

SUMMARY
Endocannabinoids are well established as inhibitors of chemical synaptic transmission via presynaptic activation of the cannabinoid type 1 receptor (CB1R). Contrasting this notion, we show that dendritic release of endocannabinoids mediates potentiation of synaptic transmission at mixed (electrical and chemical) synaptic contacts on the goldfish Mauthner cell. Remarkably, the observed enhancement was not restricted to the glutamatergic component of the synaptic response but also included a parallel increase in electrical transmission. This novel effect involved the activation of CB1 receptors and was indirectly mediated via the release of dopamine from nearby varicosities, which in turn led to potentiation of the synaptic response via a cAMP-dependent protein kinase-mediated postsynaptic mechanism. Thus, endocannabinoid release can potentiate synaptic transmission and its functional roles include the regulation of gap junction-mediated electrical synapses. Similar interactions between endocannabinoid and dopaminergic systems may be widespread and potentially relevant for the motor and rewarding effects of cannabis derivatives.
INTRODUCTION
Endocannabinoids, diffusible retrograde lipidic messengers, regulate the strength of chemical synapses (reviewed in Chevaleyre et al., 2006). The central actions of both endogenous (typically anandamide and 2-arachidonoyl-glycerol) and exogenous (i.e. derivatives of marijuana products) cannabinoids are generally mediated by activation of the cannabinoid type 1 receptor (CB1R) (Freund et al., 2003). This receptor constitutes one of the most abundant G-protein coupled receptors in the mammalian brain and, although widespread, is heavily expressed in basal ganglia and prefrontal cortices, where dopamine regulation is believed to be essential for both motor control and reward mechanisms (Van der Stelt and Di Marzo, 2003). Regardless of the nature of the synapse (excitatory or inhibitory), the involved brain structure, or the duration of the effect (short- or long-term), endocannabinoids have been mostly reported to promote depression of synaptic transmission via a presynaptic mechanism (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Gerdeman et al., 2002; Robbe et al., 2002; Alger, 2002; Brown et al., 2003; Chevaleyre et al., 2006). Thus, release of endocannabinoids and/or activation of CB1Rs are expected to trigger synaptic depression, suggesting a general role in down-regulating chemical synaptic transmission within neural circuits across the nervous system.
In contrast, no effects of the CB1 signaling system on gap junction-mediated electrical synapses have been reported. Because of their accessibility to in vivo experimentation, identifiable auditory afferents terminating as mixed synaptic contacts on the lateral dendrite of the goldfish Mauthner (M-) cell known as Large Myelinated Club endings (“Club endings”) constitute a valuable model for the study of basic mechanisms of electrical and chemical transmission in vertebrates (reviewed in Pereda et al., 2004). The M-cells are a pair of unusually large reticulospinal neurons located in the medulla of teleosts and are essential for the organization of sensory-evoked escape responses (Korn and Faber, 2005). While excitatory chemical transmission at Club endings is mediated by glutamate (Wolszon et al., 1997), electrical transmission is mediated via homotypic connexin 35 (Cx35) gap junction channels (Pereda et al., 2003), the fish ortholog of the widespread mammalian neuronal connexin 36 (Cx36) (O’Brien et al., 1998; Condorelli et al., 1998). Remarkably, synapses at these terminals are highly modifiable and undergo activity-dependent potentiation of both the electrical and chemical components of their postsynaptic response (Yang et al., 1990; Pereda and Faber, 1996; Smith and Pereda, 2003). This potentiation is triggered by brief bursts of afferent activity and requires the activation of NMDA receptors and Ca2+/Calmodulin-dependent kinase II (Pereda et al., 1998).
Because CB1Rs were also reported to be present in teleost fish (Yamaguchi et al., 1996; McPartland et al., 2007), including goldfish (Yazulla et al., 2000; Cottone et al., 2005; Valenti et al., 2005), we asked whether endocannabinoids and CB1Rs were involved in promoting depression at these synapses, an essential requirement for the bi-directional control of the synaptic strength at these auditory afferents. Unexpectedly, we show here that dendritic endocannabinoid release leads to potentiation of both electrical and chemical transmission at Club endings. This novel potentiating effect requires activation of CB1Rs and is indirectly mediated by dopamine release from nearby varicosities, which in turn leads to potentiation of the synaptic response.
RESULTS
CB1 receptor activation enhances mixed synaptic transmission
Electrical stimulation of the posterior branch of the VIIIth nerve (where auditory afferents that terminate as Club endings run) evokes a distinct excitatory synaptic potential (“mixed EPSP”; Fig. 1A) composed of an early, brief gap junction-mediated electrical component (representing the electrical coupling of the presynaptic action potentials) and a delayed, longer lasting glutamatergic response. These two components of the synaptic potential can be unambiguously identified and reliably measured during intracellular dendritic recordings due to the brief membrane time constant of the postsynaptic M-cell, estimated as ~400 μs (Fukami et al., 1965). Endocannabinoids mediate their central regulatory actions via CB1Rs, and their activation is expected to depress chemical synaptic transmission (Chevaleyre et al., 2006). Thus, we hypothesized that CB1R activation would lead to depression of the chemical component of the mixed EPSP. Instead, we found that bath application of WIN 55,212-2, a CB1R agonist, led to a long-lasting enhancement of both components (Fig. 1B–C), averaging 136.4% ± 9.9 (p < 0.01) and 149.8% ± 13.0 (p < 0.01) of control values for the electrical and chemical components, respectively (n=8). The parallel changes in both modalities of transmission cannot be ascribed to increased input resistance of the M-cell lateral dendrite, as the amplitude of the antidromically evoked M-cell spike (a reliable estimate of this cell’s resistance due to the lack of active electrogenesis in soma-dendritic membrane) remained unchanged, averaging 102.6% ± 4.0 of its control value (n=8). In addition, the observed percent increases for each synaptic component were consistently different across experiments (ANOVA, F: 54.85, F crit: 3.93, p < 0.001) indicating that both forms of transmission were, although simultaneously affected, differentially modified by CB1R activation. The enhancements were blocked by pretreatment with a combination of the CB1R antagonists AM251 (AM; 4 μM) and SR141716 (SR; 4 μM), confirming that the observed effects were mediated via CB1R activation (Fig. 1D; electrical: 106.2% ± 5.7, p > 0.1; chemical: 111.7% ± 7.3, p > 0.1; n=8). As with previous studies in this preparation (Pereda and Faber, 1996; Wolszon et al., 1997), we chose to use a combination of blockers to overcome possible differences in drug potency related to structural differences between the mammalian CB1R and its teleost orthologues, which share homology of 59% to 72% (Yamaguchi et al., 1996; McPartland et al., 2007). Also, application of CB1R antagonists (Fig. 1E) did not evoke changes in the amplitude of either component of the mixed EPSP (electrical: 98.4% ± 7.5, p > 0.5; chemical: 110.0% ± 13.2, p > 0.1; n=8), indicating that endocannabinoids are not tonically released in this system.
Figure 1. Cannabinoids evoke long-term potentiation of electrical and chemical transmission at Club endings.

(A) Experimental arrangement. Electrical stimulation of the posterior branch of the VIIIth nerve (VIIIth nerve), where Club endings (Club endings, Mixed synapse) run, elicits a mixed excitatory postsynaptic potential (mixed EPSP) (B) Bath application of WIN 55,212-2 (WIN; 500 nM) results in a long-term enhancement of both components of the synaptic response. Top left, superimposed responses obtained 5 min before and 30 min after bath application of WIN (in all figures, traces represent the average of at least 10 responses). Top right, the antidromic spike (AD spike) height (a measure of M-cell input resistance) was slightly reduced. Bottom: time course of both components of the VIIIth nerve synaptic response (each point is the average of 10 single synaptic responses). (C) Averaged time course of the electrical and chemical components of the mixed EPSP after bath application of WIN (500 nM; n=8). (D) Pre-treatment with a combination of the CB1R antagonists AM251 and SR141716 (AM + SR; 4 μM ea) blocked WIN-evoked potentiation (n=8). (E) Application of AM and SR had no effect on the amplitude of the electrical or chemical components of the mixed EPSP (n=8).
The effects of bath application of WIN could result from activation of CB1Rs expressed on remotely located neurons, so we asked if local extracellular application to the surrounding area of the M-cell dendrite could evoke similar enhancements of the synaptic response. For this purpose, a second recording pipette with a solution containing WIN was positioned in the vicinity of these terminals in the distal portion of the lateral dendrite (Fig. 2A). Brief local application of WIN was sufficient to trigger a long-lasting enhancement of the mixed EPSP (Fig. 2A; electrical: 146.5% ± 10.5, p < 0.01; chemical: 148.0% ± 13.0, p < 0.05; n=6). These increases did not result from continued activation of CB1Rs, a possibility raised by the lipophilic nature of WIN, because they were not modified by subsequent bath application of the CB1R antagonists AM and SR (n=3; not shown).
Figure 2. Brief local activation of CB1R agonist is sufficient to trigger long-term potentiation of the mixed EPSP.

(A) Experimental arrangement for local drug application in the vicinity of Club endings contacts on the distal portion of the lateral dendrite and labeling with anti-Cx35 antibodies. WIN (5 μM) was pressure ejected for 15 s. Traces represent the synaptic responses obtained 10 min before and 30 min after local application. Top: time course of both components of the mixed EPSP. Bottom: averaged time course of both components for 6 experiments with local WIN application. (B) Immunochemical analysis reveals the presence of CB1Rs in the vicinity of the M-cell lateral dendrite. Laser scanning confocal immunofluorescence in a double-immunolabeling experiment with anti-CB1 1-77 (red; Alexa fluor 594), and anti-Cx35/36 (green, Alexa fluor 488) antibodies. The image reconstructs a long stretch of the lateral dendrite (average of 3 confocal Z-sections, totaling 1.5 μm) showing abundant presence of CB1Rs (red) in the vicinity of the Club endings revealed by Cx35 labeling (green). (C) Higher magnification image of the area delimited by the dotted box in B, illustrating the close proximity of CB1Rs to Club endings. (D) Lateral view (average of 3 confocal Z-sections totaling 1.5 μm) shows labeling for CB1 in the immediate proximity of both the M-cell dendrite and a Club ending. The position of the M-cell membrane (dotted lines) was estimated from the DIC image of the section.
Local WIN application suggested that endocannabinoids could mediate their actions via activation of CB1Rs located in the proximity of these terminals. Immunohistochemical studies with polyclonal antibodies raised against either the first 14 (anti-CB1 1-14), the first 77 amino acids (anti-CB1 1-77) or the carboxy-terminal (anti-CB1-CT) of the rat CB1R showed distinct punctate labeling near the distal portion of the lateral dendrite (Figs. 2 and 7A) in the immediate vicinity of Club endings. This relationship is illustrated in Fig. 2B, where the position of Club endings along the membrane of the M-cell lateral dendrite is revealed by an antibody against Cx35 (anti-Cx35/36). Interestingly, CB1-labeled puncta (anti-CB1 1-77), although very close, do not appear to be on either the Club ending or the M-cell membranes (Fig. 2C), a distribution more clearly observed in side views of these terminals (Fig. 2D).
Figure 7. CB1Rs are closely associated to dopaminergic fibers.

(A) Laser scanning confocal immunofluorescence with double-immunolabeling by polyclonal anti-CB1-CT (red, Alexa fluor 594) and monoclonal anti-TH (green; Alexa fluor 488) antibodies. Image is the average of 31 confocal Z-sections (totaling 6.2 μm). (B) Magnification of the boxed area from A illustrates close association between CB1Rs and dopaminergic fibers, compatible with presynaptic localization of CB1Rs on the membrane of the varicosities (image is the average of 9 Z-sections, totaling 1.8 μm).
Dopamine mediates WIN-evoked potentiation
The distribution of CB1-labeled puncta resembles the dopaminergic varicosities that surround Club endings at the distal portion of the M-cell lateral dendrite (Fig. 3A). Previous studies revealed that dopaminergic varicosities lie in close proximity (within a few μm) to the M-cell and Club ending membranes and that brief local dopamine application evokes long-lasting enhancements of both components of the synaptic response (Pereda et al., 1992). These enhancements require activation of dopamine D1/5 receptors (D1/5 Rs) that act via a postsynaptic PKA phosphorylation pathway (Pereda et al., 1994). Thus, we hypothesized that the synaptic enhancements evoked by WIN application are mediated via dopamine release from nearby varicosities. Pretreatment with the D1/5 R antagonist SCH-23390 (50 μM) prevented WIN-evoked enhancements of the mixed EPSP (Fig. 3B; electrical: 94.4% ± 7.4, p > 0.1; chemical: 98.2% ± 10.4, p > 0.5; n=8). Consistent with this hypothesis, WIN-evoked enhancements of the mixed EPSP were also blocked by intracellular application of PKI 5-24 (PKI), a specific PKA inhibitory peptide (Fig. 3C; electrical: 89.4%, ± 6.6, p > 0.1; chemical: 88.4% ± 6.0, p > 0.1; n=5). Furthermore, the synaptic enhancement evoked by bath application of WIN occluded the effects of locally-applied dopamine (Fig. 3D; n=3), suggesting that both forms of potentiation share common mechanisms. In contrast, WIN-evoked enhancements did not occlude the potentiation triggered by stimulating the posterior VIIIth nerve with brief (10 ms) high frequency (500 Hz) trains (Fig. 3D), which is known to follow a different, Ca2+-dependent intracellular signaling pathway (Pereda et al., 1998).
Figure 3. Dopamine mediates WIN-evoked potentiation of synaptic transmission at Club endings.
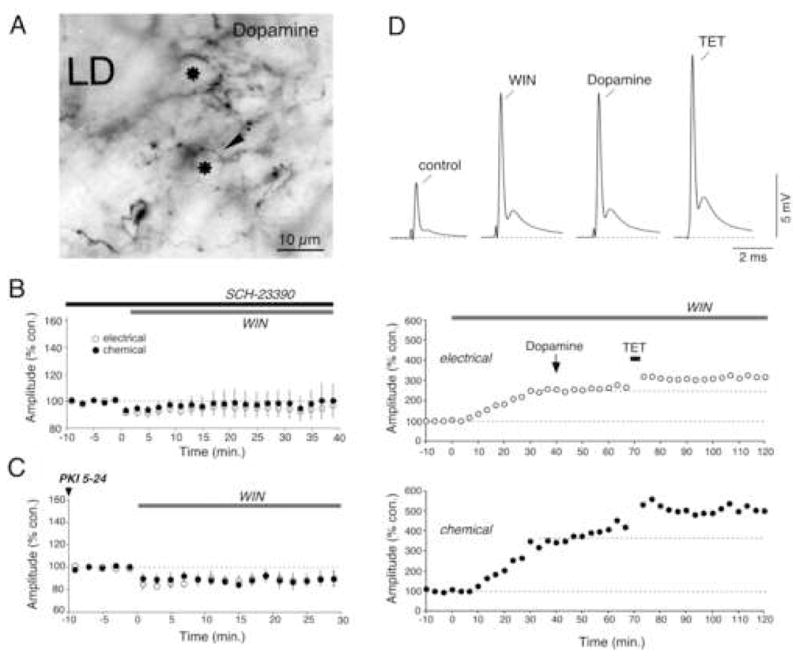
(A) Dopaminergic innervation of the M-cell lateral dendrite (LD). Immunoreactive profiles obtained with an anti-dopamine antibody under Nomarski optics reveal the presence of numerous varicose fibers (arrow) lying between the Club endings, which appear in cross section (asterisk). (B) Averaged time course of both components of the mixed EPSP in 8 experiments in which WIN application followed pretreatment with the D1/5 antagonist SCH-23390 (50 μM). (C) Averaged time course for 5 experiments in which the PKA inhibitor PKI (500 μM) was intradendritically injected prior to bath application of WIN. (D) WIN-evoked potentiation occludes dopamine-evoked potentiation (10 mM). Top: Mixed EPSPs obtained at control (control) and after each experimental manipulation (WIN, Dopamine, TET) in one representative experiment. Below: time course of the amplitudes of the electrical and chemical components of the mixed EPSP. Bath application of WIN triggers a long-lasting potentiation of the mixed EPSP that occludes the effects of local dopamine application. In contrast, WIN-evoked potentiation did not occlude activity-dependent potentiation triggered by discontinuous high frequency stimulation of the VIIIth nerve (TET: burst of 6 pulses at 500 Hz, every 2 s over 4 minutes).
Endocannabinoids are released from the lateral dendrite of the Mauthner cell
Because endocannabinoids have limited diffusion properties, estimated to be ~10–20 μm in rodent hippocampus at 20–22°C (Wilson and Nicoll, 2001; Chevaleyre and Castillo, 2003), we hypothesized that the source of their production must lie in the vicinity of dopaminergic varicosities. Furthermore, because the synthesis and release of endocannabinoids usually takes place at postsynaptic cells, often following activation of group I metabotropic glutamate receptors (Maejima et al., 2001; Varma et al., 2001; Ohno-Shosaku et al., 2001; Galante and Diana, 2004), we hypothesized that endocannabinoids are released from M-cell dendrites. Accordingly, immunochemical analysis with confocal microscopy and an mGluR1α antibody (anti-mGluR1) revealed strong labeling at Club ending synapses (Fig. 4A). Interestingly, labeling was not restricted to the periphery of these mixed contacts, where chemical synapses are located (Pereda et al., 2003), suggesting the existence of an extrasynaptic distribution of mGluR1 receptors (i.e.; beyond the limits of chemical synaptic domains but within the boundaries of the contact; Fig. 4B–C). Despite the presence of mGluR1 receptors, local application of (RS)-3,5-dihydroxyphenylglycine (DHPG; 1 mM), a group I mGluR receptor agonist, did not result in an enhancement of the mixed EPSP (Fig. 4D; electrical: 90.8% ± 10.4, p > 0.1; chemical: 87.8% ± 11.1, p > 0.1; n=6). Endocannabinoids are also known to be released following postsynaptic depolarizations (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Kreitzer and Regehr, 2001). However, as with DHPG applications, strong dendritic depolarizations (5 pulses of 5 s duration and 90 nA of intensity) expected to bring the membrane potential to about 0 mV (Experimental Procedures) did not elicit changes in the amplitude of either component (Fig. 4E; electrical: 108.0% ± 8.9, p > 0.1; chemical: 92.9% ± 6.3, p > 0.1; n=6).
Figure 4. Endocannabinoids are released from the Mauthner cell lateral dendrite.

(A) Immunochemical analysis reveals the presence of mGluR1 at Club endings. Laser scanning confocal immunofluorescence image obtained with polyclonal anti-mGlur1 (green, Alexa fluor 488); average of 11 confocal Z-sections totaling 5.5 μm. Club endings (arrowheads), unambiguously identified because of their larger size, exhibit punctate labeling for mGluR1. (B, C) Higher magnification images showing the distribution of mGluR1 at two Club endings. Note that the labeling is not restricted to the periphery of the contact, where chemical synapses are localized. (D–G) Both mGluR1 activation and dendritic depolarization are required for endocannabinoid release. (D) Time course of the electrical and chemical components of the mixed EPSP in experiments with local application of the mGluR I agonist DHPG (1 mM) in the vicinity of the M-cell dendrite (n=8). (E) Time course of experiments with dendritic depolarization of the M-cell (5 pulses, 5 s duration each, 90 nA of current, “depo”; n=6). (F) Combination of local application of DHPG and dendritic depolarization triggered long-lasting enhancement of the mixed EPSP (n=6). (G) CB1R antagonists AM and SR prevented the potentiation triggered by DHPG application and dendritic depolarization (n=6).
However, recent evidence shows that these manipulations can cooperate for the release of endocannabinoids (Ohno-Shosaku et al., 2002). We found that when local application of DHPG was followed by dendritic depolarization, it successfully triggered lasting potentiations of the mixed EPSP (Fig. 4F; electrical: 151.1% ± 11.5, p < 0.01; chemical: 148.1% ± 15.0, p < 0.05; n=6). This protocol was more efficient (occurring in 100% of the trials) when dendritic depolarizations were applied 10 minutes after local application of DHPG (Fig. 4F). This is likely due to the diffusion time required for this agonist to reach a significant number of the synaptic contacts, which are distributed along a ~250 μm length of the M-cell lateral dendrite. Consistent with this potentiation being mediated by endocannabinoid release, the observed effects were blocked by pretreatment with the CB1R antagonists AM and SR (Fig. 4G; electrical: 108.7% ± 4.6, p > 0.1; chemical: 105.6% ± 6.1, p > 0.1; n=6).
Given the properties of the mixed synaptic responses, the combined requirement of mGluR activation and dendritic depolarization suggested that patterns of activity of Club endings could be responsible for endocannabinoid release under natural conditions. Club endings represent the overwhelming majority of the excitatory inputs in this portion of the M-cell lateral dendrite, and the electrical component of their mixed EPSP evokes a large dendritic depolarization that is immediately followed by a glutamatergic response (the depolarization produced by the electrical component is generally 4–5 times bigger than that of the glutamatergic response; Fig. 5A). Therefore, we asked if patterns of stimulation of the posterior VIIIth nerve could trigger long-term enhancement of the synaptic response. We found that repetitive activity at 100 Hz, a frequency known to promote endocannabinoid release in other systems (Maejima et al., 2001; Chevaleyre and Castillo, 2003; Galante and Diana, 2004; Brenowitz and Regehr, 2005), evoked long-term potentiation of the mixed EPSP (Fig. 5B; electrical: 161.1% ± 14.0, p < 0.05; chemical: 178.6% ± 18.2, p < 0.05; n=5; antidromic spike was unchanged: 97.7% ± 8.0). This prolonged pattern of activity resembled the conditions found for endocannabinoid release (Fig. 4F) and was radically different from that used for the induction of a distinct “long-term potentiation”, which required brief bursts of 10 ms duration at 500 Hz (6 pulses) over several minutes (Yang et al., 1990). Furthermore, the potentiation triggered by this stimulating paradigm involved endocannabinoid release, as the observed effects were blocked by pretreatment with AM and SR (Fig. 5C; electrical: 112.2% ± 17.7, p > 0.5; chemical: 114.0% ± 16.3, p > 0.1; n=5). Thus, our results indicate that endocannabinoids are produced and released on demand from the M-cell lateral dendrite as a result of the activity of Club endings, leading to long-term potentiation of both components of the synaptic response via CB1R activation.
Figure 5. Endocannabinoid release is triggered by synaptic activity.

(A) Repetitive stimulation (100 Hz) of the posterior VIIIth nerve provides both strong depolarization via gap junctions and glutamate release. (B) Repetitive stimulation of the posterior VIIIth nerve (100 Hz during 1 s × 5) evoked robust potentiation of both components of the mixed EPSP (n=5). (C) Pre-treatment with CB1R antagonists AM and SR (4 μM) blocked synaptic potentiation (n=5). (D) Intradendritic injections of THL (100 μM), a diacylglycerol lipase (DGL) inhibitor, prevented synaptic potentiation (n=6). (E) Dopamine D1/5R activation is required for synaptic potentiation triggered by repetitive stimulation. Pretreatment with the D1/5 antagonist SCH-23390 (50 μM) blocked synaptic potentiation (n=5).
The endocannabinoid 2-arachidonoyl-glycerol (2-AG) has been implicated in several forms of synaptic plasticity in the mammalian nervous system (Chevaleyre et al., 2006). Therefore, we asked if this endocannabinoid was also responsible for the observed potentiations at goldfish mixed synapses. To explore this possibility we tested the effects of tetrahydrolipstatin (THL; 100 μM), an inhibitor of diacylglycerol lipase (DGL), which converts diacylglycerol into 2-AG, on the ability of the 100 Hz pattern of stimulation to evoke long-term changes in the synaptic strength. Intradendritic injections of THL prevented potentiation of the mixed EPSP (Fig. 5D; electrical: 107.3% ± 6.6, p > 0.1; chemical: 102.9% ± 7.0, p > 0.5; n=5).
Our data suggest that the CB1R mediates its effects via the dopamine release from nearby varicosities. To confirm this, we tested the effects of pretreatment with the D1/5 R antagonist SCH-23390 (50 μM) on the ability of the 100 Hz stimulation protocol to evoke long-term changes in the synaptic strength at these terminals. As illustrated in Fig. 5E, SCH-23390 prevented the potentiation of the mixed EPSP (electrical: 92.3% ± 3.7, p > 0.1; chemical: 92.3% ± 4.3, p > 0.1; n=5). Thus, activity-dependent production of the endocannabinoid 2-AG at the M-cell lateral dendrite is sufficient to trigger local dopamine release, which in turn evokes a long-term potentiation of the mixed synaptic response.
CB1 receptors are closely associated with dopaminergic varicosities
Endocannabinoids are expected to suppress transmitter release by acting on presynaptic CB1Rs. In contrast, our data strongly suggest that activation of these receptors leads to increased dopamine release. A mechanism compatible with the well-established conventional presynaptic effects of endocannabinoids would be that dopamine release is enhanced as a result of suppression of GABA release from nearby inhibitory terminals. This phenomenon, known as “dis-inhibition”, is proposed to occur at soma of dopaminergic neurons in the ventral tegmental area of the rat (Szabo et al., 2002) and substantia nigra of the mouse (Wallmichrath and Szabo, 2002). Supporting this possibility, Club endings and dopaminergic varicosities are closely surrounded in the long and smooth (spineless) lateral dendrite of the M-cell by a large number of inhibitory terminals (“small vesicle boutons”) (Tuttle et al., 1987), which are predominantly GABAergic (Triller et al., 1993). As in other systems, CB1Rs could act presynaptically on these terminals to inhibit release (Freund et al., 2003). Given the presence of numerous inhibitory terminals in the lateral dendrite, spillover of GABA could act to influence transmitter release from nearby dopaminergic varicosities. Indeed, spillover of transmitter has been shown to occur between inhibitory synapses on the M-cell (Faber and Korn, 1988). Experimentally, these terminals can be activated by antidromically stimulating the M-cell axon, as they participate in a feed-back inhibitory circuit (Fig. 6A). Since the equilibrium potential of chloride (Cl−) in the M-cell lies close to its resting potential, visualization of the feed-back inhibitory synaptic potential requires intracellular Cl− loading (Fig. 6B), often difficult to maintain at constant levels. A more accurate assay for the strength of these inhibitory connections is to measure the change in membrane conductance (“shunt”) produced by the opening of ligand-gated Cl− channels. The conductance change (“fractional conductance”) is proportional to the ratio between the amplitude of the M-cell antidromic spike in the absence and presence of the inhibitory synaptic potential (Oda et al., 1995). For this purpose, a second antidromic stimulating pulse was applied 6 ms after the first, conditioning pulse (Fig. 6C). We asked if this ratio could be affected by local application of WIN or dendritic release of endocannabinoids (DHPG + depolarization) and found that neither manipulation led to changes in fractional conductance, averaging 105.8% ± 9.3, p > 0.5 and 100.5% ± 7.6, p > 0.9, respectively (n=6).
Figure 6. Inhibitory synaptic transmission to the M-cell in the vicinity of Club endings is not modified by CB1R activation.
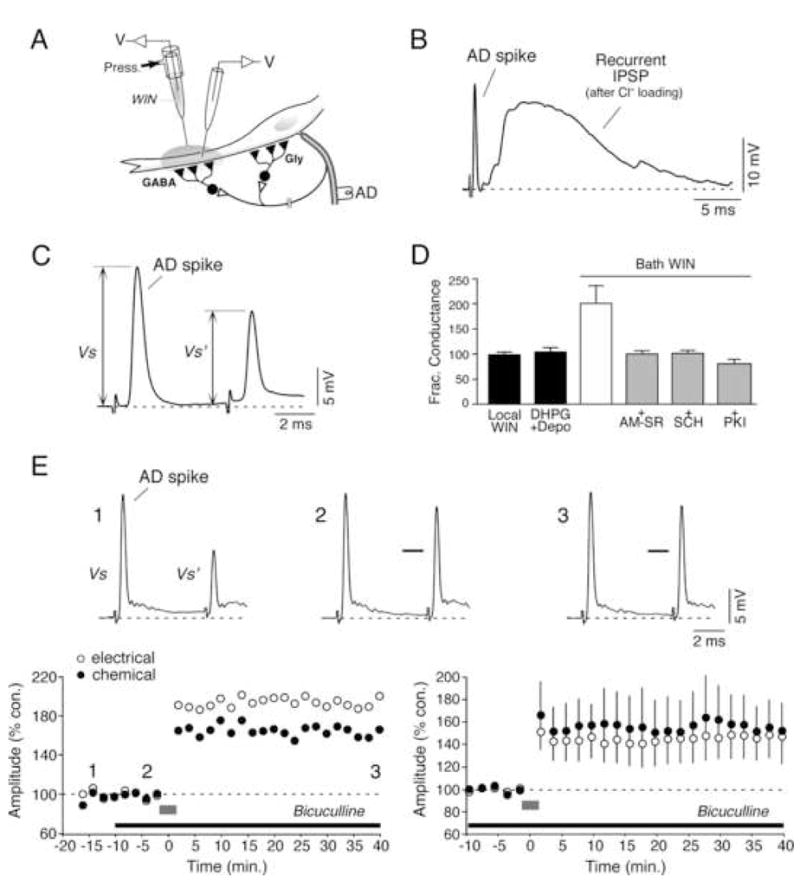
(A) Experimental arrangement. The cartoon illustrates the relative distribution of inhibitory interneurons on the M-cell soma (mostly glycinergic) and lateral dendrite (mostly GABAergic), which are recurrently activated by antidromic stimulation (AD). (B) Cl− loading of the M-cell reveals the presence of a powerful recurrent inhibitory synaptic potential in response to antidromic stimulation (AD spike). (C) The strength of the feed-back inhibition was quantified by determining the fractional conductance (expressed as % control). (D) Neither local application of WIN (Local WIN) nor local release of endocannabinoids triggered by DHPG and dendritic depolarization (DHPG+Depo) affected the strength of recurrent inhibition. In contrast, bath application of WIN (Bath WIN) led to a marked increase in recurrent inhibition suggesting that inhibitory synapses in the soma are sensitive to endocannabinoid modulation. This enhancement was prevented by application of CB1R antagonists AM and SR, the dopamine D1/5R antagonist SCH-23390 and the PKA blocker PKI. (E) Bath application of the GABAA receptor antagonist bicuculline (75 μM) prior to repetitive stimulation (100 Hz) of the posterior VIIIth nerve does not prevent the induction of activity-dependent synaptic potentiation. Bottom left: time course of the mixed EPSP from a single experiment during baseline recordings following repetitive stimulation of the posterior VIIIth nerve (gray bar). Note the lack of modification in baseline following the application of bicuculline and prior to repetitive stimulation. Top: responses to antidromic stimulation of the M-cell axon obtained at the time points 1 (−13 min), 2 (−4 min.) and 3 (40 min.) indicated in the graph. Bicuculline led to a suppression of recurrent inhibition at time points 2 and 3, indicated by the increase in the amplitude of the second antidromic action potential (bars at the second antidromic spikes in 2 and 3 indicate the amplitude prior to drug application). Bottom right: averaged time course of 5 experiments in which pre-treatment with bicuculline did not prevent the potentiation triggered by repetitive stimulation at 100Hz.
The absence of effect on dendritic inhibition did not result from the inability to detect changes in synaptic inhibition, as this ratio was increased after bath application of WIN, indicating an unexpected enhancement of feed-back inhibitory transmission. Because dendritic applications did not induce changes in the inhibitory synaptic response, we concluded that the observed enhancement must result from changes in feed-back inhibitory neurons innervating the M-cell soma (located ~500 μm from the dendritic injection site), which are predominantly glycinergic (Fig. 6A) (Triller et al., 1993). Remarkably, this effect was not observed in the presence of CB1R antagonists (AM-SR), the D1/5R antagonist SCH-23390, or following intracellular injections of PKI; fractional conductance averaged 99.2% ± 6.4% (p > 0.5), n=8; 100.2% ± 6.2 (p > 0.5), n=8, and 80.0% ± 7.9 (p > 0.05), n=5, respectively (Fig. 6D), suggesting that the observed enhancement of synaptic inhibition was also mediated by dopamine release via a postsynaptic mechanism. In agreement, immunochemical analysis showed that both dopaminergic fibers and CB1Rs were distributed in the vicinity of the M-cell soma (not shown).
The lack of effect of cannabinoids on dendritic inhibition, largely GABAergic (Triller et al., 1993), suggests that dis-inhibition mechanisms are unlikely to underlie dopamine release from nearby varicosities. To directly determine if dis-inhibition is involved in promoting the dopamine release, we studied the effect of the GABAA antagonist bicuculline on both the amplitude of the mixed EPSP and the ability of repetitive stimulation of the VIIIth nerve at 100 Hz to trigger potentiation. If dopamine release results from a reduction of GABA inhibitory influences, the addition of bicuculline is expected to induce an enhancement of the mixed EPSP. In contrast, we found that bath application of bicuculline did not lead to significative changes in the baseline amplitude of either component of the mixed EPSP (Fig. 6E, bottom left). Furthermore, pretreatment with bicuculline did not prevent activity-dependent potentiation of the electrical and chemical components of the synaptic response evoked by repetitive stimulation of the VIIIth nerve at 100 Hz (148.9% ± 16.6, p < 0.5 and 156.4% ± 19.4, p < 0.5, respectively; n=5; Fig. 6E, bottom right). In contrast, bicuculline abolished recurrent inhibition (Fig. 6E, top).
Thus, our results indicate that dis-inhibition of dopaminergic varicosities near the lateral dendrite does not constitute the mechanism underlying dopamine release. However, recent evidence demonstrates that CB1Rs are presynaptically distributed in the axons and varicosities of other aminergic neurons (Oropeza et al., 2006), suggesting that their activation could be responsible for the release of noradrenaline (Oropeza et al., 2005). Therefore, we investigated the colocalization of CB1Rs and dopaminergic varicosities in the M-cell lateral dendrite via double-immunolabeling with anti-CB1-CT and anti-tyrosine hydroxylase (anti-TH) antibodies (Fig. 7A). We found close association and overlap of CB1 labeled puncta and TH-labeled dopaminergic fibers and varicosities in a pattern consistent with localization of these receptors on dopaminergic varicosities or processes immediately adjacent to them (Fig. 7B). The observed distribution suggests that activation of CB1Rs could directly promote the dopamine release via unconventional presynaptic mechanisms. As illustrated in Fig. 7A, most of the CB1 labeled puncta were not associated with TH-labeled fibers, indicating a wide distribution of this ubiquitous receptor in other cell types of the goldfish hindbrain. Nevertheless, the incidence of the association between CB1 and dopaminergic varicosities was certainly underestimated, as anti-TH immunofluorescence proved to have a lower efficiency (~20%) in detecting these fibers when compared with that of anti-dopamine antibodies combined with peroxidase methods (Fig. 3A; also Pereda et al., 1992).
Cx35 is a direct target of PKA phosphorylation at Club endings
Cx35 contains several PKA phosphorylation sites, and PKA can directly phosphorylate Cx35 to modify its function (Mitropoulou and Bruzzone, 2003; Ouyang et al., 2005; Urschel et al., 2006). We asked whether Cx35 could be similarly phosphorylated at Club endings. For this purpose, Western blots were obtained from small areas of the goldfish hindbrain containing the M-cells with a polyclonal antibody raised against a PKA phosphorylation site at serine 110 of Cx35 (anti-Cx35/P110). This antibody labeled Cx35 (two closely spaced bands at 32–33 kDa) and labeling was lost upon alkaline phosphatase digestion, suggesting that this connexin is also the target of PKA phosphorylation in neurons of this region that includes the M-cells (Fig. 8A). Accordingly, immunohistochemical studies using the anti-Cx35/P110 antibody revealed abundant punctate labeling at Club endings, which were unambiguously identified by the presence of labeling with the monoclonal anti-Cx35/36 antibody (Fig. 8B,C).
Figure 8. PKA phosphorylates connexin 35 in Club endings.
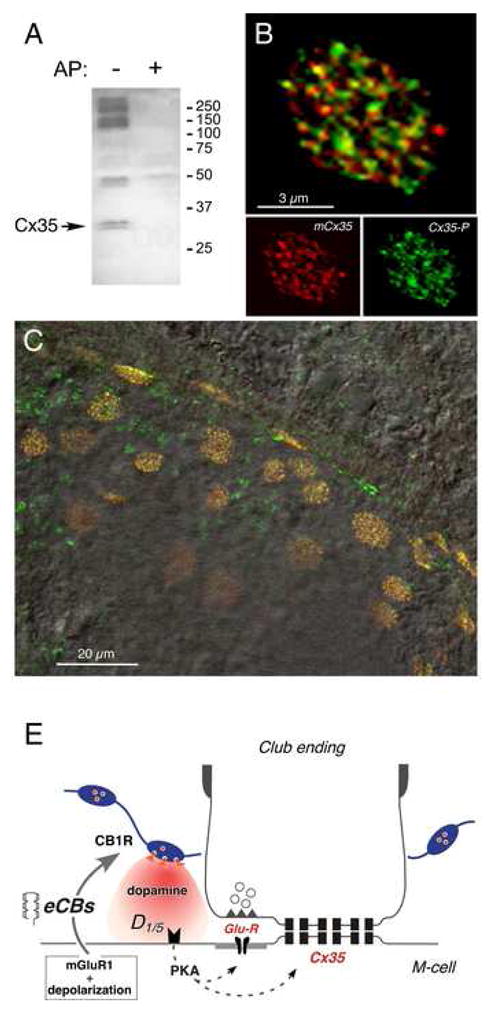
(A) Polyclonal anti-Cx35/P110 antibody recognizes Cx35 as a pair of bands at 32–33 kDa (arrow, left lane, AP-) in membranes from a small area of goldfish hindbrain where M-cells are located. Labeling is lost in membranes digested with alkaline phosphatase (right lane, AP+). The high molecular weight phospho-proteins labeled non-specifically by the antibody are not associated with Cx35 (Kothmann et al., 2007). 50 μg/lane crude membrane protein. (B) Laser scanning confocal immunofluorescence of a single Club ending with double-labeling by polyclonal anti-Cx35/P110 (green, Alexa fluor 488), and monoclonal anti-Cx35/36 (mCx35; red, Alexa fluor 594) antibodies. Top: superimposition of individual mCx35 and Cx35-P images (bottom), average of 3 confocal Z-sections (totaling 1.5 μm). (C) Lower magnification image showing extensive co-localization at individual Club endings in a section of the M-cell lateral dendrite. A DIC image of this region is superimposed. (D) Model for endocannabinoid-mediated potentiation of electrical and chemical synaptic transmission at Club endings. Synaptic activity leads to mGluR activation paired with postsynaptic membrane depolarization, triggering endocannabinoid (eCB) release from the postsynaptic M-cell dendrite, which activates CB1Rs on dopaminergic fibers. CB1R activation leads to dopamine release which, by activating postsynaptic D1/5 receptors, increases PKA activity responsible for simultaneous potentiation of electrical (Cx35) and glutamatergic (GluR) synaptic transmission.
DISCUSSION
Contrasting with their well-defined role in promoting depression of synaptic transmission, our results show that endocannabinoids evoked long-term enhancement of synaptic transmission at mixed contacts between Club endings and the M-cell lateral dendrite. As previously demonstrated in other cell types, endocannabinoids are released from the postsynaptic dendrite (Freund et al., 2003); in this case, release requires simultaneous mGluR1 activation and postsynaptic depolarization. Such requirements are ideally suited to the characteristics of the synaptic potential evoked by Club endings, where glutamate release is always accompanied by the large dendritic depolarization produced by the spread of the presynaptic action potentials via the numerous gap junctions present at these terminals. Accordingly, we show that activity of these afferents leads to endocannabinoid release and subsequent potentiation of synaptic transmission, suggesting that this signaling system is likely to participate in the regulation of the synaptic strength of these afferents under natural conditions. Club endings provide the M-cell with essential auditory information for the initiation of an escape response (Korn and Faber; 2005). Activity-dependent release of endocannabinoids may have an important functional role, as the increased synaptic gain of these VIIIth nerve synapses will sensitize this vital behavior, lowering its threshold to auditory stimuli.
The enhancements in transmission observed following endocannabinoid release and CB1R activation cannot be ascribed to possible interactions with other well known potentiating mechanisms present at Club endings (Yang et al., 1990), as those do not require PKA activation (Kumar and Faber, 1999; Pereda et al., 1998). Furthermore, the stimulation pattern used to trigger the observed endocannabinoid-mediated potentiation was radically different from that used for the induction of the previously described “long-term potentiation” of both components of the mixed synaptic response (Yang et al., 1990). In contrast to that used to trigger endocannabinoid release, this protocol consists of brief (10 ms) high frequency (500 Hz) trains of stimuli applied every 2 seconds during 4 minutes, a pattern known to optimize activation of NMDA receptors that are required for the induction of these synaptic changes (Yang et al., 1990; Pereda et al., 1996). This potentiation requires an intracellular increase in Ca2+ that in turn leads to the activation of Ca2+/calmodulin dependent kinase II (Pereda et al., 1998) and was not prevented by intracellular injection of PKA inhibitors or bath application of dopamine D1/5R antagonists (Kumar and Faber, 1999). Thus, our results suggest that several activity-dependent mechanisms might converge for the regulation of the synaptic strength at these auditory afferents.
Endocannabinoids regulate the strength of electrical synapses
Our data demonstrate that electrical synapses, whose functional roles in the mammalian CNS have been increasingly recognized (Connors and Long, 2004), are the target of endocannabinoid control. Most of the known effects of cannabinoids on gap junctional communication are so far restricted to non-neural tissue (Brandes et al., 2002; Upham et al., 2002). In the nervous system the endogenous cannabinoid anandamide was reported to decrease junctional conductance between cultured astrocytes, although this effect was not mediated via activation of CB1Rs (Venance et al., 1995). Given that electrical transmission at these terminals is mediated by Cx35 (Pereda et al., 2003), the fish ortholog of the widespread neuron-specific Cx36 (O’Brien et al., 1998; Condorelli et al., 1998), our results suggest that mammalian electrical synapses could be similarly regulated. Endocannabinoids, in particular 2-AG, have a well-established role in modulating chemical synaptic transmission (Alger, 2002; Freund et al., 2003; Chevaleyre et al., 2006). The present results show that their modulatory role is not restricted to a specific modality of synaptic transmission, and they suggest a general role for the endocannabinoid system in shaping interneuronal communication in general.
CB1R-evoked dopamine release potentiated both forms of transmission via postsynaptic activation of PKA. Ionotropic glutamate receptors are known to be targets of PKA modulatory action in a variety of experimental preparations where PKA phosphorylation mediates changes of synaptic transmission (Swope et al., 1999). Both Cx35 and its mammalian ortholog Cx36 have also been shown to be targets of PKA phosphorylation (Mitropoulou and Bruzzone, 2003; Ouyang et al., 2005; Urschel et al., 2006). We show here that Cx35 is phosphorylated at regulatory PKA phosphorylation sites in Club endings. Thus, endocannabinoid release is likely to potentiate electrical transmission via a direct action on this connexin.
Synaptic potentiation is mediated via the release of dopamine
The effects of cannabinoids required both CB1 and D1/5 receptor activation, indicating that potentiation is mediated indirectly via dopamine release. We previously showed the presence of a rich dopaminergic innervation in the vicinity of the M-cell lateral dendrite and demonstrated that local dopamine application is sufficient to trigger a lasting potentiation of both components of the synaptic response (Pereda et al., 1992). We show now that dopamine release is under the regulatory control of local factors such as dendritic endocannabinoid production. Thus, by the action of neurotransmitter modulators, endocannabinoids can amplify and promote regulatory actions that go beyond their conventional effects on synaptic transmission. Furthermore, because endocannabinoids are thought to travel no more than 10–20 μm, dopamine release could also act to spatially amplify their regulatory effects, as this less lipophilic neurotransmitter would diffuse a greater distance along the dendritic tree.
We found a close association of CB1Rs with dopaminergic varicosities, suggesting their localization on these terminals. While this possibility is consistent with the classical distribution of these receptors on presynaptic terminals, it is in contradiction with the conventional suppressive effects of CB1Rs on synaptic transmission (Freund et al., 2003), as activation of these receptors would lead instead to increased dopamine release. Recently, similar presynaptic distribution of CB1Rs was reported in the axons and varicosities of other monoaminergic neurons (Oropeza et al., 2006), where they have been proposed to promote release of noradrenaline (Oropeza et al., 2005). Taken together, these findings raise the possibility that CB1Rs located on monoaminergic varicosities could be coupled to alternative signaling pathways, making them capable of enhancing transmitter release. Consistent with this possibility, recent evidence shows that CB1Rs are not exclusively coupled to Gi/o (which is in turn negatively coupled to adenylate cyclase) but also to Gq/11 (Lauckner et al., 2005) and Gs (Glass and Felder, 1997), suggesting that intracellular signaling triggered by activation of these receptors could be more diverse than originally anticipated. Supporting these observations, studies in slices of globus pallidus (Maneuf and Brotchie, 1997), cultured striatal neurons (Glass and Felder, 1997; Jarrahian et al., 2004) and chinese hamster ovary cells (Bonhaus et al., 1998) reported that CB1R activation can lead to increases in the intracellular concentration of cAMP. Furthermore, activation of CB1Rs in neuroblastoma cells was shown to promote an increase in cytoplasmic Ca2+ levels through a pathway involving both Gs and PKA (Rubovitch et al., 2002; Bash et al., 2003). Thus, the ability of these receptors to promote increases in cAMP and intracellular Ca2+ opens the possibility that similar mechanisms could underlie enhanced transmitter release from dopaminergic varicosities. Finally, CB1Rs could enhance dopamine release by modifying presynaptic excitability and/or the properties of presynaptic action potentials, as these receptors have been shown to target K+ currents in goldfish retinal bipolar cells (Fan and Yazulla, 2005).
An alternative possibility that we considered was that CB1Rs located on nearby inhibitory terminals or processes that are presynaptic to dopaminergic varicosities act to regulate GABA release. This mechanism seems highly unlikely given the lack of effect on dendritic inhibition and the absence of direct contacts with either the M-cell or presynaptic terminals in serial EM reconstructions of dopaminergic varicosities (Pereda et al., 1992). This possibility was directly ruled out in experiments where activity-dependent potentiation was observed in the presence of blockers of GABAergic transmission, indicating that inhibition is not required for the induction or expression of this potentiation. Thus, our results indicate that dopamine release is not mediated by “dis-inhibition” of dopaminergic varicosities, and therefore, activation of the CB1R must necessarily increase dopamine release directly.
Enhancement of inhibitory synaptic transmission
Interestingly, our results show that while inhibitory synapses on the M-cell lateral dendrite are insensitive to CB1R activation, those targeting the soma were enhanced by CB1R activation. This finding represents a novel endocannabinoid regulatory action, as inhibitory transmission in hippocampus and neocortex is consistently reported to be depressed by CB1R activation (Freund et al., 2003; Chevaleyre et al., 2006). Because inhibitory synapses on the soma are predominantly glycinergic (Triller et al., 1993), the results suggest the existence of a differential sensitivity of the predominantly GABAergic dendritic inhibitory synapses to endocannabinoid modulation. Our results show that this enhancement of somatic inhibition is also mediated by dopamine and a PKA-dependent postsynaptic mechanism. Consistent with these findings, intracellular injections of cAMP or aminophylline (that inhibits phosphodiesterase and therefore cAMP degradation) in the M-cell soma have been shown to enhance feed-back inhibition (Wolszon and Faber, 1989). Because dendritic release of endocannabinoids did not affect feedback inhibition (consistent with limited diffusion of these lipidic messengers), a second source of endocannabinoid release seems to be required to modify inhibitory synaptic transmission at the soma. Thus, our findings suggest that neurons could be endowed with multiple sites of endocannabinoid production to modulate the strength of synaptic inputs at specific cellular compartments.
Endocannabinoids and dopamine function
Behavioral, biochemical and electrophysiological data have increasingly implicated the involvement of dopamine in the central actions of cannabinoid compounds (Van der Stelt and Di Marzo, 2003). However, the sites and cellular mechanisms by which cannabinoids and dopamine systems interact remain to be determined. We describe here a novel form of interaction between these two systems in which dendritic endocannabinoid production is capable of inducing local release of dopamine from nearby varicosities. Localized interactions between cannabinoid and dopaminergic systems could constitute a more general property relevant to structures where dopamine and endocannabinoid signaling co-exist, such as basal ganglia (Marsicano and Lutz, 1999; Hohmann and Herkenham, 2000) and prefrontal cortex (Herkenham et al., 1990), and contribute to the motor and addictive effects of the marijuana products. These interactions might also be relevant to the retina, which contains dopamine receptors, abundant gap-junctions and CB1Rs (Fan and Yazulla, 2005).
EXPERIMENTAL PROCEDURES
Electrophysiology
Intracellular in-vivo recordings from adult goldfish (Carassius auratus) were performed as previously described (Pereda et al., 1994). Briefly, responses to VIIIth nerve or spinal cord stimulation were recorded from the lateral dendrite of the M-cell (300–400 μm from the axon hillock) with glass microelectrodes (4–7 MΩ) containing 5M K-Acetate, pH 7.2. Due to the brief membrane time constant of the M-cell (~400 μs), both components of the synaptic potential can be unambiguously identified and reliably measured, as the brief duration of the electrical component does not significantly influence the peak of the subsequent chemical response (peak amplitudes were detected within a range of points using Igor software). Synaptic potentials were monitored every 4 seconds and traces were averaged in sets of 10 or more. Amplitude changes were estimated by comparing averages obtained at baseline and 30 minutes after each experimental protocol. Results are expressed as mean ±SEM. Student’s t test was used to assess statistical significance of the data, unless otherwise stated (level of significance: 5%). The amplitude of the M-cell antidromic action potential was routinely monitored and taken as an indicator of the cells’ input resistance. The strength of the feed-back inhibition was quantified by measuring amplitude changes in a second antidromic action potential (6 ms interval) superimposed on the inhibitory response (the reduction in amplitude of the second action potential is proportional to the membrane shunting produced by the opening of ligand-gated Cl- channels) and expressed as the ratio between the amplitudes of the first, conditioning, action potential (V) and the second, or “test”, action potential. This ratio, “fractional conductance”, was defined as (V/V′)-1 (Oda et al., 1995). In order to activate the auditory afferents, a bipolar stimulating electrode (FHC, Brunswick, ME) was placed on the posterior VIIIth nerve. To obtain “long-term potentiation” of the mixed EPSP, high frequency stimulation of the posterior VIIIth nerve, consisting of 6 pulses at 500 Hz applied every 2 seconds, was applied over 4 minutes (“TET” in Fig 3D). In the case of dendritic depolarizations, the protocol consisted of a total of 5 pulses of 90 nA, 5 s duration each, applied every 4 s through the recording electrode; this amount of current is expected to bring the membrane potential to approximately 0 mV (estimated input resistance ~1 MΩ) (Pereda et al., 1992; Wolszon et al., 1997).
Drug Application
WIN 55,212-2 (500 nM; Tocris), SR141716 (4 μM; NIMH Drug Supply program), AM251 (4 μM; Tocris), SCH-23390 (50 μM; Biomol) and bicuculline methiodide (75 μM; Sigma) were bath-applied by superfusion of the brain after dilution in artificial CSF. WIN 55,212-2, SR141716, AM251 and Tetrahydrolipstatin (THL) were prepared from 5 mM stocks in DMSO. For extracellular local application, a second microelectrode (12–16 MΩ) was positioned 300–400 μm from the axon hillock and 20–30 μm above the M-cell lateral dendrite. WIN (5 μM), (RS)-3,5-Dihydroxyphenylglycine (DHPG; 1 mM) or dopamine (10 mM; Calbiochem) were dissolved in 130 mM NaCl and 10 mM HEPES, pH 7.2 and pressure-ejected (Picospritzer II, Parker Instrumentation, Cleveland, OH; 5 pulses of 3 s duration each, 20 psi). Extracellular application of this vehicle solution has been used in previous studies (Pereda et al., 1992; Pereda et al., 1994; Kumar and Faber, 1999) and does not produce changes in recorded synaptic potentials. Ejection from the tip of the microelectrode was routinely verified at the end of each experiment. In the case of intracellular injections, microelectrodes (4–6 MΩ) contained the PKA inhibitor 5–24 (500 μM; Calbiochem) or the DGL inhibitor THL (100 μM; Sigma) in 0.5 M KCl and 10 mM HEPES, pH 7.2. This vehicle solution does not interfere with the induction of plastic changes (Pereda et al., 1998; Kumar and Faber, 1999).
Immunohistochemistry
Fish were perfused intracardially with 4% paraformaldehyde. The brain was removed and kept overnight in 4% paraformaldehyde, 30% sucrose phosphate buffer and sectioned with a cryostat (18–50 μm). The primary antibodies were as follows: monoclonal anti-connexin 35 (1:250; “anti-Cx35/36”, MAB3045, Chemicon); polyclonal anti-connexin 35 phosphorylated at serine 110 (1:1000, “anti-Cx35/P110”, John O’Brien) (Kothmann et al., 2007); polyclonal anti-mGluR1α (1:200; “anti-mGluR1”, B1551, Chemicon); monoclonal anti-tyrosine hydroxylase (1:500; “anti-TH”, MAB318, Chemicon); polyclonal anti-CB1 carboxy terminus (1:750; “anti-CB1-CT”, Ken Mackie) (Hajos et al., 2000); polyclonal anti-CB1 residues 1-77 (1:100; “anti-CB1 1-77”, ab3559, Novus) (Cottone et al., 2005; Valenti et al., 2005) and polyclonal anti-CB1 residues 1-14 (1:100; “anti-CB1 1-14”, 101500; Cayman) (McIntosh et al., 1998; Yazulla et al., 2000). Primary antibodies were detected using Alexa Fluor 594 (Molecular Probes A11005) and/or Alexa Fluor 488 (Molecular Probes A11001). Control sections were obtained in the absence of primary antibodies. The sections were examined using a confocal microscope (Olympus Fluoview 500) and analyzed using ImageJ (NIH) and Adobe Photoshop software. The presence of dopamine was revealed with a polyclonal anti-dopamine antibody (Geffard, Bordeaux, France) and an immunoperoxidase reaction using a secondary biotinylated anti-rabbit and avidin-biotin-HRP complex (ABC Vectastain Kit, Vector, USA).
Immunoblotting
For Western blots, crude goldfish plasma membranes were prepared by differential centrifugation of fresh tissue homogenates from small portions of goldfish hindbrain containing the M-cells (Pereda et al., 2003). Tissues were homogenized by sonication in ice-cold 0.32 M sucrose, 10 mM Tris-Cl, pH 7.2, 2 mM EGTA, 5 mM MgCl2, 1% phosphatase inhibitor cocktail (Sigma), 1% protease inhibitor cocktail for His-tagged proteins (Sigma). The suspension was centrifuged for 5 min at 1000 × g to remove large particulates, and the supernatant was centrifuged for 1 hr at 100,000 × g to pellet membranes. The membrane pellet was resuspended in the homogenization medium without protease or phosphatase inhibitors plus 0.5% Igepal CA-630. Protein concentration was measured by the BCA assay (Pierce).
For each membrane preparation, a portion was digested with bacterial alkaline phosphatase. The membrane preparations were diluted to 4 mg protein/ml with alkaline phosphatase assay buffer (final composition: 10 mM Tris-Cl, 10 mM MgCl2, 0.5% Igepal CA-630, 30% membrane homogenization buffer) and digested with 25 units alkaline phosphatase (Fermentas, Hanover, MD) per mg protein for 2 hr at 50°C. The samples were then diluted to 2 mg protein/ml with 2x SDS sample buffer. Comparable non-digested samples were prepared at the same concentration with alkaline phosphatase buffer but no enzyme.
For Cx35 immunoblots, 50 μg aliquots of membrane protein were dissolved in reducing SDS sample buffer and resolved on 12% gels. Proteins were transferred at 18 V overnight to polyvinylidenedifluoride membranes. Blots were blocked with 5% non-fat dry milk in TBST (137 mM NaCl, 3 mM KCl, 25 mM Tris-Cl, pH 7.4, 0.1% Tween-20) for 1 hr at room temperature. Blots were incubated with affinity purified phospho-Ser110 specific antibodies (“anti-Cx35/P110”, John O’Brien) (Kothmann et al., 2007) diluted to 0.8 μg/ml in high-salt TBST (500 mM NaCl, 3 mM KCl, 25 mM Tris-Cl, pH 7.4, 0.1% Tween-20) plus 2% nonfat dry milk. Blots were also probed with a monoclonal anti-Cx35/36 antibody (MAB3045, Chemicon) at 1 μg/ml under the same conditions. Blots were developed with peroxidase-linked secondary antibodies (Pierce) and detected by chemiluminescence.
Acknowledgments
We thank Carl Castillo, Carmen Flores and Smaranda Ene for assistance with immunochemical experiments and M.V.L. Bennett and P.E. Castillo for helpful comments on the manuscript. We also thank V. Chevaleyre for useful discussions. Supported by NIH grants NS0552827 and DC03186 to A. Pereda, DA11322 to K. Mackie and EY12857 to J. O’Brien.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Bash R, Rubovitch V, Gafni M, Sarne Y. The stimulatory effect of cannabinoids on calcium uptake is mediated by Gs GTP-binding proteins and cAMP formation. Neurosignals. 2003;12:39–44. doi: 10.1159/000068915. [DOI] [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther. 1998;287:884–888. [PubMed] [Google Scholar]
- Brandes RP, Popp R, Ott G, Bredenkotter D, Wallner C, Busse R, Fleming I. The extracellular regulated kinases (ERK) 1/2 mediate cannabinoid-induced inhibition of gap junctional communication in endothelial cells. Br J Pharmacol. 2002;136:709–716. doi: 10.1038/sj.bjp.0704776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron. 2005;45:419–431. doi: 10.1016/j.neuron.2004.12.045. [DOI] [PubMed] [Google Scholar]
- Brown SP, Brenowitz SD, Regehr WG. Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci. 2003;6:1048–1057. doi: 10.1038/nn1126. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron . 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-Mediated Synaptic Plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Parenti R, Spinella F, Trovato Salinaro A, Belluardo N, Cardile V, Cicirata F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci. 1998;10:1202–1208. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- Cottone E, Campantico E, Guastalla A, Aramu S, Polzonetti-Magni AM, Franzoni M. Are the cannabinoids involved in bony fish reproduction? Ann N Y Acad Sci. 2005;1040:273–276. doi: 10.1196/annals.1327.041. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Synergism at central synapses due to lateral diffusion of transmitter. Proc Natl Acad Sci USA. 1988;85:8708–8712. doi: 10.1073/pnas.85.22.8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan SF, Yazulla S. Reciprocal inhibition of voltage-gated potassium currents (I K(V)) by activation of cannabinoid CB1 and dopamine D1 receptors in ON bipolar cells of goldfish retina. Vis Neurosci. 2005;22:55–63. doi: 10.1017/S0952523805221089. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Fukami Y, Furukawa T, Asada Y. Excitability changes of the Mauthner cell during collateral inhibition. J Gen Physiol. 1965;48:581–600. doi: 10.1085/jgp.48.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Diana MA. Group I metabotropic glutamate receptors inhibit GABA release at interneuron-Purkinje cell synapses through endocannabinoid production. J Neurosci. 2004;24:4865–4874. doi: 10.1523/JNEUROSCI.0403-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of cannabinoid CB(1) receptor mRNA in neuronal subpopulations of rat striatum: a double-label in situ hybridization study. Synapse. 2000;37:71–80. doi: 10.1002/(SICI)1098-2396(200007)37:1<71::AID-SYN8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- Korn H, Faber DS. The Mauthner cell half a century later: a neurobiological model for decision-making? Neuron. 2005;47:13–28. doi: 10.1016/j.neuron.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Kothmann WW, Li X, Burr GS, O’Brien J. Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis Neurosci. 2007;24:363–375. doi: 10.1017/S095252380707037X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Faber DS. Plasticity of first-order sensory synapses: interactions between homosynaptic long-term potentiation and heterosynaptically evoked dopaminergic potentiation. J Neurosci. 1999;19:1620–1635. doi: 10.1523/JNEUROSCI.19-05-01620.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci USA. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Brotchie JM. Paradoxical action of the cannabinoid WIN 55,212-2 in stimulated and basal cyclic AMP accumulation in rat globus pallidus slices. Br J Pharmacol. 1997;120:1397–1398. doi: 10.1038/sj.bjp.0701101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- McIntosh HH, Song C, Howlett AC. CB1 cannabinoid receptor: cellular regulation and distribution in N18TG2 neuroblastoma cells. Brain Res Mol Brain Res. 1998;53:163–173. doi: 10.1016/s0169-328x(97)00294-5. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Glass M, Matias I, Norris RW, Kilpatrick CW. A shifted repertoire of endocannabinoid genes in the zebrafish (Danio rerio) Mol Genet Genomics. 2007;277:555–570. doi: 10.1007/s00438-007-0207-3. [DOI] [PubMed] [Google Scholar]
- Mitropoulou G, Bruzzone R. Modulation of perch connexin35 hemi-channels by cyclic AMP requires a protein kinase A phosphorylation site. J Neurosci Res. 2003;72:147–57. doi: 10.1002/jnr.10572. [DOI] [PubMed] [Google Scholar]
- O’Brien J, Bruzzone R, White TW, Al-Ubaidi MR, Ripps H. Cloning and expression of two related connexins from the perch retina define a distinct subgroup of the connexin family. J Neurosci. 1998;18:7625–7637. doi: 10.1523/JNEUROSCI.18-19-07625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Charpier S, Murayama Y, Suma C, Korn H. Long-term potentiation of glycinergic inhibitory synaptic transmission. J Neurophysiol. 1995;74:1056–1074. doi: 10.1152/jn.1995.74.3.1056. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Oropeza VC, Page ME, Van Bockstaele EJ. Systemic administration of WIN 55,212-2 increases norepinephrine release in the rat frontal cortex. Brain Res. 2005;1046:45–54. doi: 10.1016/j.brainres.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Oropeza VC, Mackie K, Van Bockstaele EJ. Cannabinoid receptors are localized to noradrenergic axon terminals in the rat frontal cortex. Brain Res. 2006;1127:36–44. doi: 10.1016/j.brainres.2006.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Winbow VM, Patel LS, Burr GS, Mitchell CK, O’Brien J. Protein kinase A mediates regulation of gap junctions containing connexin35 through a complex pathway. Brain Res Mol Brain Res. 2005;135:1–11. doi: 10.1016/j.molbrainres.2004.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A, Triller A, Korn H, Faber DS. Dopamine enhances both electrotonic coupling and chemical excitatory postsynaptic potentials at mixed synapses. Proc Natl Acad Sci USA. 1992;89:12088–12092. doi: 10.1073/pnas.89.24.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, Nairn AC, Wolszon LR, Faber DS. Postsynaptic modulation of synaptic efficacy at mixed synapses on the Mauthner cell. J Neurosci. 1994;14:3704–3712. doi: 10.1523/JNEUROSCI.14-06-03704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, Faber DS. Activity-dependent short-term enhancement of intercellular coupling. J Neurosci. 1996;16:983–992. doi: 10.1523/JNEUROSCI.16-03-00983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, Bell TD, Chang BH, Czernik AJ, Nairn AC, Soderling TR, Faber DS. Ca2+/calmodulin-dependent kinase II mediates simultaneous enhancement of gap-junctional conductance and glutamatergic transmission. Proc Natl Acad Sci USA. 1998;95:13272–13277. doi: 10.1073/pnas.95.22.13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A, O’Brien J, Nagy JI, Bukauskas F, Davidson KG, Kamasawa N, Yasumura T, Rash JE. Connexin35 mediates electrical transmission at mixed synapses on Mauthner cells. J Neurosci. 2003;23:7489–7503. doi: 10.1523/JNEUROSCI.23-20-07489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, Rash JE, Nagy JI, Bennett MV. Dynamics of electrical transmission at club endings on the Mauthner cells. Brain Res Brain Res Rev. 2004;47:227–244. doi: 10.1016/j.brainresrev.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubovitch V, Gafni M, Sarne Y. The cannabinoid agonist DALN positively modulates L-type voltage-dependent calcium-channels in N18TG2 neuroblastoma cells. Brain Res Mol Brain Res. 2002;101:93–102. doi: 10.1016/s0169-328x(02)00174-2. [DOI] [PubMed] [Google Scholar]
- Smith M, Pereda AE. Chemical synaptic activity modulates nearby electrical synapses. Proc Natl Acad Sci USA. 2003;100:4849–4854. doi: 10.1073/pnas.0734299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci. 2002;15:2057–2061. doi: 10.1046/j.1460-9568.2002.02041.x. [DOI] [PubMed] [Google Scholar]
- Triller A, Sur C, Korn H. Heterogeneous distribution of glycinergic and GABAergic afferents on an identified central neuron. J Comp Neurol. 1993;338:83–96. doi: 10.1002/cne.903380107. [DOI] [PubMed] [Google Scholar]
- Tuttle R, Masuko S, Nakajima Y. Small vesicle bouton synapses on the distal half of the lateral dendrite of the goldfish Mauthner cell: freeze-fracture and thin section study. J Comp Neurol. 1987;265:254–274. doi: 10.1002/cne.902650209. [DOI] [PubMed] [Google Scholar]
- Upham BL, Rummel AM, Carbone JM, Trosko JE, Ouyang Y, Crawford RB, Kaminski NE. Cannabinoids inhibit gap junctional intercellular communication and activate ERK in a rat liver epithelial cell line. Int J Cancer. 2002;104:12–18. doi: 10.1002/ijc.10899. [DOI] [PubMed] [Google Scholar]
- Urschel S, Hoher T, Schubert T, Alev C, Sohl G, Worsdorfer P, Asahara T, Dermietzel R, Weiler R, Willecke K. Protein kinase A-mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem. 2006;281:33163–33171. doi: 10.1074/jbc.M606396200. [DOI] [PubMed] [Google Scholar]
- Valenti M, Cottone E, Martinez R, De Pedro N, Rubio M, Viveros MP, Franzoni MF, Delgado MJ, Di Marzo V. The endocannabinoid system in the brain of Carassius auratus and its possible role in the control of food intake. J Neurochem. 2005;95:662–672. doi: 10.1111/j.1471-4159.2005.03406.x. [DOI] [PubMed] [Google Scholar]
- Van der Stelt M, Di Marzo V. The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: implications for neurological and psychiatric disorders. Eur J Pharmacol. 2003;480:133–150. doi: 10.1016/j.ejphar.2003.08.101. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21(RC188):1–5. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance L, Piomelli D, Glowinski J, Giaume C. Inhibition by anandamide of gap junctions and intercellular calcium signaling in striatal astrocytes. Nature. 1995;376:590–594. doi: 10.1038/376590a0. [DOI] [PubMed] [Google Scholar]
- Wallmichrath I, Szabo B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience. 2002;113:671–682. doi: 10.1016/s0306-4522(02)00109-4. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wolszon LR, Faber DS. The effects of postsynaptic levels of cyclic AMP on excitatory and inhibitory responses of an identified central neuron. J Neurosci. 1989;9:784–797. doi: 10.1523/JNEUROSCI.09-03-00784.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolszon LR, Pereda AE, Faber DS. A fast synaptic potential mediated by NMDA and non-NMDA receptors. J Neurophysiol. 1997;78:2693–2706. doi: 10.1152/jn.1997.78.5.2693. [DOI] [PubMed] [Google Scholar]
- Yamaguchi F, Macrae AD, Brenner S. Molecular cloning of two cannabinoid type 1-like receptor genes from the puffer fish Fugu rubripes. Genomics. 1996;35:603–605. doi: 10.1006/geno.1996.0406. [DOI] [PubMed] [Google Scholar]
- Yang XD, Korn H, Faber DS. Long-term potentiation of electrotonic coupling at mixed synapses. Nature. 1990;348:542–545. doi: 10.1038/348542a0. [DOI] [PubMed] [Google Scholar]
- Yazulla S, Studholme KM, McIntosh HH, Fan SF. Cannabinoid receptors on goldfish retinal bipolar cells: electron-microscope immunocytochemistry and whole-cell recordings. Vis Neurosci. 2000;17:391–401. doi: 10.1017/s0952523800173079. [DOI] [PubMed] [Google Scholar]