Fig. 1b), indicating retention of the two markers. The presence of both PCR fragments D and E in single clones is consistent with their origin from recombination between LoxP sites in trans and to their having one chromosome with an interstitial deletion and the second chromosome with an interstitial duplication. Clones obtained from the two lines yielding many more PuroR clones were sensitive to hygromycin and G418 and negative for PCR E. These results are consistent with these clones arising from recombination between LoxP sites in cis and to their having only a chromosome with a deletion. The large difference observed in the frequencies of PuroR clones with the various lines (1c1, 3c4, 3b3, 4b1, 4b4, 3c1 vs. 4c1, 4d1) indicates that Cre-mediated recombination between the LoxP sites in cis is much more frequent than when they are in trans configuration, consistent with previous observations (23). The differences in the frequencies of PuroR clones observed among lines transfected with pCMV-Cre vs. pCAG-Cre also suggest that Cre may be produced in larger amounts when driven by the pCAG promoter than when driven by the cytomegalovirus (CMV) promoter.
Table 1.
Antibiotic resistance and PCR analysis of clones obtained from transfections of doubly targeted ES cell lines with Cre expression plasmids
| Idd–Arvcf knockout cell line | No. of clones
|
|||||
|---|---|---|---|---|---|---|
| CMV-Cre
|
CAG-Cre
|
|||||
| 1c1, 3c4, 3b3 | 4c1 | 4b1 | 4b4 | 3c1 | 4d1 | |
| PuroR | 0 | 84 | 3 | 5 | 5 | >1,000 |
| HygR, NeoR | 0 | 0/24 | 2/2 | 1/1 | NT | 0/42 |
| PGK-Puro PCR+ | 0 | 24/24 | 2/2 | 1/1 | NT | 41/42 |
| Hyg-Neo PCR+ | 0 | 0/24 | 2/2 | 1/1 | NT | 0/42 |
NT, not tested.
Interphase fluorescence in situ hybridization with BACs RPCI22-174J11 and RPCI-213A6, lying inside and outside, respectively, the deleted/duplicated region was used to confirm the chromosomal rearrangements. In situ hybridization of nuclei from cell line IIb4, which is sensitive to hygromycin and G418, exhibited colocalization of the two probes in one region of the nucleus and a solitary signal with BAC 213A6 in another region (Fig. 2b), consistent with this line having only a chromosome with an interstitial deletion. On the other hand, cell line Vc1, which is HygR and NeoR, exhibited colocalization of one copy of BAC 213A6 and two copies of BAC 147J11 in one region of the nucleus and a solitary signal in another region (Fig. 2d; see also an example of the analysis in G2 cells of each line in Fig. 2 c and e). This result implies that cell line Vc1 has a deleted chromosome 16 and one with an interstitial duplication.
Figure 2.
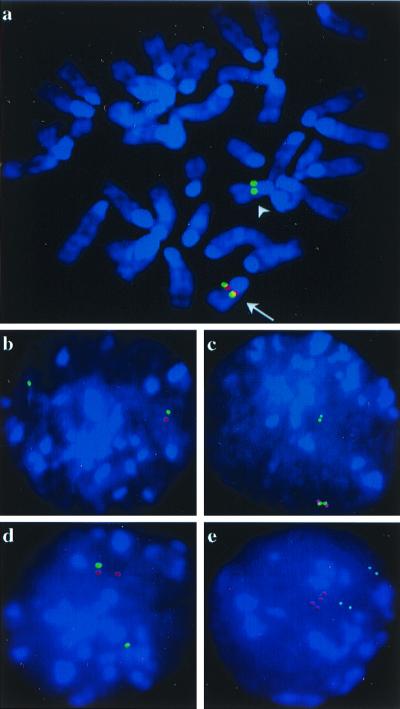
Fluorescence in situ hybridization analysis of ES cell clones with Cre-mediated chromosome rearrangements. (a) Simultaneous hybridization with BACs 174J11 and 213A6 to metaphase spreads of ES cell line Vc1. The outside probe BAC 213A6 (green) hybridized to both copies of MMU16, and the inside probe BAC 174J11 (red) colocalized with the outside probe only on one homolog (arrow). The presence or absence of a duplication could not be resolved on metaphase chromosomes. (b and c) Simultaneous hybridization to interphase nuclei of cell line IIb4. The inside probe (red) was colocalized with the outside probe (green) in 74% (66/89) of the nuclei. In 24% of the nuclei, duplicate signals representing hybridization to G2 cells were observed. (d and e) Simultaneous hybridization to interphase nuclei of cell line Vc1. The inside probe (two red dots) colocalized with the outside probe in 57% of nuclei (49/86). In 31% of the nuclei duplicate signals from G2 cells were observed.
Generating Mice with Altered Dosage of the Region Lying Between Idd and Arvcf.
Several ES cell clones containing either a deletion or a deletion and a duplication were injected into C57BL/6 blastocysts. Clones Va1, Vc1, and IIb4 generated chimeras ranging in chimerism between 30% and 80%, as judged by coat color. These chimeras were mated to C57BL/6 animals, and their offspring were genotyped by PCR analysis of tail DNA. Mice heterozygous for the deletion of part of the MMU16 region were detected in offspring of chimeras obtained from clone IIb4. This result indicates that mice hemizygous for at least 16 genes from this region are viable. Mice heterozygous for the interstitial deletion were interbred. Both wild-type (37.2%, 67/180) and heterozygous (62.8%, 113/180) mice were obtained, but no homozygous animals were represented among the offspring. Analysis of E9.5 embryos from such matings failed to detect any homozygous mutant embryos. The number of resorptions at this stage (10/30) suggests a postimplantation lethality due to an essential gene(s) between Idd and Arvcf.
An equal number of mice with either a duplication or a deletion of this region were obtained by mating chimeras derived from cell lines Va1 and Vc1 (Fig. 3), indicating that mice with a trisomy of at least 16 genes from this region are viable. Furthermore, by interbreeding animals that contain both the deleted and the duplicated chromosomes, we obtained animals homozygous for the chromosome with the duplicated region at the expected Mendelian ratio (62/99 heterozygotes for the deletion and the duplication, 37/99 homozygotes for the duplication, no homozygotes for the deletion). Mice homozygous for the chromosome with the duplication were interbred and produced normal-sized litters. Thus mice with three and even four copies of 16 known genes in this region are viable and fertile.
We also generated mice with a deletion of the region between Idd and Arvcf by carrying out Cre-mediated recombination directly in animals. To obtain a recombination between the two loci with the trans configuration, two lines of mice were established from ES cells that had a LoxP site inserted at either the Idd locus or the Arvcf locus. CMV-Cre (24) or ZP3-Cre (25) transgenes were introduced into the Idd modified line by matings. Then mice heterozygous for the Idd modified allele and transgenic for one of the transgenes were mated to mice heterozygous for the Arvcf modified allele to generate mice containing three modifications. Heterozygotes in which the modified alleles at Idd and Arvcf are in the cis configuration were obtained by extensive breeding and identification of recombinant chromosomes containing the two modified alleles. The CMV-Cre and ZP3-Cre transgenes were introduced into their progeny by further mating. One animal with a deletion was obtained from 32 offspring of CMV-Cre transgenics containing the Idd and Arvcf modified alleles in cis. No deleted chromosomes were detected in more than 200 offspring of 10 mating pairs containing CMV-Cre and the two modified alleles in trans, suggesting that as in ES cells, recombination of LoxP sites in cis is much more efficient than when the sites are in trans. The ZP3-Cre transgene appears to be much more efficient than the CMV-Cre transgene, inasmuch as we obtained 16 of 51 offspring (31.3%) with a deleted chromosome from matings with the two modified alleles in cis. The higher efficiency of the ZP3-Cre transgene is consistent with a high level of expression of the ZP3 promoter during oogenesis. These results indicate that large chromosomal deletions can be obtained directly at very high efficiencies in living animals when the LoxP sites are inserted in cis.
Characterization of Mice with Partial Monosomy or Trisomy of MMU16.
Lindsay et al. (13) recently reported that E18.5 embryos with a hemizygous deletion from Es2 to Ufd1l frequently exhibit cardiovascular abnormalities and that mutant pups show a somewhat increased mortality at birth. Because the deletion described above, from Idd to Arvcf, overlaps significantly the interval from Es2 to Ufd1l, we were interested to learn whether mice with the Idd to Arvcf deletion have similar cardiovascular abnormalities, as well as any other VCFS/DGS phenotypes. By backcrossing F1 mice with the deletion to C57BL/6 animals we obtained heterozygous mutant and wild-type progeny at the expected Mendelian ratio (186 wild type vs. 184 heterozygotes). Mice heterozygous for the Idd-to-Arvcf deletion exhibited normal birth weight and size and were indistinguishable from their wild-type littermates. Among the few rare dead pups identified, cleft palate was never observed. Body weight; liver, kidney, and adrenal weights; the gross appearance of organs; as well as heart shape and size in heterozygous mutants were within the normal range. Histological examination of all organs, including the thymus, parathyroid glands, and the heart, from 4- to 5-month-old mutant (n = 5) and control (n = 3) littermates did not show significant abnormal findings. Special attention was given to the heart by preparing serial sections (5 μm in thickness), across approximately 80–100 μm of tissue. In this manner, it was possible to visualize the septa and the great vessels along with the valves. We did not detect any cardiovascular abnormalities such as interrupted aortic arch, ventricular septal defect, or any other cardiac or conotruncal defects in the five heterozygotes investigated by necropsy and by microscopic examination of histological sections of the conotruncal region. To examine the architecture of the major blood vessels of the heart, E18.5 heterozygous (22/34) and wild-type (12/34) embryos were perfused by intracardiac injection with corrosion cast. All embryos demonstrated a normal pattern of the outflow tracts and major vessel branching from the aortic arch region (Fig. 4b). These results indicate that monosomy for at least 16 genes present in the VCFS/DGS deleted region did not impair normal development of the heart and the vascular pattern of large vessels immediately cranial to the heart and derived from aortic arches during embryogenesis.
Figure 4.
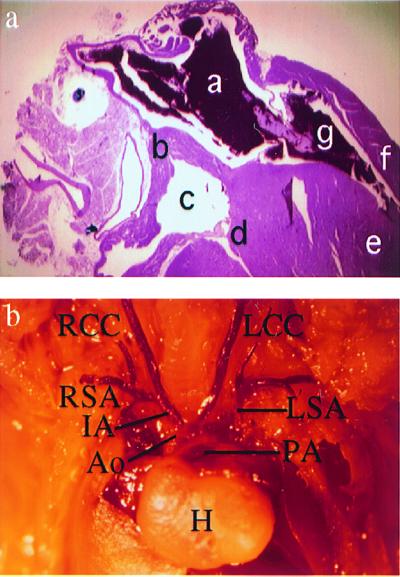
(a) Cardiovascular anatomy in mice with a deletion of the Idd–Arvcf interval. A section of the adult heart from a mouse with a deletion of the Idd–Arvcf interval shows the normal anatomy of the atrioventricular region. a, Right atrium; b, atrial septum; c, left atrium; d, mitral valve; e, ventricular septum; f, ventricular free wall; g, tricuspid valve. (b) Corrosion cast of a heart, major branches from the aortic arch, and pulmonary artery in an E18.5 embryo with a deletion of the Idd–Arvcf interval, showing normal development of the major vessels. H, heart; Ao, aorta; IA, innominate artery; LCC and RCC, left and right common carotid arteries; LSA and RSA, left and right subclavian arteries; PA, pulmonary artery.
Patients with tetrasomy and trisomy of HSA22q11 have been described in cat eye and derivative 22 syndromes, which are characterized by developmental abnormalities, including congenital heart defects, renal malformations, and male genital anomalies. Because the extra copies of the 22q11 region present in these patients include the 1.5-Mb region that is deleted in some VCFS/DGS patients (7), we also examined mice heterozygous for the chromosome with a duplication. No consistent differences were found between wild-type and heterozygous animals by examination of birth weight, growth curves, organ sizes, and weights, or hematoxylin/eosin-stained paraffin-embedded sections from 40 different tissues collected from six 4- to 5-month-old mutant and control littermates. These results indicate that trisomy for 16 genes present in the VCFS/DGS deleted region did not impair normal mouse development.
Discussion
We have generated mice with a 550-kb deletion corresponding to a part of human chromosome 22q11 deleted in VCFS/DGS. Although several investigations have attempted to define a critical region in VCFS/DGS and a large number of genes in the deleted region have been identified to date, no single gene has been implicated as a cause of the disorder. In an attempt to localize candidate genes, two different interstitial deletions of MMU16 were recently described. One is a conventional ≈150-kb deletion that results in hemizygosity of seven genes from Znf74 l to Ctp (26). Mice hemizygous for this deletion do not have any phenotype similar to those seen in VCFS/DGS patients, although they exhibit increased prepulse inhibition of the startle response, a phenotype that may indicate behavioral abnormalities. Mice with this deletion in the homozygous state do not survive much beyond implantation. A larger deletion, estimated to be 1.2 Mb in size and spanning 22 genes between Es2 and Ufd1 l, was described by Lindsay et al. (13). Three of the genes (Idd, Tsk1, and Tsk2) in this deletion are common to the deletion generated by Kimber et al. (26). Hemizygosity for this deletion causes fetal cardiovascular defects similar to those seen in VCFS/DGS patients and may be responsible for the reduced survival rate of these mice at birth. Homozygosity for this deletion also leads to embryonic lethality. The mice that we describe in this report are hemizygous for 16 genes included in the Idd–Arvcf interval. Thirteen of these genes are included in the deletion described by Lindsay et al. (13).
The fact that mice hemizygous for a deletion spanning the sequence from Idd to Arvcf do not show reduced survival rates or any of the cardiovascular anomalies seen in the larger deletion of Lindsay et al. (13) suggests that none of the shared 13 genes, singly or in combination with one another, is responsible for the cardiovascular defects seen in VCFS/DGS patients. That leaves 9 other genes encompassed by the deletion of Lindsay et al. (13) as candidates for cardiovascular defects in VCFS/DGS patients. Mice with mutations in Comt (27) and Ufd1l (13) do not have any VCFS/DGS phenotypes, despite the claim that mutations in UFD1L are responsible for the VCFS phenotype in one patient (28). Recessive mutations in GP1Bβ are responsible for Bernard–Soulier disorder in humans, which is characterized by giant platelets, but these patients do not have any heart abnormalities (29). That leaves 6 other genes, haploinsufficiency of which may result in cardiovascular defects. It is also possible that hemizygosity of more than one gene is required to produce VCFS/DGS cardiovascular defects, and such genes might be located far apart within the VCFS/DGS deleted region. An attempt to complement the large deletion or other deletions of that nature in mice with the duplication chromosome that we describe here might reveal such a possibility.
Duplications and triplications of a large region of chromosome 22q11 are responsible for cat eye and derivative 22 syndromes (30). A part of the region that is duplicated in these two syndromes is represented in mice with the duplication chromosome described here (7, 31–33). However, mice with three or even four copies of the 16 known genes in this region are viable and normal, suggesting that the gene(s) responsible for the major developmental abnormalities in cat eye and derivative 22 syndromes may be located in another portion of human chromosome 22q11.
The engineering of chromosomes with a deletion and a duplication in ES cells was facilitated by the use of a dominant selectable marker system. In our system, recombination between the two LoxP sites results in a functional puromycin resistance gene. Reconstruction of a functional gene has been used previously as a strategy to select for Cre-mediated LoxP recombination events. In a series of reports, Bradley and colleagues (23, 34) described the use of an HPRT minigene to select for deletions of different sizes. However, the use of the HPRT minigene requires using HPRT-deficient ES cells. Because most ES cell lines are wild type, it is desirable to have a dominant selectable marker system that can be used in a variety of cell types. Clerc and Avner (35) attempted to use a phleomycin-resistance selectable marker, but they were not successful in obtaining a 65-kb deletion, because of problems with the drug selection. The puromycin-selectable system we used is robust, nonleaky, and reliable. We also compared two different Cre expression vectors and found that pCAG-Cre is much more efficient than pCMV-Cre in mediating recombination between LoxP sites in ES cells.
We also generated deletions in vivo, using two different Cre transgenes, CMV-Cre and ZP3-Cre, and found that ZP3-Cre was much more efficient, yielding 16 of 51 (31.3%) of offspring with a deletion. Because of the restricted expression of ZP3-Cre in the female germ line, the use of this transgene is well suited to generate LoxP-mediated rearrangements in vivo. Thus it is possible to generate numerous deletions or inversions in animals in which LoxP sites have been introduced into homologous chromosomes at distinct locations, bringing the LoxP sites together by meiotic recombination and mating with Cre-expressing transgenic mice. Such an approach should have important applications in discovering the causes of other human genomic disorders involving large chromosomal deletions or duplications, such as Williams syndrome, Smith–Magenis syndrome, and, most significantly, Down syndrome, which together constitute 0.7% of live births (reviewed in ref. 36).
Acknowledgments
We thank Eric Bouhassira for providing the CMV-Cre and CAG-Cre plasmids, Mark Lewandoski for the ZP3-Cre transgenic mouse line, and Harry Hou for ES cell injections. We are grateful to Bernice Morrow, Winfried Edelmann, and Jörg Heyer for helpful discussions. This research was supported by National Institutes of Health Grant HD34980.
Abbreviations
- VCFS
velocardiofacial syndrome
- DGS
DiGeorge syndrome
- MMU16
Mus musculus chromosome 16
- HSA22q11
Homo sapiens chromosome 22q11
- BAC
bacterial artificial chromosome
- NeoR
neomycin (or G418) resistance or resistant
- HygR
hygromycin resistance or resistant
- PuroR
puromycin resistance or resistant
- ES cells
embryonic stem cells
- En
embryonic day n
- HPRT
hypoxanthine phosphoribosyltransferase
- PGK
phosphoglycerate kinase
- CMV
cytomegalovirus
References
- 1.DiGeorge A. J Pediatr. 1965;67:907. doi: 10.1016/s0022-3476(65)80399-7. [DOI] [PubMed] [Google Scholar]
- 2.Shprintzen R J, Goldberg R B, Lewin M L, Sidoti E J, Berkman M D, Argamaso R V, Young D. Cleft Palate J. 1978;15:56–62. [PubMed] [Google Scholar]
- 3.Goldberg R, Motzkin B, Marion R, Scambler P J, Shprintzen R J. Am J Med Genet. 1993;45:313–319. doi: 10.1002/ajmg.1320450307. [DOI] [PubMed] [Google Scholar]
- 4.Edelmann L, Pandita R K, Morrow B E. Am J Hum Genet. 1999;64:1076–1086. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrow B, Goldberg R, Carlson C, Dasgupta R, Sirotkin H, Collins J, Dunham I, Odonnell H, Scambler P, Shprintzen R, et al. Am J Hum Genet. 1995;56:1391–1403. [PMC free article] [PubMed] [Google Scholar]
- 6.Iselius L, Lindsten J, Aurias A, Fraccaro M, Bastard C, Bottelli A M, Bui T H, Caufin D, Dalpra L, Delendi N, et al. Hum Genet. 1983;64:343–355. doi: 10.1007/BF00292366. [DOI] [PubMed] [Google Scholar]
- 7.Funke B, Edelmann L, McCain N, Pandita R K, Ferreira J, Merscher S, Zohouri M, Cannizzaro L, Dalpra L, Delendi N, et al. Am J Hum Genet. 1999;64:747–758. doi: 10.1086/302284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Lievre C S, Le Douarin N M. J Embryol Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- 9.Kirby M L, Waldo K L. Circulation. 1990;82:332–340. doi: 10.1161/01.cir.82.2.332. [DOI] [PubMed] [Google Scholar]
- 10.Dunham I, Shimizu N, Roe B, Chissoe S, Hunt A, Collins J, Bruskiewich R, Beare D, Clamp M, Smink L J, et al. Nature (London) 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 11.Lund J, Chen F, Hua A, Roe B, Budarf M, Emanuel B S, Reeves R H. Genomics. 2000;63:374–383. doi: 10.1006/geno.1999.6044. [DOI] [PubMed] [Google Scholar]
- 12.Amati F, Conti E, Novelli A, Bengala M, Diglio M, Marino B, Giannotti A, Gabrielli O, Novelli G, Dallapiccola B, et al. Eur J Hum Genet. 1999;7:903–909. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- 13.Lindsay E A, Botta A, Jurecic V, Carattini R S, Cheah Y C, Rosenblatt H M, Bradley A, Baldini A. Nature (London) 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 14.Ioffe E, Liu Y, Bhaumik M, Poirier F, Factor S, Stanley P. Proc Natl Acad Sci USA. 1995;92:7357–7361. doi: 10.1073/pnas.92.16.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazan J, Dubay C, Pankowiak M, Becuwe N, Weissenbach J. Genomics. 1992;12:183–189. doi: 10.1016/0888-7543(92)90364-x. [DOI] [PubMed] [Google Scholar]
- 16.Araki K, Imaizumi T, Okuyama K, Oike Y, Yamamura K. J Biochem. 1997;122:977–982. doi: 10.1093/oxfordjournals.jbchem.a021860. [DOI] [PubMed] [Google Scholar]
- 17.Robertson E. Teratocarcinomas and Embryonic Stem Cells. Oxford, U.K.: IRL; 1987. [Google Scholar]
- 18.Puech A, Saint J B, Funke B, Gilbert D J, Sirotkin H, Copeland N G, Jenkins N A, Kucherlapati R, Morrow B, Skoultchi A I. Proc Natl Acad Sci USA. 1997;94:14608–14613. doi: 10.1073/pnas.94.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Telenius H, Pelmear A H, Tunnacliffe A, Carter N P, Behmel A, Ferguson S M, Nordenskjold M, Pfragner R, Ponder B A. Genes Chromosomes Cancer. 1992;4:257–263. doi: 10.1002/gcc.2870040311. [DOI] [PubMed] [Google Scholar]
- 20.Cherif D, Der S H, Berger R. Hum Genet. 1994;93:1–6. doi: 10.1007/BF00218903. [DOI] [PubMed] [Google Scholar]
- 21.Botta A, Lindsay E A, Jurecic V, Baldini A. Mamm Genome. 1997;8:890–895. doi: 10.1007/s003359900606. [DOI] [PubMed] [Google Scholar]
- 22.Sutherland H F, Kim U J, Scambler P J. Genomics. 1998;52:37–43. doi: 10.1006/geno.1998.5414. [DOI] [PubMed] [Google Scholar]
- 23.Ramirez-Solis R, Liu P, Bradley A. Nature (London) 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- 24.White J K, Auerbach W, Duyao M P, Vonsattel J, Gusella J F, Joyner A L, MacDonald M E. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 25.Lewandoski M, Wassarman K M, Martin G R. Curr Biol. 1997;7:148–151. doi: 10.1016/s0960-9822(06)00059-5. [DOI] [PubMed] [Google Scholar]
- 26.Kimber W L, Hsieh P, Hirotsune S, Yuva-Paylor L, Sutherland H F, Chen A, Ruiz-Lozano P, Hoogstraten-Miller S L, Chien K R, Paylor R, et al. Hum Mol Genet. 1999;8:2229–2237. doi: 10.1093/hmg/8.12.2229. [DOI] [PubMed] [Google Scholar]
- 27.Gogos J A, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, Karayiorgou M. Proc Natl Acad Sci USA. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D. Science. 1999;283:1158–1161. doi: 10.1126/science.283.5405.1158. [DOI] [PubMed] [Google Scholar]
- 29.Kunishima S, Lopez J A, Kobayashi S, Imai N, Kamiya T, Saito H, Naoe T. Blood. 1997;89:2404–2412. [PubMed] [Google Scholar]
- 30.Schinzel A, Schmid W, Fraccaro M, Tiepolo L, Zuffardi O, Opitz J M, Lindsten J, Zetterqvist P, Enell H, Baccichetti C, et al. Hum Genet. 1981;57:148–158. doi: 10.1007/BF00282012. [DOI] [PubMed] [Google Scholar]
- 31.Edelmann L, Spiteri E, McCain N, Goldberg R, Pandita R, Duong S, Fox J, Blumenthal D, Lalani S R, Shaffer L G, Morrow B E. Am J Hum Genet. 1999;65:1608–1616. doi: 10.1086/302689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edelmann L, Pandita R K, Spiteri E, Funke B, Goldberg R, Palanisamy N, Chaganti R S, Magenis E, Shprintzen R J, Morrow B E. Hum Mol Genet. 1999;8:1157–1167. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- 33.Shaikh T, Budarf M, Celle L, Zackai E, Emanuel B. Am J Hum Genet. 1999;65:1595–1607. doi: 10.1086/302666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng B, Sage M, Sheppeard E, Jurecic V, Bradley A. Mol Cell Biol. 1999;20:648–655. doi: 10.1128/mcb.20.2.648-655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clerc P, Avner P. Nat Genet. 1998;19:249–253. doi: 10.1038/924. [DOI] [PubMed] [Google Scholar]
- 36.Lupski J R. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]