

Abstract
Viruses have recently proven useful for the detection of target analytes such as explosives, proteins, bacteria, viruses, spores, and toxins with high selectivity and sensitivity. Bacteriophages (often shortened to phages), viruses that specifically infect bacteria, are currently the most studied viruses, mainly because target-specific nonlytic phages (and the peptides and proteins carried by them) can be identified by using the well-established phage display technique, and lytic phages can specifically break bacteria to release cell-specific marker molecules such as enzymes that can be assayed. In addition, phages have good chemical and thermal stability, and can be conjugated with nanomaterials and immobilized on a transducer surface in an analytical device. This Review focuses on progress made in the use of phages in chemical and biological sensors in combination with traditional analytical techniques. Recent progress in the use of virus—nanomaterial composites and other viruses in sensing applications is also high-lighted.
Keywords: bacteria, biosensing, phages, sensors, viruses
1. Introduction
Chemical and biological sensors are analytical devices that can provide quantitative or semiquantitative information by exploiting a chemical or biological recognition element.[1-4] In the sensors, the recognition element is either integrated within or is closely associated with a transducer interface that can convert chemical, physical, or biological interactions into a measurable output. During the past two decades, sensors, especially biosensors, have become necessary for detecting different analytes such as explosives,[5,6] proteins,[7,8] DNA,[9,10] cancer markers,[11,12] bacteria,[13,14] viruses,[15,16] and toxins[17,18] in food processing, environmental monitoring, clinical diagnostics, and the fight against bioterrorism.[3,19,20] In terms of the transducer properties, biosensors can be classified as optical, thermometric, piezoelectric, magnetic, micromechanical, and electrochemical.[21,22] Since the transducers normally do not have specificity against a target analyte, how to introduce target-specificity to the sensors is a daunting challenge.[22] In addition, the samples to be analyzed are always complicated by the broad range of concentration over which the analytes might be present, the limited volumes of the samples, and the heterogeneous nature of the samples.
As a result, there is a pressing need for the development of target-selective sensors. Viruses, which can specifically infect their host, are being employed to overcome such a challenge. Although the natural function of the viruses is to store and transport genetic material, they have recently been demonstrated to act as templates for the synthesis and assembly of nanomaterials,[23-27] as vehicles for targeted drug and gene delivery,[28,29] and as probes for sensing and imaging.[30-32] Three types of viruses may be used as platforms for these applications: bacteriophages, plant viruses, and animal viruses.[33,34] The use of viruses in sensing is being actively developed, and many opportunities are left for scientists to explore in this area.
Of the different viruses being studied for chemical and biological sensing, bacteriophages (shortened to phages)—viruses that specifically infect bacteria—have proved to be the most powerful candidates for several reasons. The most important reason is that a well-established technique, called phage display,[35-38] can be used to identify target-recognizing peptides or proteins from a combinatorial phage-displayed random peptide or protein library, which in turn results in the selection of target-specific virus particles. In addition, phages are robust, thermally and chemically stable, easy to conjugate with other motifs such as biomolecules or nanoparticles, and can be immobilized on a transducer surface. Therefore, we review here the use of phages in sensing applications.
Phages can be engineered to bear target-specific peptides or proteins for biorecognition. However, they do not themselves have physical properties that can be exploited to generate a readable output upon binding to a specific target analyte. Thus, the current application of phage in developing sensors relies on the integration of the probe (target-specific phage or phage-derived peptides or proteins) into an analytical device that can convert the recognition process between the probe and a target analyte into a readable output. Therefore, we focus in this Review on how phages can be integrated into and coupled with traditional analytical techniques to develop advanced sensors with high selectivity and sensitivity for different applications. We will also discuss the application of other viruses in the development of sensors, and finally we propose some future directions for the area of virus-based sensors.
2. Phage as a Bacteria-Specific Virus and Phage Display
2.1. Phages
Phages—bacteria-specific viruses—are made of an outer protein coat that encases genetic material (DNA or RNA).[39,40] There are two major types of phage in nature: lytic and nonlytic. When the lytic phages such as the T4 and MS2 phage infect bacteria, bacterial cells are lysed (that is, broken open) and killed after the virions are replicated.[41] When the bacterial cells are destroyed, the new phages will infect new host cells. It is the lytic phage that has been proposed for use as an antibacteria treatment strategy (phage therapy).[41,42] However, the nonlytic phages such as fd and M13 phage (Figure 1) do not break the host bacterial cells when they infect their host cells. Instead, the phages undergo lysogeny—the phage genome is integrated with the host DNA and replicated. The host cells thus continue to survive and reproduce, and the phages are reproduced and amplified. Therefore, nonlytic phages such as M13 phage are often used in recombinant DNA engineering.
Figure 1.

Structure of filamentous phage (M13 or fd) and the genetically engineered version. The phage DNA is encased in a capside made of four minor coat proteins (pIII, pIX, pVI, and pVII) located at one of the two distal ends of the phage and one major coat protein (pVIII), several thousand copies of which constitute the side wall of the phage. The arrows indicate different modes of displaying a peptide or protein on the surface of the phage by fusing it to pIII or pIX (at the tip) and/or pVIII (on the side wall). One, two, or three peptides or proteins can be displayed on the surface of the phage, thereby leading to the site-specific modification of the phage surface by genetically altering the phage DNA. The foreign peptide or protein fused to the coat protein is highlighted in red.
For chemists and materials scientists, it will be very helpful to consider the morphological differences in the different types of phages when designing phage-based nanosyntheses and sensor applications. In terms of morphology, there are three different types of phage:[39] 1) filamentous (rodlike) phages such as fd and M13 (Figures 1 and 2); 2) icosahedral phages such as MS2 and Φ174; and 3) prolate icosahedral phages with helical tails such as T4 and T7. All three types of phages have nanometer-sized structures. For example, the filamentous fd and M13 phages are nanofibers about 1 μm long and 7 nm wide (Figures 1 and 2), the icosohedral MS2 phage is a nanosphere with a diameter of about 23–27 nm,[39,43,44] and the T7 phage is made of a spherical protein coat with an inner diameter of 55 nm and a tail (19 nm in diameter and 28.5 nm long) attached to the spherical capsid.[39,40,45] The size of the phages along with the possibility to site-specifically genetically modify their surfaces by displaying a peptide or protein places the phages (Figure 1) in a unique position for templating the synthesis and assembly of nanomaterials as well as in the development of advanced nanosensors.
Figure 2.
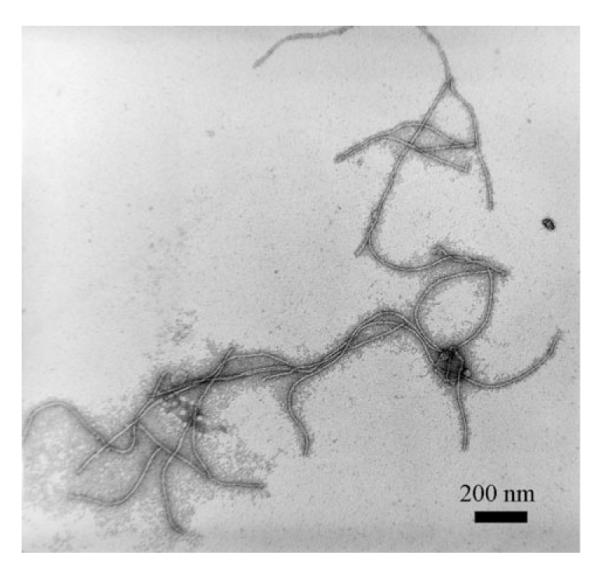
Transmission electron microscopy (TEM) image of stained M13 phage. The morphology of the fd and the M13 phage is almost the same in the TEM images.
2.2. Filamentous Phage as a Genetically Modifiable Biomolecular Nanofiber
Filamentous M13 and fd phages are the most successful phages used in templated nanosynthesis and the development of biosensors. This results from the fact that the well-established phage display technique is mainly based on filamentous phages. M13 and fd phages both have an almost identical structure and morphology (Figures 1 and 2). They have a cylindrical structure composed of about 3000 highly ordered copies of the major coat protein (pVIII) which surrounds a circular single-stranded DNA genome (Figure 1).[46] The difference between M13 and fd phages lies in the 12th amino acid of the 50 amino acid long pVIII: the N in M13 is substituted by D in the fd phage. The single-stranded DNA encodes pVIII on the major coat and four other structural proteins at two tips: pIII, pVI, pVII, and pIX. At one tip of the cylinder there are five copies each of pIII and pVI, while at the other tip there are five copies each of pVII and pIX. By inserting DNA that encodes foreign peptides into the genes of the coat proteins, the peptides are themselves displayed on the outer surface of the phage at the tips (for example, pIII or pIX display) and/or along the length of the filamentous scaffold (pVIII display; Figure 1). This is the principle of displaying a peptide or protein on a phage; namely, by DNA engineering, a peptide or protein can be site-specifically displayed at the tip and/or along the side wall of the phage through fusion to the minor and/or major coat protein. This leads to the single, double, or triple display of different peptides or proteins on a single virus particle (Figure 1). Therefore, the filamentous phage is in fact a nanofiber (Figure 2) whose surface chemistry can be site-specifically modified by genetic means (Figure 1).[36]
2.3. The Phage Display Technique for Genetically Identifying a Target-Specific Peptide or Protein
Foreign peptides or proteins can be displayed at the tip (for example, pIII) and/or along the side walls (namely, pVIII) of the filamentous phage simply by inserting DNA that encodes the foreign peptides or proteins (Figure 1). Thus, a library made of billions of M13 or fd phage clones can be constructed where each phage clone displays a unique foreign peptide or protein sequence.[36] Such a library can be used to identify a peptide or protein (out of the billions of candidates) that can specifically bind to a target (for example, a protein, a cell, or a crystal) through a biological evolutionary selection process called biopanning (Figure 3).[35,47,48]
Figure 3.

Schematic illustration of the biopanning procedure used in phage display to identify a target-specific binding peptide or protein. a) The analyte (blue circle) is immobilized on the dish; b) a library made of billions of phage clones is allowed to interact with the analyte; c) the unbound phages are washed away with a buffer and the bound phages remain attached to the target; d) the binding phages are eluted away from the analyte with a buffer; e) the binding phages are amplified by infecting bacteria in the culture to obtain an enriched library, which is used as an input for (b) in the next round of selection; f) after several rounds of selection (b→c→d→e→b), the best target-binding phage clones are propagated. g) The coding region of the phage DNA is sequenced to obtain the sequence of the target-specific binding peptide or protein. h) The structure of landscape phage showing the fusion of a foreign peptide to each of several thousand copies of pVIII. PCR = polymerase chain reaction.
Currently, there are two major types of phage-displayed random peptide or protein libraries:[35,49] One is the pVIII library, such as the landscape phage library,[50] where each peptide sequence is displayed along the side wall by fusion to several thousand copies of pVIII (Figures 1 and 3).[51] It should be noted that the fusion of longer peptides to pVIII may not favor the assembly of pVIII along DNA to form a mature phage particle. Thus, there is a limitation on the length of the peptide that can be fully displayed on the major coat. Depending on the sequence of the peptide, a peptide with up to 12 amino acids may be displayed on the major coat.[23] The alternative is the pIII library,[23,24,52] where each peptide or protein is displayed at the pIII tip through fusion to the five copies of pIII at the tip (Figure 1).[48] The landscape phage library was pioneered by Petrenko, Smith, and co-workers.[51,53] Figure 3 illustrates the biopanning process, whereby a peptide that can specifically bind to a target can be identified from a landscape phage library.
The general procedure used in biopanning for selecting a target-specific phage from a random phage library includes the following steps (Figure 3). First, a library containing billions of phage clones with each phage clone carrying a unique peptide or protein (either at the tip or on the side wall of the phage, Figure 1) is allowed to interact with an immobilized target. During interaction, some phages will bind to the target and others will not. The unbound phage is then washed away using a washing buffer such as a detergent. An elution buffer is then used to elute the bound phage from the immobilized target. The eluate contains the binding phage, which will be amplified through infecting E. coli bacteria, thereby leading to the formation of an enriched library (namely, with reduced phage clones), which is an output in biopanning. To find the best target-specific phage (namely, carrying the best target-specific peptide or protein), the enriched library is treated as a new input and allowed to interact with the same target again. After several rounds, an enriched library with significantly reduced phage clones is sequenced to determine the peptides or proteins that are carried by the phage clones and survive in the biopanning process. As the peptides or proteins carried by the phage clones are fused to the coat protein of phages, they are genetically encoded by the DNA inside the phage. This allows DNA sequencing to be used to determine the sequence of the target-specific binding peptides or proteins displayed on the surface of the phage. In essence, the phage display technique is an evolutionary selection process. In theory, a specific target will correspond to the identification of a unique peptide or protein specific to this target. A change in the target will result in a change in the sequence of the target-specific peptide or protein. The phage display technique has been summarized in several reviews[35,54,55] and books.[36-38] It has been applied successfully in the development of vaccines and antibodies, the study of protein—protein interactions, and protein engineering.[35,37,49,56]
2.4. Phage-Templated Synthesis of Nanostructures
Filamentous phages are unique templates for nanosynthesis and assembly. There are two reasons for this:[23,24] First, phages can express foreign peptides or proteins on the surface through insertion of genes that encode the foreign peptides or proteins into phage DNA (Figure 1), and then serve as a platform to test the nucleation of nanocrystals. Second, similar to wild-type phage,[57,58] nanoparticle-coated phages have a uniform size and shape, and thus can self-assemble into a 3D free-standing material with different architectures (for example, through the formation of a lyotropic liquid crystal).[23,24,48] Peptides, which were identified by a combinatorial library screening (biopanning) and specific to inorganic materials such as ZnS or CdS, were displayed on the major coat of the M13 phage, which resulted in the site-specific nucleation of nanocrystals with controlled organization and orientation of the crystal along the length of the phage (Figure 4).[23,24] More importantly, such mineralized phage can be assembled into a 3D ordered, nanostructured bio-inorganic hybrid material (Figure 5).[23,48] This example shows that phages are powerful templates for the controlled synthesis and assembly of nanomaterials. Engineering DNA allows different material-nucleating peptides to be displayed on the major coat so that different nanocrystals can be nucleated and organized on the phage.
Figure 4.

Top: Wild-type phage (left) can be genetically engineered to display crystal-specific peptide on major coat pVIII (middle), which can further induce the biomimetic mineralization of nanocrystals on the major coat (right). Bottom: High-resolution TEM images of phage nanofibers with ZnS nanocrystals nucleated on the side wall. The insets are electron diffraction patterns of the mineralized nanofibers, and indicate a preferred crystal orientation.[23,24]
Figure 5.
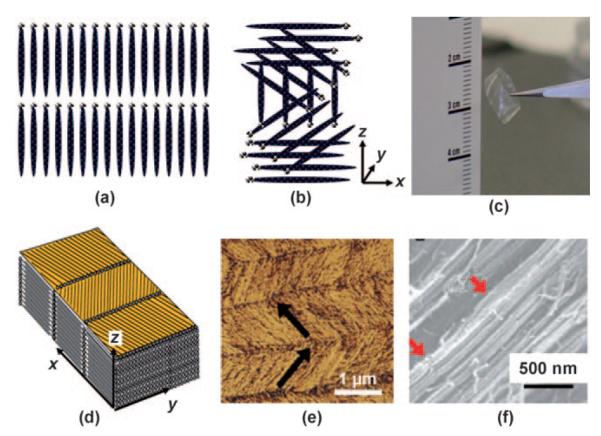
Examples of nanostructures that can be assembled from mineralized phage nanofibers through the formation of liquid crystals: a) cholesteric, b) smectic. c) Photograph of a liquid-crystalline thin film. d) Schematic illustration of the thin film in (c). e) AFM image of the top surface of the thin film, and f) SEM image of the side surface. The location of nucleated nanocrystals will be dictated by that of the binding or nucleating peptide on the phage.[48]
3. Virus and Sensors: The Case of Phages
3.1. Phages in Sensor Development
Although phage display has been applied since the 1980s for the identification of target-specific peptide or protein probes, the use of phages in the sensor area is new. Target specificity is always a key issue in the development of chemical and biological sensors.[59] Phages can be employed to identify target-specific peptides or proteins and can be engineered to become specific to a target analyte. Lytic phages can specifically kill bacteria and cause the release of the contents of the bacterial cell into the medium; thus, lytic phages may be applied to specifically detect bacteria. Since phages lack the physical properties for generating a readable output, the phage and the target-specific peptides or proteins identified by phage display have to be combined with an analytical technique that can convert the biorecognition of a target by a phage into a readable signal.
Phages have several desirable chemical and biological properties for the development of a real-time sensor to rapidly and selectively detect and monitor antigen molecules. First, phage display can be applied to select peptides or proteins that can specifically recognize a target and display the target-specific peptide or protein on the surface of the phage, thereby enabling the phage to become a target-specific probe. Second, nonlytic phages such as M13 and fd can be cost-efficiently mass-produced by infecting bacteria. Third, phages are stable in a variety of harsh conditions, such as under acidic or basic pH ranges, and in the presence of nucleases or proteolytic enzymes.[60] Fourth, phages are also thermally stable (up to 180°C),[61] and chemically stable (for example, in non-aqueous media).[62,63] Such excellent stability along with the genetically tunable target specificity enables the use of phages as a powerful biorecognition probe in biosensors.
So far, phages have been used in four ways in the development of sensors: 1) nonlytic phages (M13 or fd) selected by phage display and displaying the target-specific peptides or proteins as a target-recognizing sensing probe serve as an antibody substitute; 2) target-recognizing peptides, proteins, or antibodies identified by phage display are chemically synthesized or genetically produced, and directly used as probes; 3) lytic phages (T4 or T7) can act as bacteria-sensing probes by breaking its bacteria host strain specifically and releasing the cell-specific contents from the bacteria, thereby leading to the detection of the specific bacteria strain; and 4) the phage nanofibers can be conjugated with other organic or biological molecules and/or assembled with other nanomaterials to form a composite device that can be responsive to some external stimuli.
3.2. Sensors Based on Phages and Bioanalytical Methods
Phage-based biorecognition has to be combined with analytical methods to develop a working sensor. In the following sections, we will review how a sensor can be developed by combining phages and phage-derived proteins with traditional analytical methods, including the quartz crystal microbalance (QCM) and enzyme-linked immuno-sorbent assay (ELISA) as well as optical and electrochemical methods. The detection of a great variety of target analytes through such a combination is summarized in Tables 1-4.
Table 1.
Summary of phage-enabled QCM sensors (n.a. indicates not available).
| Probe | Analyte | Detection limit |
Ref. |
|---|---|---|---|
| branched polymers of maltose | M13 phage displaying maltose-binding protein on pIII |
20 phages | [64] |
| anti-pVIII antibody | M13 phage | 106 pfu mL−1 | [65] |
| fd phages displaying VTPPTQHQ on pVIII from a landscape f8/8 phage library through affinity-selection |
Salmonella typhimurium | 102 cells mL−1 | [66] |
| fd phages displaying β-gal-binding peptide on pVIII | β-gal from E. coli | 3 pm | [67] |
| spheroid fd phage displaying S. typhimurium-binding peptide on pVIII | Salmonella typhimurium | 102 cells mL−1 | [68] |
| MOMP[a] fragment from Legionella pneumophila | M13 phage displaying anti-MOMP fragment |
n.a. | [69] |
MOMP = major outer membrane protein.
Table 4.
Phage-based electochemical biosensors.
| Probe | Analyte(s) | Detection methods | Detection limit | Ref. |
|---|---|---|---|---|
| lytic phage-mediated release of the β-D-gal as a cell marker |
E. coli K-12 E. coli MG1655 |
release of enzymatic marker; amper- ometry |
1 CFU/100 mL | [130] |
| M13 phage with a gene for alkaline phosphatase |
E. coli TG-1 | amperometry | 1 CFUmL−1 | [131] |
| anti-MS2 IgG functionalized para- magnetic bead |
bacteriophage MS2 | paramagnetic bead based electro- chemical immunoassay |
1.5×1010 MS2 phage particles mL−1 |
[132] |
| anti-MS2/OVA IgG functionalized paramagnetic microbeads |
MS2 phage and ovalbumin | amperometry | 1.6×1011 MS2 phage particles mL−1; 470 (ng ovalbumin) mL−1. |
[133] |
| phage-displayed antibodies | lactose L. monocytogenes enzyme MtKatG |
amperometry | n.a. | [134] |
| phage displaying target- specific peptides |
hPRL-3 and the mammary adeno- carcinoma cell (MDAMB231) |
potentiometry, LAPS |
0.04 nM hPRL-3 105 MDAMB231cellsmL−1 |
[135] |
| M13 phage immobilized on elec- trode |
PSMA[a] | EIS | 120 nM | [136] |
| M13 phage immobilized on elec- trode |
anti-M13 antibody | EIS | 20 nM | [137] |
| M13 phage immobilized on elec- trode |
anti-M13 antibody | EIS, QCM | 7 nM | [138] |
| phage-displayed antibody | hα1 acid glycoprotein, ricin, M13 phage, fluorescein, Bacillus globigii |
redox enzyme amplified electro- chemical detection |
attomole concentration | [139] |
| DNA probe | phage | electric DNA chips bead-based sandwich hybridization with enzyme-labeled probes |
107 phages | [140] |
| phage-displayed α- or β-glucosi- dase |
Bacillus cereus and Mycobacterium smegmatis |
amperometry with disposable screen- printed electrodes |
10 viable cellsmL1 | [141] |
PSMA = prostate-specific membrane antigen.
3.2.1. QCM Biosensors Based on Phages
A QCM can respond sensitively to nanogram-changes in mass. A QCM sensor is made of a disk-shaped, AT-cut piezoelectric quartz crystal, which is coated on both sides with metallic electrodes (such as gold). A reduction in the resonance frequency of the piezoelectric crystal will be observed in response to the adsorption of a small amount of mass onto the electrode surface, with the frequency reduction being proportional to the adsorbed mass. The change in the resonance frequency (Δf) of the crystal is directly related to the mass change (Δm) according to the Sauerbrey Equation [Eq. (1)].[70]
| (1) |
Herein, n is the overtone number, μ is the shear modulus of the quartz (2.947 × 1011 gcm−1 s−2), f0 is the frequency of the resonator, A is the piezoelectrically active crystal area, and ρ is the density of the quartz (2.648 gcm−3). Thus, the QCM is actually a very sensitive balance that can measure mass on the nanogram scale and can be used for the real-time analysis of biomolecular interactions.[71] A QCM sensor can be used to evaluate the binding between the analyte to be detected and a sensing probe immobilized on the surface of the piezoelectric crystal. Such QCM sensors have been developed for immunoassays[72,73] as well as for bacterial,[74-83] virus, and toxin detection.[82,84-86]
The main drawback of this method is that it lacks selectivity. Thus, target-specific phages or proteins identified by the phage display technique can be immobilized on the QCM transducer surface to eliminate this disadvantage (Table 1). Figure 6 illustrates schematically the set-up of a quartz crystal microbalance with phage immobilized on the piezoelectric transducer.[64]
Figure 6.
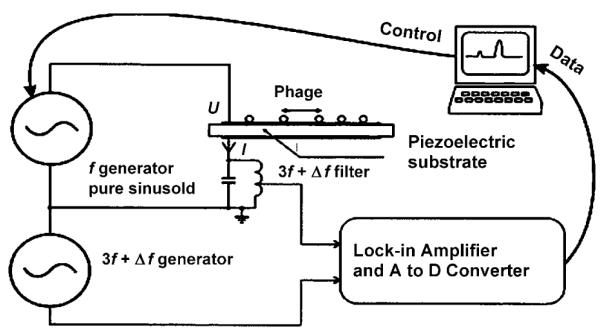
Schematic representation of the set-up of the quartz crystal microbalance.[64] The target-specific phage can be immobilized on the surface of the piezoelectric substrate.
QCM sensors can be used to detect bacteria if the target-specific phage is immobilized on the QCM transducer (Figure 6). The sensitive detection of bacteria is difficult to achieve by the traditional QCM method in the absence of phage. Furthermore, the direct binding of bacteria to the transducer will cause internal friction or the trapping of water by the cells, which results in a dampening of the oscillation of the crystals.[87] Consequently, there is a need to find a method to allow the strong attachment of bacteria to the QCM transducer surface for the detection of bacteria by using a QCM sensor—for example, the immobilization of bacteria-recognizing phages on the QCM transducer surface. The rapid detection of S. typhimurium bacteria in solution was achieved by using fd phage, which was affinity-selected by the phage display technique (Figure 3) and physically adsorbed onto the piezoelectric transducers, as a detection probe.[66] First, approximately 3 × 1010 phage particles per cm2 were adsorbed onto the surface of the QCM transducers over 1 h at room temperature. The specific binding of bacteria by the phage immobilized on the transducers then caused a change in the resonance frequency of the QCM sensors. It was found that the phage-bearing QCM sensors had a very short response time (less than 180 s) and a detection limit as low as 102 bacteria cells per mL.[66]
There are several ways to immobilize phages on the QCM transducer surface, for example, by physical adsorption, by use of the Langmuir—Blodgett (LB) technique, and by molecular self-assembly. Among them, physical adsorption is the simplest and most cost-efficient. However, the sensor performance is comparable for the three different immobilization techniques.[67] For example, affinity-selected β-galactosidase-binding fd phages were physically adsorbed onto the QCM sensor for the detection of β-galactosidase (β-gal).[67] The use of the phage as a probe resulted in the detection limit of the sensor being as low as a few nanomoles and in short response time of approximately 100 s. In addition, the sensor showed an amazing selectivity for β-gal over BSA, even when the concentration of BSA in the mixture was about 2000 times that of β-gal.[67] In another example, phages affinity selected for streptavidin or S. typhimurium by using the phage display technique were immobilized on the QCM sensors by using the LB technique. It was found that the QCM sensor was very selective and the sensor responded to increasing concentrations of target analytes (streptavidin or S. typhimurium) rapidly and with high sensitivity. Instead of directly using affinity-selected phage as probes in QCM sensors, phage display selected peptides were successfully immobilized on the QCM sensors to detect cell-surface cross-species markers in tissue homogenates.[93]
QCM sensors can also be used to directly detect phage from a solution if the probes that can bind to the phage are immobilized on the transducers.[64,65] A molecular layer containing branched polymers of maltose was chemically coated on the surface of the QCM transducer through a Au−S bond.[64] This maltose-bearing QCM sensor can specifically bind M13 phage that displays a maltose-binding protein at the tip (pIII). When the maltose-binding phages were detached from the surface of the transducer because of the breaking of the specific binding interaction between the phages and maltose immobilized on the sensor surface by oscillating the sensor surface, the resultant accoustic emission could be used for the sensitive detection of the maltose-binding phage; furthermore, the non-maltose-binding phages were found not to interfere with the detection.[64] It is claimed that the QCM sensor can detect as few as 20 phages, thereby indicating the potential application of the QCM for the sensitive and low-cost detection of viruses.[64] Similarly, in an immunoassay exploiting the specific interaction between the anti-M13 phage antibody and the M13 phage, the anti-M13 antibody was first immobilized onto the chemically milled shear mode quartz crystal transducer. The antibody-bearing QCM sensor was then used to detect the M13 phage directly and specifically. The specific attachment of the M13 phage onto the sensor led to an increase in the signal to noise ratio by a factor of more than 6 and the detection limit for M13 phage being improved by a factor of 200.[65]
The above results indicate that phage-based QCM sensors can be used for the detection of bacteria and viruses in food safety as well as for defense and homeland security applications.[68] Different spectroscopic techniques such as infrared and fluorescence spectroscopy, flow cytometry, chromatography, and chemiluminescence techniques have been used for these purposes.[76] Some advances have been made in developing sensors for the detection of pathogenic bacteria in clinical diagnostics and environmental monitoring,[94] but it is still a challenge to selectively detect food-borne pathogenic bacteria. Sensors based on phages and analytical techniques such as QCM could allow the faster and more selective detection of bacteria.[95]
Furthermore, phage display can be used to identify an antibody fragment that specifically binds to the antigen. It is important to determine the affinity of the recombinant antibody fragment to its antigen. QCM sensors can be used for this purpose. When the phage antibodies are bound to the antigen immobilized on a QCM sensor, there will be a mass-dependent decrease in the resonance frequency. In this way, the apparent affinity of a single-chain Fv (scFv) antibody against the antigen can be determined. It was found that the apparent affinity obtained by this method was consistent with the affinity of the scFv fragment estimated by other methods, such as conventional equilibrium ELISA and surface plasmon resonance methods.[96,97]
3.2.2. Phages in ELISA Biosensors
ELISA is generally used for detecting biological agents and agent-specific antibodies.[117] There are two types of ELISA: In indirect ELISA, the target antigen is captured by one antibody and detected by a second antibody, while in direct ELISA, the antigen in the sample solution will compete with labeled antigen to bind to the antibodies. Phage probes can serve as antibody substitutes in direct ELISA (Table 2). For example, fd phage probes that can capture their targets, such as β-gal,[67] streptavidin, and neutravidin,[88] can be used in direct ELISA. Consequently, direct ELISA using phage probes as antibody substitutes can specifically detect bacteria,[67,88] spores,[80] and viruses.[105,118]
Table 2.
Phage-based ELISA sensors (n.a. indicates not available).
| Probe | Analyte(s) | Detection limit | Ref. |
|---|---|---|---|
| fd phage displaying B. anthracis spore binding peptide on pVIII |
Bacillus anthracis spores | n.a. | [80] |
| fd phage displaying target-binding peptides on pVIII (selected by phage display) |
streptavidin, avidin, and β-gal | n.a. | [88] |
| T7 phage displaying mAbs F4, F5, and LT1 binding heptapeptide (selected by phage display) |
mAbs F4, F5, and LT1 (directed against the large T-antigen of mouse polyomavirus) |
n.a. | [89] |
| M13 phage displaying HBsAg binding heptapeptide on pIII (selected by phage display) |
hepatitis B surface antigen (HBsAg) | Kd = 2.9 nM | [90] |
| human scFv antibody (selected by phage display) |
protein antigens (e.g. ErbB2) | Kd = 220 pM—4 nM | [91] |
| anti-atrazine antibodies (selected by phage display) |
atrazine | 1–2 ppt | [92] |
Landscape phages are fd phages that display a foreign peptide on each copy of the major coat protein constituting the side walls of a single phage particle (Figure 1). Compared to tip-engineered phages (Figure 1), where proteins are fused to the five copies of the minor coat proteins at the tip of the phage particles, the landscape phage has thousands of copies of the highly ordered peptide on the side walls of the fd phages. Thus, the affinity-selected landscape phages from a landscape phage library (Figure 3) can serve as an antibody substitute in ELISA that is more convenient than immuno-globulin.[88] For example, the most specific spore-binding phage, which was selected from a landscape phage library by using the phage display technique and which displayed a peptide (EPRLSPHS) on its major coat, showed a binding affinity to spores of B. anthracis Sterne 3.5- to 70-fold higher than that to spores of other Bacillus species.[80] This result indicates that these phages could be used in continuous monitoring devices and biosorbents.[80]
3.2.3. Optical Biosensors Based on Phages
There are two types of optical sensing. In the first type, sensing is achieved by measuring the change in intensity at a particular wavelength, and include techniques such as UV/Vis spectrometry, fluorescence/phosphorescence spectrometry, bio/chemiluminescence, and IR spectrometry. In the other type, sensing is achieved by detecting the changes in the chemical properties upon changing the wavelength, and include techniques such as surface plasmon resonance (SPR), fluorescence resonance energy transfer (FRET), and colorimetry. In this section, we will focus on optical biosensors based on immunofluorescence, FRET, and SPR (Table 3).
Table 3.
Phage-based optical biosensors (n.a. indicates not available).
| Probe | Analyte(s) | Detection methods | Detection limit | Ref. |
|---|---|---|---|---|
| calcium-sensitive X-rhod dye connected with a fused dye-binding peptide (selected by phage display) |
Ca2+ | calcium ion sensitive fluorescence | 100 nM | [98] |
| fluorescent bacteriophage LG1 | E. coli O157:H7 | FBA flow cytometry |
2.2 CFU g−1 artificially conta- minated ground beef |
[99] |
| fluorescent bacteriophage LG1 | E. coli O157:H7 | immunomagnetic separation with fluo- rescent stained bacteriophage assay and flow cytometry |
104 cells mL−1 in broth | [100] |
| recombinant A511:luxAB phage | Listeria cells | direct epifluorescent-filter technique | n.a. | [101] |
| M13 phage displaying TNT binding pep- tide (selected by phage display) |
2,4,6-trinitrotoluene (TNT) |
ELISA, fluoroimmunoassay | 10 μgmL−1 | [102] |
| recombinant anti-TNT antibodies (isolated from an antibody phage library) |
2,4,6-trinitrobenzene (TNB) |
ELISA, fluoroimmunoassay | 1 ngmL−1 | [103] |
| Cy5 dye labeled M13 phage displaying SEB binding peptide on pIII |
staphylococcal enter- otoxin B (SEB) |
ELISA, fluoroimmunoassay | 1.4 ngmL−1 | [104] |
| M13 phage displaying HBsAg binding peptide on pIII |
hepatitis B surface antigen (HBsAg) |
phage-Linked immunoabsorbant assay | approx. 1 ng | [105] |
| human anti-HBsAg antibody (selected by phage display) |
hepatitis B surface antigen (HBsAg) |
ELISA nitrocellulose filter assay |
n.a. | [106] |
| M13 phage tagged anti-HSV antibodies | herpes simplex virus (HSV) |
phage-linked immunoabsorbant assay | n.a. | [107] |
| phage displaying morphine-binding pep- tide (selected by phage display) |
morphine | FRET | 5 ngmL−1 | [108] |
| phage displaying CPMV binding peptide on pIII (selected by phage display) |
cowpea mosaic virus (CPMV) |
SPR | n.a. | [109] |
| β-gal binding landscape phage | β-gal | SPR | 1 pM | [110] |
| phage Lm P4:A8 displayed scFv antibody (fused to pIII) |
L. monocytogenes actin polymerization pro- tein (ActA) |
SPR |
L. monocytogenes, 2×106 CFCmL−1; ActA, 4.5 nM. |
[111] |
| lytic phage | S. aureus ssp. aureus | SPR | 104 CFUmL−1 | [112] |
| anti-aflatoxin B1 scFv antibodies (selected by phage display) |
aflatoxin B1 | SPR | 3 ngmL−1 | [113] |
| affibody ligands (selected by phage dis- play) |
HIV-1 envelope glyco- protein gp120 |
SPR |
Kd ≈100 nM for the univalent affibody and 10 nM for the bivalent affibody |
[114] |
| lytic phage-mediated release of the enzyme AK as a cell marker |
Salmonella enteritidis E. coli |
bioluminescence | 103 CFUmL−1 | [115] |
| λ phage with a luxI-based acyl-homoserine lactone synthase attached |
E. coli | bioluminescence bioreporter |
contaminated lettuce leaf washings, 130 CFUmL−1 |
[116] |
3.2.3.1. Immunofluorescence Assays
The immunofluorescence assay exploits fluorescence spectrometry and specific biorecognition to obtain quantitative results. The lytic phage will destroy bacterial cells and cause the cell components to be released when it infects the cells. If the cell-specific components released from the cells can be detected, the lytic phage can be used as a probe to detect bacteria. One method takes advantage of the fluorescence properties of the released cell components. Blasco et al developed a sensitive and rapid assay to detect bacteria by using E. coli and Salmonella newport as the model bacteria.[119] In their assay, lytic phage specifically lysed the bacteria and the released cell content was detected by ATP bioluminescence.[119] The detection sensitivity was enhanced when the bacteria’s adenylate kinase was used as the cell marker instead of ATP, with this method detecting fewer than 103 bacterial cells in less than 1 h.[119] Wu et al used a bioluminescent method to detect adenylate kinase released from lysed bacteria and determined the number of bacteria lysed by phages that were specific to Salmonella enteritidis or E. coli.[115] It was found that the release of adenylate kinase was greatest when the multiplicity of infection was in the range of 10 to 100. This assay allows for the detection of bacteria faster (within 2 h) and at lower levels (103 CFU mL−1).[115] The specific detection of bacteria by lytic phage eliminates the need for time-consuming conventional microbiological methods. It also removes the need for using selective media, because the selectivity of phages for bacteria is due to the natural, specific attachment of the phage to its specific receptor.[32,120]
A fluorescent bacteriophage assay (FBA) was developed to detect E. coli O157:H7 in ground beef and raw milk.[99] This two-step assay includes the use of an immunomagnetic method to purify the target organism from a mixed culture and the use of a highly specific fluorescently labeled phage to target the E. coli O157:H7 cells.[99] The FBA could detect between 10 and 102 CFU mL−1 in artificially contaminated raw milk after a 10 h enrichment step. This study shows, therefore, that the FBA can be used to rapidly detect E. coli O157:H7 in food and milk.[99] The FBA was also employed to detect E. coli O157:H7 at a concentration of 104 cells mL−1 in broth.[100] A modified direct epifluorescent-filter technique (DEFT) showed that a bacteria detection limit as low as 102–103 cells mL−1 could be achieved with the FBA.[100]
In another study, phage display was applied to select a 12-mer peptide that could bind to staphylococcal enterotoxin B (SEB), which is a causative agent of food poisoning.[104] Nine SEB-binding phage clones were selected, and showed a consensus sequence of Trp-His-Lys at the N-terminal end. Fluorescently labeling the SEB-binding phages with Cy5 led to a detection limit of 1.4 ng of SEB per sample well in a fluorescence-based immunoassay.[104] Similarly, a number of biosensors were later developed to simultaneously detect Bacillus globigii, MS2 phage, and SEB by a similar principle.[121] These studies show that phage-based immunofluorescence assays can be applied to selectively detect species with high selectivity for food analysis and clinical diagnosis applications.
Phage-based immunofluorescence assays can also be used for the sensitive detection of explosives.[102] First, phage display was used to identify peptides specific to the 2,4,6-trinitrotoluene (TNT) derivative 2,4,6-trinitrobenzene. The specific phage was then integrated into an immunosensor for the specific detection of TNT.[102] The specific phage was chemically labeled with the fluorescent dye cyanine 5 (Cy5), and integrated with a flow-through sensor. The fluorescence sensor could specifically detect 10 μgmL−1 TNT with a high signal to noise ratio in a seawater sample (added to the sensor by repeated injections).[102] This was the first example where a dye-labeled specific phage was used for the detection of TNT in a continuous-flow sensor,[102] and with a very low detection limit of 1 ng mL−1.[103] These results show that phage-based sensors can be developed to detect TNT and related compounds as well as other explosives. More interestingly, it was recently found that the explosive-binding peptides selected in the liquid phase by phage display showed selective binding of the same explosive in the gas phase.[122]
A hybrid material made of target-specific phages and fluorescent species can result in a novel type of target-specific fluorescent probe for sensing applications. For example, a ligand-displayed T7 ghost phage can be filled with a fluorescent inorganic europium complex to form a hybrid nanoparticle.[45] A target-specific peptide (ligand) can be genetically selected by using phage display and displayed on the virus surface. This method offers a new approach to fluorescently labeling a target-specific virus particle and/or to biofunctionalize the fluorescent nanoparticles.[45] When radioactive or magnetic particles are used to fill the ghost phage, the hybridized phages will serve as a new type of functional probes for imaging and bioassays.[45]
3.2.3.2. Fluorescence Resonance Energy Transfer
The observation of FRET processes has become a popular method for studying biomolecular interactions, structure, and dynamics.[123,124] To generate a measurable FRET signal, the spacing between two fluorophores should be between 2 and 6 nm. Thus, the spacing between two antibody Fab fragments, which form an immune complex (IC) binding pair, are ideal for use in a FRET assay. Phage display selected specific binding protein can be integrated with a FRET assay to develop a single-step immunoassay for detecting small analytes such as morphine. For example, anti-morphine and anti-IC antibody fragments, which were selected from phage display antibody libraries, were fluorescently labeled with donor (europium) and acceptor (Cy5) fluorophores, respectively, to develop a FRETassay.[108] This phage-enabled FRET assay is rapid, sensitive, specific, easy to perform, and can be applied to detect other small analytes. By this FRET assay, a morphine concentration of 5 ng mL−1 could be detected without showing cross-reactivity to codeine or heroin.[108] The anti-IC Fab antibody specifically recognizes the immunocomplex formed between the M1 Fab and morphine instead of the immunocomplexes between the M1 Fab and the structurally similar substances heroin or codeine.[91]
3.2.3.3. Surface Plasmon Resonance Sensors
SPR results from the light-induced excitation of surface plasmons and is related to changes in the refractive index induced by the molecules near the metallic surface.[125] SPR has been exploited in the development of sensors because it enables the sensitive, rapid, real-time, and label-free detection of biomolecules in a small sample volume. Recently, Haes et al reviewed optical nanosensors based on SPR.[126] The set-up of a SPR sensor is shown schematically in Figure 7.[110] SPR-based biosensors (for example, from BIA-core) have proven useful in studying biomolecular interactions in real time.[113,127] Phages can enable the development of SPR sensors for target-specific detection in two ways:[110,114,113,127-129] by immobilizing the target-specific phage on the sensor chip[110,127] or by immobilizing the target-specific protein selected by phage display on the sensor chip.[113,114]
Figure 7.

Top: The flow-through mode setup consisting of a source point and a waste collector for solutions flowing in and out of the SPR system, a peristaltic pump, and personal computer (PC) for collecting data and offline analysis. Phage is immobilized on the gold-coated SPR sensor chip in the system. Bottom: Typical dose/response curves obtained from a phage-immobilized SPR sensor in the flow-through mode. Curve A represents the responses obtained from the β-gal-specific phage 1G40 immobilized in one of the two channels of the sensor against graded concentrations of β-gal (0.0032—210 nM). Curve B represents the responses obtained from the wild-type phage F8-5 immobilized in the second channel. Even at the highest concentration of β-gal tested (210 nM), the response obtained from the β-gal-specific phage is approximately ten times greater than that obtained from the wild-type phage.[110] RU = resonance units.
When target-specific phage is directly integrated into the SPR sensor, the sensor performance is significantly improved. For example, the β-gal-specific landscape phage identified by the phage display technique (Figure 3) was immobilized on the gold surface of an SPREETA SPR sensor chip through physical adsorption.[110] In a reference channel, a nonspecific wild-type phage was immobilized on the surface of the sensor chip. The optical response of the sensor was then monitored to study the binding of different concentrations of β-gal to the sensor chip in both channels (Figure 7). The SPR sensor chip with the immobilized β-gal-specific phage responded selectively to the β-gal in the channel, whereas that with an immobilized wild-type phage did not (Figure 7). The sensor could detect β-gal at a concentration between 10−3 nM and 10−1 μm. From the time-dependent optical response of the sensor, the SPR sensor could also determine the mean Kd value (1.3 ± 0.001 nM) and binding valences (1.5±0.03) when a flow-through mode was used.[110]
In another study,[127] a phage displaying a scFv antibody to the virulence factor actin polymerization protein (ActA), was immobilized on the SPR sensor chip by physical adsorption. This SPR sensor chip was then used to specifically detect the common food-borne bacteria L. monocytogenes. This study showed that the SPR sensor had a detection limit for L. monocytogenes cells of 2 × 106 CFU mL−1. The Kd value for the interaction between the phage-displayed scFv and soluble ActA in solution was determined experimentally to be 4.5 nm.[127] The ability to measure Kd values and binding valences is one of the major advantages of phage-based SPR sensors over other types of sensors. Alternatively, a target-specific peptide or protein selected by phage display can be immobilized on the sensor chip to enable the biorecognition that will result in detection.[114] For example, ScFv antibodies specific to aflatoxin B1 (AFB1) could be selected from a phage display library. The scFv antibodies could then be used to develop SPR-based inhibition immunoassays for the sensitive and selective detection of AFB1.[113] The proposed SPR biosensor is much more rapid and simpler than conventional ELISA analysis.
SPR sensors can also be used to verify the target-binding specificity of phages or proteins selected by the phage display technique. For example, glutathione peroxidase (GPX), an antioxidant enzyme, is very important in scavenging reactive oxygen species. Human GPX catalytic antibodies could be selected from the phage-displayed library by biopanning, with three haptens, S-2,4-dinitrophenyl tert-butyl ester, S-2,4-dinitrophenyl tert-hexyl ester, and S-2,4-dinitrophenyl cyclo-hexyl ester, used as the target.[128] Each antigen was then immobilized on the gold surface of the SPR sensor chip by chemical methods to form a membrane. The binding specificity could be evaluated by challenging the antigen-containing membrane with different phage antibodies and it was found that the membrane specifically binds the corresponding phage antibodies.[128]
3.2.4. Phages in Electrochemical Biosensors
Biosensors based on electrochemical methods have been widely studied because they are simple, rapid, highly sensitive, amenable to miniaturization, and can be operated in turbid media.[142–146] Such electrochemical sensors are proposed for the detection of bacteria, viruses, and toxins.[147] The surface of the electrodes in an electrochemical sensor can be coated with a variety of materials such as polymers, sol—gel, ionophores, enzymes, antibodies, DNA, bacteria, viruses, and toxins, which has led to the development of various electrochemical sensors.[145,148,149] The immobilization of a target-specific phage probe onto the surface of the working electrodes of electrochemical sensors can enable the target-specific detection of target analytes (Table 4).
3.2.4.1. Amperometric Sensors
Phages are electrochemically inactive species. However, cell-lytic phages can cause the release of intrinsic cell-marker enzymes when it meets a specific target bacterial cell; such enzymes can further catalyze some substrates to produce electrochemically active species. This process has been used in the design of an electrochemical immunoassay for the detection of bacteria or lytic phages.[132,150,139,150] For example, infection by a virulent phage (phageλvir) and the cell-marker activity of the intracellular enzyme β-D-gal are both specific for the bacterial strain E. coli (K-12, MG1655).[130] The lytic phage specifically recognizes, infects, and lyses this bacterial strain, which results in the release of the δ-D-gal. The enzymatic activity of the δ-D-gal was then measured amperometrically by using p-aminophenyl-δ-D-galactopyranoside (PAPG), which is an electro-inactive species, as a substrate. p-Aminophenol (PAP), the product of this enzymatic reaction, was then oxidized electrochemically to create a current that can be measured, thereby leading to the detection of the specific bacterial strain with a detection limit as low as 1 CFU/100 mL within 6 to 8 h.[130]
A similar concept can be applied to detect phages.[132,150] For example, MS2 phage is a stimulant for biothreat viruses, such as smallpox, and thus developing a method for detecting this phage is of paramount important. An electrochemical immunoassay based on the application of paramagnetic beads was designed to detect MS2 phage by amperometry (Figure 8).[132] A biotinylated rabbit anti-MS2 IgG was first attached to a streptavidin-coated bead, which then captured the phage. A rabbit anti-MS2 IgG-β-gal conjugate was then attached to the other side of the phage. The β-gal initiates the conversion of PAPG into p-aminophenol (PAP), which can be oxidized electrochemically to p-quinone imine (PQI). The current arising from this reaction is directly proportional to the concentration of the antigen in the sample, and can be measured by rotating disk electrode (RDE) amperometry by using an interdigitated array electrode. The detection limit of this electrochemical method can reach 3.2× 1010 viral particles per mL (RDE amperometry at an applied potential of + 290 mV versus Ag/AgCl and 3000 rpm).[132] A fully automated fluidic system can be designed for a bead-based immunoassay with amperometric detection of the MS2 phage and ovalbumin (OVA).[150]
Figure 8.

Schematic illustration of the bead-based immunoassay for bacteriophage MS2. Biotinylated rabbit anti-MS2 IgG (biotin-1° Ab) is attached to the streptavidin-coated microbead. Bacteriophage MS2 is sandwiched between the biotin-1° Ab and rabbit anti-MS2 IgG labeled with β-galactosidase (conjugate). The enzyme label converts p-amino-phenyl-β-D-galactopyranoside (PAPG) into p-aminophenol (PAP). PAP is oxidized through a two-electron reaction to p-quinone imine (PQI) at an electrode with an applied potential of + 290 mV versus Ag/AgCl and PQI can be reduced to PAP at −300 mV.[132]
In addition to the use of lytic phages in developing an amperometric sensor, a reporter phagemid system can also be employed for the specific amperometric detection of bacteria.[131] The M13KO7 helper phage and the commercial plasmid pFLAG-ATS-BAP were used to construct the phagemid system. This phagemid system includes a gene that encodes for alkaline phosphatase (ALP), which serves as a reporter gene.[131] The ALP can react with p-aminophenyl phosphate, which acts as a substrate in the enzymatic reaction that occurs in the periplasmic space of the bacterial cells.[131] PAP, which is the product of the enzymatic reaction, can leave the cells and be oxidized at the working electrode at an applied potential of 220 mV versus Ag/AgCl. The generated current signals detection of the bacteria. The ALP activity can be measured by using an electrochemical cell designed for the specific amperometric detection of bacteria in the presence of phage. The phagemid-based amperometric sensor can reach a lower detection limit of 1 CFU mL−1 E. coli TG1 in less than 3 h. The use of ALP activity resulted in the sensitivity of the sensor being increased by 10-fold compared to the lytic phage based amperometric detection. It is possible to apply such a phage-based amperometric sensor to rapidly detect any strain of bacteria as long as the appropriate phage and reporter gene are used.[131]
3.2.4.2. Light-Addressable Potentiometric Sensors
Light-addressable potentiometric sensors (LAPSs) are semiconductor-based chemical or biological sensors that have an electrolyte-insulator-semiconductor structure.[151] Many measuring points are integrated on the sensing surface in the LAPS which are individually accessed by a light beam. When the measuring points are modified with different materials, the sensor can be used to detect multiple analytes. LAPS can be used to monitor the extracellular action potential of single living cells upon stimulus. Since the phage probes are stable under the testing conditions and have a lower cost than antibodies, a target-specific phage can be used to modify the measuring points of the LAPS to enable the selective detection of the target analyte. In phage-based LAPS, specific phages are covalently immobilized on the surface of a Si3N4 chip in a back-illuminating system. The applicability of phage-based LAPSs was successfully demonstrated by detecting two analytes at different concentrations. One is the human phosphatase of regenerating liver-3 (hPRL-3) at concentrations of 0.04 to 400 nm, and another is the mammary adenocarcinoma cell (MDAMB231) at concentrations of 0 to 105 cells mL−1.[135] The cells gave a maximum potentiometric signal of about 10 and 60 μV, respectively, in response to the binding to the specific phages immobilized on the surface of the sensor chip. Phage-based LAPS is a cell assay that is better suited to the detection of cancer cells than cancer biomarkers.[135] It could find applications in the early diagnosis and result in an improvement in the efficiency of clinical treatment.
3.2.4.3. Electrochemical Impedance Spectroscopy
Electrochemical impedance spectroscopy (EIS) is a method for studying the interfacial properties of electrodes,[119] and has been used in combination with phages for the development of a biosensor (Figure 9).[137,138,152,153] A layer of M13 phages was first covalently conjugated to the surface of the gold electrode to form a covalent virus layer (CVL).[152] The impedance of the CVL-coated electrode was then measured as a function of the concentration of anti-M13 phage antibody (p-Ab) in a buffer solution at a frequency range between 0.1 Hz and 1 MHz; this resulted in the recording of the electrochemical response of the CVL to p-Ab. The change in the resistive impedance (ΔZre, relative to the initial impedance of the original CVL coated electrode) measured at high frequencies (between 4 and 140 kHz) was directly proportional to the concentration of p-Ab in the solution (Figure 10).[137] When a nonbinding antibody (n-Ab) such as the anti-FLAG M2 antibody was used as an analyte, ΔZre was almost independent of the antibody concentration. Therefore, the phage-based EIS can be employed to selectively detect p-Ab with high sensitivity (Figure 10). This method is very reproducible.[137] When prostate-specific membrane antigen (PSMA) was used as the target in the biopanning to identify a PSMA-binding M13 phage, the phage in the CVL was able to specifically bind PSMA. The EIS biosensor using a CVL made of PSMA-binding phage can be applied to selectively detect PSMA.[136] Since target-specific proteins can be genetically identified by phage display and displayed on the surface of phages, the use of phage in EIS can result in the biosensors having enhanced affinity and specificity for the detection of almost any target analyte.[154]
Figure 9.

Virus electrode: A 3 mm diameter gold disk electrode onto which engineered M13 phage particles are covalently attached (steps 1–3 shown in (e)). The status of this electrode was determined by measuring its resistive impedance ZRe at high frequency (2–500 kHz) versus a large-area platinum electrode immersed in the same salt-based buffer solution. Prior to the attachment of phage particles (a), the ZRe of the system, at all frequencies, was relatively low. b) A dense virus layer was then covalently bonded to the gold surface according to steps 1–3 shown in (e). This produces a dense phage layer that completely insulates the gold surface electrically from contact with the buffer solution. c) Exposure of this virus electrode to a “negative” antibody (n-Ab, blue) causes no change to either the imaginary component of impedance ZIm or to ZRe. d) Exposure to a “positive” antibody (p-Ab, red) that is selectively recognized and bound by the phage causes a significant increase in ZRe at high frequency, but little change in ZIm (at any frequency). e) Schematic view of the step involved in the preparation of the virus electrode. 1) An electrochemically activated gold electrode was exposed to thioctyl N-hydroxysuccinimide (NHS) NHS ester to form a thiol—gold bonded self-assembled monolayer (SAM). 2) M13 phage was covalently tethered to the self-assembled monolayer, through formation of amide bonds between free amines on the phage and the activated carboxylate. 3) Gaps in the monolayer and unreacted NHS esters were capped with BSA. 4) The virus electrode is ready to be used for analyses.[136]
Figure 10.
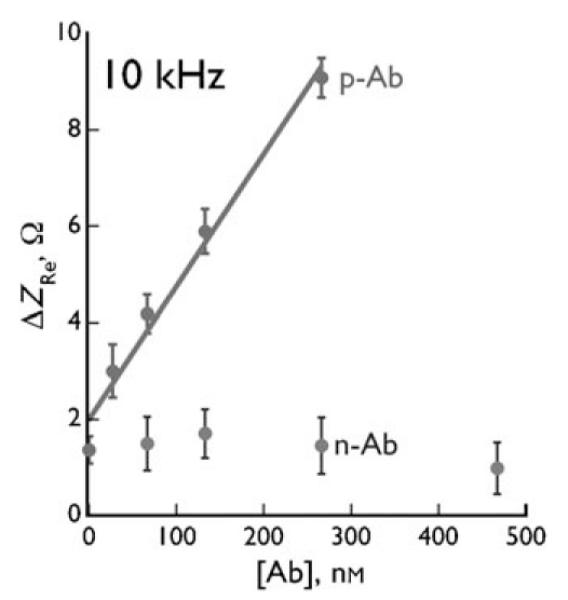
Resistive impedance change ΔZre versus the concentration of anti-phage antibody (p-Ab) and a nonbinding antibody (n-Ab).[137]
3.3. Phages in Humidity Sensing: Applications of Virus—Nanomaterial Composites
Gold nanoparticles (AuNPs) exhibit interesting localized SPR which can be exploited in optical sensors.[155] Virus-templated assembly of nanoparticles[23,24,48] and layer-by-layer assembly (LBL)[156-159] have been used separately to prepare bio-nanoarchitectures. Recently, we integrated both approaches to fabricate nanocomposite films from anionic AuNPs and cationic M13 viruses genetically engineered to display a positively charged peptide (tetraArg) on the side wall.[160] First, a quartz slide was precoated with two bilayers of anionic poly(vinyl sulfate) potassium salt (PVS) and cationic poly(diallyldimethylammonium chloride) (PDDA). The LBL assembly of the composite structures was achieved by immersing the quartz slide in an anionic AuNP solution and tetraArg-M13 phage solution alternately and repeatedly for 30 minutes. Interestingly, the SPR spectra were tunable to the environmental humidity.[160]
The change in the SPR spectra of the (PVS/PDDA)2(AuNPs/M13)4 nanocomposite film with the relative humidity (RH) at 25 °C is shown in Figure 11. As the humidity decreases, the λmax value of the SPR spectra is red-shifted. The response is fairly linearly dependent on the RH within a range between 19% and 86%. When the sensor was exposed to moist air at 98% RH and then to 70% RH air, the absorbance of the sensor rapidly reached a relatively stable value within 20 s (Figure 12); no further change in the SPR spectra occurred after 15 minutes. We also found that the humidity sensing is reversible in air.[160] This film composed of genetically engineered M13 phage and AuNPs exhibits unique humidity-dependent SPR spectra, and thus has led to the development of a spectrophotometric phage-based humidity sensor.[160]
Figure 11.
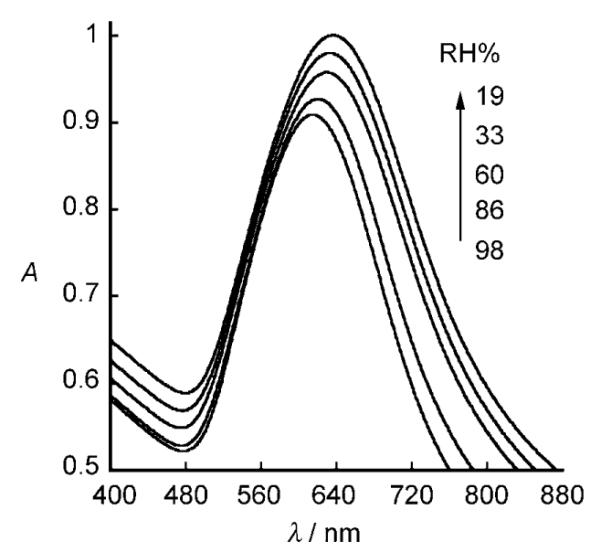
SPR spectra of (PVS/PDDA)2(AuNPs/M13)4-coated quartz slide as a function of ambient relative humidity.[160]
Figure 12.
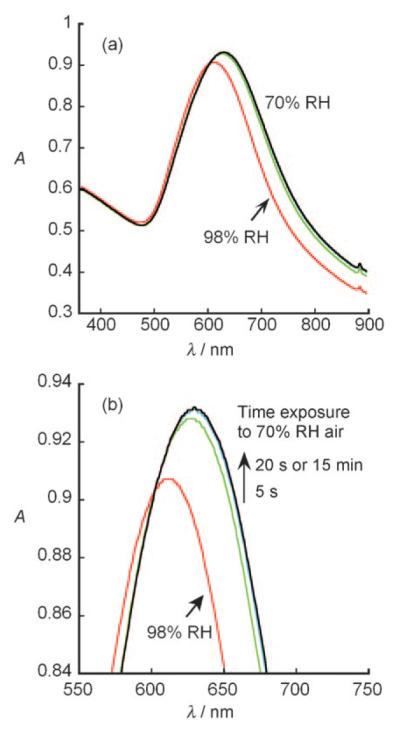
Response time of the humidity sensor of the (PVS/PDDA)2(AuNPs/arg-M13)4 film on a quartz substrate. a) SPR spectra of the film in air with 98% RH and at a different time after being switched to air with 70% RH. b) Enlargement of the absorbance peaks in (a). The time on the arrow in (b) shows the time at which the SPR spectrum of the film was recorded after the film was switched from 98% to 70% RH.[160]
The humidity-dependent SPR spectra of the phage—AuNP nanocomposite films can be explained by considering two well-known phenomena: 1) the SPR spectra of the AuNPs are dependent on the interparticle spacing, and 2) the phage bodies flatten as the humidity decreases. During the LBL assembly, the AuNPs are organized along the phage through electrostatic interactions. When the phage—AuNP nanocomposite film is dried in air, the reduction in the amount of water inside the film results in a flattening of the AuNPs-wrapped cylinder-like M13 phage. This flattening leads to a reduced spacing of the AuNPs along both the vertical (for AuNPs above and beneath a cylinder lying on the film) and horizontal (for AuNPs bound to two neighboring cylinders) directions (Figure 13). This reduced spacing causes the red-shift of the maximum absorbance of the SPR spectra of the film. Conversely, an increased AuNP separation under high humidity will cause a blue-shift of the SPR spectra. The phage—AuNP nanocomposite film exhibited better response to the water content than a composite made of amphiphilic triblock copolymer and gold nanorods.[160]
Figure 13.
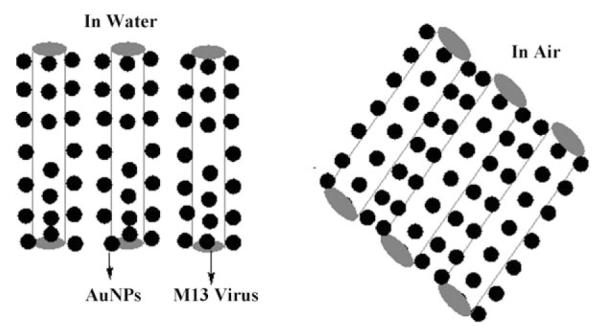
Schematic illustration of the change in the arg-M13/AuNP nanocomposite in water and in air (not scaled). In air, the rodlike virus is flattened to some extent, resulting in a change of its cross-section from a circle to an oval shape, that is, the height of the virus is changed. The side wall of each virus is made of about 2700 copies of protein pVIII, and thus the pVIII conformation will be changed if the virus is flattened.
4. Other Viruses for Sensing Applications
Similar to phages, other viruses, including the tobacco mosaic virus (TMV),[161] cowpea chlorotic mottle virus (CCMV),[162] red clover necrotic mosaic virus (RCNMV),[163] and cowpea mosaic virus (CPMV),[164] have been used for templating the synthesis of nanomaterials. Inorganic materials can be formed either outside the coat or inside the core of rodlike viruses such as TMV. For example, copper nanowires (3 nm wide and up to 150 nm long) were prepared by electroless deposition within the central channel of TMV particles.[25] Nanomaterials can be synthesized either outside the coat or inside the core of icosohedral viruses such as CCMV. For example, the gated entrapment of inorganic minerals and an organic polymer can occur inside the CCMV, thereby leading to the formation of hybrid materials.[162]
There are, however, very few reports on using other viruses for sensing applications. TMV viruses coated with palladium nanoparticles were proposed as a sensing material to develop surface acoustic wave (SAW) hydrogen sensors by using two-port SAW resonators at 315 MHz.[165] It was found that the adsorption and desorption of hydrogen from the Pd-TMV sensing layer resulted in the perturbation of the SAW device, which further caused variation in the wave velocity and resonant frequency of the device, and which is directly related to the hydrogen concentration. Hydrogen with a concentration as low as 0.2–2.5% can be detected with a response time of 30 seconds. In this gas sensor, TMV serves as a scaffold to organize and support the palladium nano-particles.
The modification of other viruses have afforded new hybrid materials that could be used for future sensing studies. Functional molecules can be conjugated to viruses for the detection and imaging of biological analytes. For example, the surface-exposed carboxylate groups on CPMV particles can be modified to increase the number and types of addressable surface groups on the surface.[166] The carboxylate groups could be modified with the fluorescent dye N-cyclohexyl-N’-(4-(dimethylamino)naphthyl)carbodiimide or conjugated with approximately 180 redox-active methyl(aminopropyl)-viologen moieties. Such virus-based novel constructs can be applied to develop novel electron-transfer mediators in redox catalysis, biosensors, and nanoelectronic devices.[166] It was also reported that fluorescent dyes can be conjugated to the subunits of CPMV to form a virus particle with a high density of fluorescent dyes on the surface.[164] Thus, the modified viruses can be used for the imaging of deep tissue inside living organisms. The fluorescently labeled CPMV does not show any detectable quenching, and can thus serve as very bright particles for high-resolution intravital imaging of vascular endothelium for periods of at least 72 h. They were used to image the vasculature and blood flow in living mouse and chick embryos at a depth of up to 500 μm.[164]
Recently, the rodlike TMVs were used as templates to grow conducting polymers such as polyaniline on the coat of the viruses.[167] The virus templating results in the polyaniline nanowires formed being monodisperse. Although the biosensing applications of such virus—polymer composites have not been demonstrated, the use of conducting polymers as a sensing and biosensing material has been well established[168—170] and such virus—polymer composites should be explored to develop new types of sensors. On the basis of these results, it can be imagined that a sensor with improved performance could be designed if other functional molecules or nanomaterials were conjugated to the viruses and combined with modern analytical methods.
Nanoparticles (NPs), quantum dots (QDs), nanowires, and nanotubes have extraordinary physical and chemical properties,[171-175] which allow them to serve as active components or sensing materials in sensors with enhanced sensitivity.[144,176,177] AuNPs can selectively bind to the ends of TMV to form gold—virus—gold dumbbells.[26] The selective attachment of AuNPs to the viruses may indicate some future possibilities in applying the composite to sensing. This can be supported by the fact that colorimetric biosensors can be designed by using nucleic acid enzyme-directed assembly of nanoparticles.[178]
Exploring the physical and chemical properties of the virus—nanomaterial composites could lead to the design of novel sensors based on other viruses. Scientists can learn from the success of the phage-based sensors for this unexplored area. This is possible because, similar to phages, other viruses can also be genetically modified to display a specific peptide or protein on the surface and form a complex with other nanomaterials. In addition, the inorganic nanomaterials can confer physical or chemical properties to the composite, which can lead to the generation of measurable signals upon binding of the target analyte to the composite. For example, Medintz et al. described methods for conjugating luminescent QDs to CPMV by taking advantage of biotin—avidin binding.[179] Specifically, the CPMV was biotinylated by conjugating biotin groups to the lysine residues on the surface of the virus and then immobilized onto a substrate through neutravidin—biotin interactions. The biotin groups exposed on the surface of the immobilized CPMV could be used as sites for capturing QDs that were conjugated with both avidin and a second protein such as a maltose-binding protein.[179] The resultant virus—QD conjugate may find possible application in the detection of maltose.
Recent progress has also been made in using enveloped viruses for the development of novel bio-nanomaterials that can be used for biosensing.[180–184] For example, lipid-enveloped rubella virus (RV) can bind to its host cell surface and induce endocytosis and fusion with the late endosomal membrane. The RV membrane protein E1, which forms a complex with another outer membrane protein of the RV (called E2), mediates the binding and fusion process. A low pH value can trigger the fusion competence of the E1 protein. RVs lacking the virus genome, described as rubella-like particles (RLPs), can be used to decorate LBL colloids.[184] Toward this end, a polyelectrolyte multilayer is first assembled onto a colloidal core through an LBL process, followed by adding a lipid bilayer onto the polyelectrolyte layer. The resulting particles are then interacted with RLPs at a low pH value to trigger the fusion of the RLPs with the lipid bilayers. The RLPs are electrostatically attached to and then fused with the lipid bilayers, thereby leaving the RV envelope proteins E1 and E2 displayed on the surface. After removal of the colloidal core, a capsule is formed with virus proteins displayed on the surface. This strategy can be used to form virus-coated LBL colloids, which can be used as a multiplex suspension array for the detection and quantification of virus-specific antibodies.[182] For example, authentic viruses (chimeric and wild-type) such as baculoviruses and influenza A viruses were coated onto lipid-bilayer-surrounded color-coded beads to catch and quantify virus-specific antibodies in a flow-cytometric analysis. Immunofluorescence was used to detect the specific binding between the viruses presented on the bead surface and the chosen antibodies. It was demonstrated that such bead-based multiplex suspension arrays could give positive results for primary antibodies diluted by up to a factor of 105. This method was competitive with ELISA for the fast and easy analysis of virus-specific antibodies,[182] and serves as an approach for the detection of major viral infections in humans.
5. Future Opportunities for the Incorporation of Viruses in Sensors
Sensors based on viruses have been actively studied in recent years and have become increasingly attractive as the area of nanotechnology has developed. Phage-based sensors are still the most popular in this field because selectivity is a very important property of a chemical and biological sensor, and phage can be engineered to bear a target-specific motif by using the well-established phage-display technique. More and more viruses are being used as templates for nanomaterials and in biosensors. We believe that the following three areas need more attention for the development of advanced real-time chemical and biological sensors with improved sensitivity and selectivity: 1) Combining viruses with new analytical methods and the fabrication of micro- and nanofunctional devices; 2) developing and applying virus-like nanomaterials (VLNs); and 3) exploiting supramolecular materials self-assembled from viruses and nanomaterials.
5.1. Integrating Viruses into New Analytical Methods and Their Incoporation into Micro- and Nanofunctional Devices
Our previous discussions indicate that viruses themselves cannot be used as sensors, but have to be used as key components, such as sensing probes. With the development of new techniques such as lab-on-a-chip, microfluidics, as well as micro- and nanofabrication, viruses can be integrated as sensing probes into new devices. For example, very recently a new sensing device, termed an optofluidic ring resonnator (OFRR), was developed by combining the ring resonator with a microfluidic system for the real-time detection of biomolecules.[185] The OFRR uses a microsized quartz capillary as the microfluidic channel, whose cross-sections form ring resonators. As a proof of concept, filamentous phage with a streptavidin-binding peptide displayed on the major coat was chemically immobilized on the inner surface of the capillary, thereby resulting in the immobilization of streptavidin-specific phage probes on the walls of the microfluidic channel. The waveguide modes (WGMs) circulate along the circumference (that is, the resonator surface). The evanescent field of the resonant WGMs can extend into the microfluidic channel and interact with the target analyte (in this case, streptavidin) in the channel. The target in the channel will be specifically bound to the phage probes immobilized on the inner channel surface. The interaction between the evanescent field of the resonant WGMs and the target analyte bound to the inner channel surface generates a readable signal (a shift in the WGM spectrum), thus enabling the detection of the target analyte without the need for fluorescent or radioactive labeling. In OFRRs, photons circulate around the circumference of the ring resonator many times, which significantly increases the interaction between the evanescent wave and the surface-bound analyte (the phage-bound streptavidin) and results in enhanced sensitivity. The detection limit for an OFRR with phage was about 100 pm streptavidin and the Kd value was 25 pm.[185] The ease of generating target-specific phage by the phage display technique means that the phage-based OFRR biosensor can be extended to detect different target biomolecules with high sensitivity, low cost, and selectivity.
Viruses can serve as templates to organize nanomaterials with different functionalities, such as magnetic and metallic nanoparticles and quantum dots (Figure 4). The virus—nanomaterial composites can serve as new building blocks for micro- or nano-functional devices. The advantages of using such virus-based composites include the monodisperse and precise size and morphology that cannot be achieved by other synthetic templates. Recently, for example, Pt nanoparticles were formed on the surface of rodlike TMVs.[186] The virus—nanoparticle nanowires were placed into the gaps between two electrodes in microelectronic devices. The memory effect of the TMVs coated with platinum nanoparticles was observed, which enabled the development of a memory device. This effect results from the entrapment of the charge in the nanoparticles assembled along TMV (which serve as data-storage media) and electron tunneling between the nanoparticles.[186] This study indicates that the virus—nanoparticle composites can be integrated into micro- and nanofunctional devices, which can enable the development of sensors based on the nanoelectronic properties of the composites. In addition, if M13 phage is used, its two distal ends can be engineered to bear proteins that can specifically recognize target materials on the devices (Figure 1). Hence, it will be possible to precisely place such virus—nanomaterial composites into the desired position (for example, in between two electrodes made by electron-beam lithography).
5.2. Developing and Applying Viruslike Nanomaterials
Viruses such as M13 or T7 phage can be viewed simply as a protein coat assembled around a nucleic acid (DNA or RNA). There is electrostatic interaction between the nucleic acid inside the virus and the protein coat, which is made of many copies of protein subunits. Thus, the formation of such a virus particle can be simplified as the assembly of protein subunits around a nuclei acid template driven by the electrostatic interaction. Therefore, if the nucleic acid in the virus is replaced by an inorganic nanomaterial (spherical nanoparticles or filamentous nanorods or nanowires) and the inorganic nanomaterial surface is functionalized to mimic the electrostatic behavior of the nucleic acid, a viruslike nanomaterial (VLN) will result from the electrostatic self-assembly of proteins around the nanomaterial. Viral protein purified from the virus particle is needed to form such a VLN. The self- assembly of viral protein around nanoparticle templates to form viruslike particles (VLPs) has recently been successfully demonstrated.[163,187-191] For example, the coat protein from a brome mosaic virus (BMV) can self-assemble around a functionalized AuNP core to form a VLP (Figure 14).[188] The functionalized AuNPs mimick the electrostatic behavior of the nucleic acid component of the native virus and initiate the assembly of proteins around the AuNPs.[188] The assembly of virus proteins around nanomaterials will allow the resultant VLNs to bear target-specific peptides or proteins on the surface.[163] Furthermore, the functionalized spherical magnetic nanoparticles could be encapsulated by viral protein cages even when the size of the nanoparticle was larger than that of the inner cavity of the native capsid of BMV.[192] Recently, Petrenko and co-workers demonstrated that phage-display-selected target-specific coat protein can be presented on the liposome surface.[193] This finding suggests that the target-specific protein identified by phage display can be presented on the nanoparticle surface for specific target binding.
Figure 14.

a) TEM images of negatively stained viruslike particles obtained from functionalized AuNPs (black centers, 12 nm diameter) and BMV capsid protein. b) Comparison of encapsidation yields for citrate—Au, Au—TEG, and native RNA. Averaged transmission electron micrograph of c) empty BMV capsid, d) citrate-coated VLP, and e) TEG-coated VLP. The averages have been obtained by superposition of 10 individual images in each case.[188]
5.3. Self-Assembled Supramolecular Materials from Viruses and Nanomaterials
It is known that rodlike viruses can self-assemble into 3D lyotropic liquid crystals as a result of their monodisperse size and anisotropic shape,[23,24,48,194] whereas spherical viruses can self-assemble into a 2D superlattice because of their uniform size and shape.[166,195,196] The ordering of the 2D and 3D viral assembly can be imparted to that of the inorganic nanomaterials if nanomaterials such as nanoparticles are assembled on the surface of the rodlike viruses or inside the core of the icosohedral viruses. In addition, the rodlike virus—nanoparticle composite can self-assemble into a 3D liquid crystal, which can be transformed into a free-standing film after evaporation of the solvent from the liquid-crystalline suspension (Figure 5).[48] These results indicate that free-standing supramolecular materials can be self-assembled from viruses alone or from composites of viruses and nanomaterials. However, to the best of our knowledge, there has been no report on the sensing applications of such virus-based free-standing supramolecular materials. Recently, the self-assembly of the virus—nanoparticle composites was applied to the construction of a lithium ion battery with enhanced performance, thus suggesting the power of using such materials in an electrochemical device.[197]
The applications of nanomaterials such as nanoparticles, nanowires, and nanotubes in sensing have been successfully demonstrated.[198,199] The application of supramolecular materials self-assembled from target-specific viruses and one or more of these nanomaterials should be pursued in the development of chemical and biological sensors. In free-standing supramolecular materials (films or bulk materials) assembled from target-specific viruses and functional nanomaterials, many physical phenomena, such as electron tunneling (when semiconducting nanoparticles are in close contact in the materials) and surface plasmon resonance (when metallic nanoparticles are in proximity in the materials), can occur as a result of the individual and collective properties of nanomaterials. The combination of the target specificity of the viruses and the physical properties of the nanomaterials in the composite materials should be explored when developing a sensor with enhanced analyte specificity and sensitivity.
6. Summary and Outlook
Viruses have been exploited in the development of chemical and biological sensors, with phages being the most successful viruses used. Nonlytic phage such as M13 phage can be engineered to display target-specific peptides or proteins by using well-established phage display techniques (for universal target detection), while lytic phage can be used to specifically kill the bacteria to release cell markers (for bacteria detection). Phages enable virus-based target-selective sensing in four ways: First, the target-specific peptides or proteins selected from a random phage-displayed peptide or protein library can be used as a sensing probe. Second, the phage that displays the target-specific peptides or proteins can be directly used as a sensing probe. Third, the lytic phage can specifically kill bacterial cells, which results in the release of cell-specific markers for the detection of bacteria. Fourth, the phage can serve as a scaffolding material to carry, support, or organize other functional molecules or nanomaterials for use in sensing devices. To develop a useful sensor, the phages, phage-derived proteins, or phage—nanomaterial composites should be coupled with or integrated into modern analytical devices that can generate and report a measurable signal for sensing.
Sensors based on viruses other than phages have not been widely studied, but more examples using other viruses can be imagined if the viruses are modified by chemical conjugation with functional molecules or assembled with inorganic nanomaterials which offer unique physical properties (optical, electronic, and magnetic). Such composites, which can offer target specificity by displaying a target-specific protein on the capsid, can form a complex that can be used as a sensing and/or transducing material.
Further advances in the field of virus-based target-selective sensing could be made by combining viruses with new analytical and micro- or nanofabricated devices, the development of viruslike sensing nanomaterials, and the use of free-standing supramolecular materials assembled from target-specific viruses and functional nanomaterials. In addition, new sensing materials and devices could be developed by learning lessons from virus-related biological processes, such as how enveloped viruses bind and fuse with cell membranes. Such a biomimetic strategy could lead to biosensors that combine the target-specific recognition of authentic viruses and the functional properties of nanomaterials. Virus-based sensors will lead to new strategies for the rapid, selective, and sensitive detection and monitoring of chemicals, explosives, proteins, bacteria, spores, viruses, and toxins for analytical chemistry, food storage and processing, medical diagnosis, and environmental protection applications as well as in the fight against bioterrorism.
Acknowledgments
We thank the National Science Foundation, the Department of Defense Congressionally Directed Medical Research Programs, the National Institutes of Health, and the Oklahoma Center for the Advancement of Science and Technology for financial support.
Biography
Chuanbin Mao received his undergraduate degree and PhD from Northeastern University in China. He then worked at Tsinghua University, first as a postdoctoral researcher and lecturer and then as an associate professor. After a short stay at the University of Tennessee, he took a postdoctoral position at the University of Texas at Austin in 2000. He became an assistant professor at the University of Oklahoma in 2005. His research interests include biotemplated nanosynthesis, biosensor applications, bio-nanotechnology, and nanomedicine.
References
- [1].Turner APF, Karube I, Wilson GS. In: Biosensors: Fundamentals and Applications. Turner APF, editor. Oxford University Press; Oxford: 1987. p. 770. [Google Scholar]
- [2].Thévenot DR, Toth K, Durst RA, Wilson GS. Biosens. Bioelectron. 2001;16:121. doi: 10.1016/s0956-5663(01)00115-4. [DOI] [PubMed] [Google Scholar]
- [3].Turner APF. Science. 2000;290:1315. doi: 10.1126/science.290.5495.1315. [DOI] [PubMed] [Google Scholar]
- [4].Willner I. Science. 2002;298:2407. doi: 10.1126/science.298.5602.2407. [DOI] [PubMed] [Google Scholar]
- [5].Steinfeld JI. Annu. Rev. Phys. Chem. 1998;49:203. doi: 10.1146/annurev.physchem.49.1.203. [DOI] [PubMed] [Google Scholar]
- [6].Yinon J. TrAC Trends Anal. Chem. 2002;21:292. [Google Scholar]
- [7].Diercksa AH, Ozinskya A, Hansen CL, Spotts JM, Rodrigueza DJ, Aderema A. Anal. Biochem. 2009;386:30. doi: 10.1016/j.ab.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tsou PH, Chou CK, Saldana S, Hung MC, Kameoka J. Nanotechnology. 2008;19:445714. doi: 10.1088/0957-4484/19/44/445714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gagnon Z, Senapati S, Gordon J, Chang H-C. Electrophoresis. 2008;29:4808. doi: 10.1002/elps.200800528. [DOI] [PubMed] [Google Scholar]
- [10].Guo Q, Yang X, Wang K, Tan W, Li W, Tang H, Li H. Nucleic Acids Res. 2009;37:e20. doi: 10.1093/nar/gkn1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brower V. J. Natl. Cancer Inst. 2009;101:11. doi: 10.1093/jnci/djp150. [DOI] [PubMed] [Google Scholar]
- [12].Gokarna A, Jin LH, Hwang JS, Cho YH, Lim YT, Chung BH, Youn SH, Choi DS, Lim JH. Proteomics. 2008;8:1809. doi: 10.1002/pmic.200701072. [DOI] [PubMed] [Google Scholar]
- [13].Panda BR, Singh AK, Ramesh A, Chattopadhyay A. Langmuir. 2008;24:11995. doi: 10.1021/la802171b. [DOI] [PubMed] [Google Scholar]
- [14].Luo PG, Stutzenberger FJ. Adv. Appl. Microbiol. 2008;63:145. doi: 10.1016/S0065-2164(07)00004-4. [DOI] [PubMed] [Google Scholar]
- [15].MacCuspie RI, Nuraje N, Lee S-Y, Runge A, Matsui H. J. Am. Chem. Soc. 2008;130:887. doi: 10.1021/ja075244z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cheng X, Chen G, Rodriguez WR. Anal. Bioanal. Chem. 2009;393:487. doi: 10.1007/s00216-008-2514-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang S, Moayeri M, Chen Z, Harma H, Zhao J, Hu H, Purcell RH, Leppla SH, Hewlett IK. Clin. Vaccine Immunol. 2009;16:408. doi: 10.1128/CVI.00412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].She RC, Durrant RJ, Petti CA. Am. J. Clin. Pathol. 2009;131:81. doi: 10.1309/AJCPMM2E7VSPHNPG. [DOI] [PubMed] [Google Scholar]
- [19].Henderson DA. Science. 1999;283:1279. doi: 10.1126/science.283.5406.1279. [DOI] [PubMed] [Google Scholar]
- [20].Cui Y, Wei Q, Park H, Lieber CM. Science. 2001;293:1289. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- [21].Blum LJ, Coulet PR. Biosensor principles and applications. CRC; Boca Raton: 1991. [Google Scholar]
- [22].Gorton L. Biosensors and Modern Biospecific Analytical Techniques. Elsevier; Dordrecht: 2005. [Google Scholar]
- [23].Mao CB, Flynn CE, Hayhurst A, Sweeney R, Qi J, Iverson B, Georgiou G, Belcher AM. Proc. Natl. Acad. Sci. USA. 2003;100:6946. doi: 10.1073/pnas.0832310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mao CB, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM. Science. 2004;303:213. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- [25].Balci S, Bittner AM, Hahn K, Scheu C, Knez M, Kadri A, Wege C, Jeske H, Kern K. Electrochim. Acta. 2006;51:6251. [Google Scholar]
- [26].Balci S, Noda K, Bittner A, Kadri A, Wege C, Jeske H, Kern K. Angew. Chem. 2007;119:3210. doi: 10.1002/anie.200604558. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:3149. [Google Scholar]
- [27].Vega RA, Maspoch D, Salaita K, Mirkin CA. Angew. Chem. 2005;117:6167. doi: 10.1002/anie.200501978. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:6013. [Google Scholar]
- [28].Carrera MRA, Kaufmann GF, Mee JM, Meijler MM, Koob GF, Janda KD. Proc. Natl. Acad. Sci. USA. 2004;101:10416. doi: 10.1073/pnas.0403795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Bioconjugate Chem. 2007;18:1140. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- [30].Souza GR, Christianson DR, Staquicini FI, Ozawa MG, Snyder EY, Sidman RL, Miller JH, Arap W, Pasqualini R. Proc. Natl. Acad. Sci. USA. 2006;103:1215. doi: 10.1073/pnas.0509739103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu C, Barnhill H, Liang X, Wang Q, Jiang H. Opt. Commun. 2005;255:366. [Google Scholar]
- [32].Petrenko VA, Sorokulova IB. J. Microbiol. Methods. 2004;58:147. doi: 10.1016/j.mimet.2004.04.004. [DOI] [PubMed] [Google Scholar]
- [33].Goicochea NL, De M, Rotello VM, Mukhopadhyay S, Dragnea B. Nano Lett. 2007;7:2281. doi: 10.1021/nl070860e. [DOI] [PubMed] [Google Scholar]
- [34].Petrenko VA. Expert Opin. Drug Delivery. 2008;5:825. doi: 10.1517/17425247.5.8.825. [DOI] [PubMed] [Google Scholar]
- [35].Smith GP, Petrenko VA. Chem. Rev. 1997;97:391. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- [36].Barbas CF, Burton DR, Scott JK. Phage display, a laboratory manual. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- [37].Sidhu SS. Phage Display in Biotechnology and Drug Discovery. CRC; Boca Raton: 2005. [Google Scholar]
- [38].Clackson T, Lowman HB. Phage display: a practical approach. Oxford University Press; Oxford: 2004. [Google Scholar]
- [39].McGrath S, Sinderen D. v. Bacteriophage: Genetics and Molecular Biology. Horizon Scientific Press; 2007. [Google Scholar]
- [40].Harris JR, Horne RW. Electron Microscopy of Proteins: Viral Structure. Academic Press; New York: 1986. [Google Scholar]
- [41].Waddell TE, Franklin K, Mazzocco A, Kropinski AM, Johnson RP. Methods Mol. Biol. 2009;501:293. doi: 10.1007/978-1-60327-164-6_25. [DOI] [PubMed] [Google Scholar]
- [42].Chen J, Novick RP. Science. 2009;323:139. doi: 10.1126/science.1164783. [DOI] [PubMed] [Google Scholar]
- [43].Hooker JM, Kovacs EW, Francis MB. J. Am. Chem. Soc. 2004;126:3718. doi: 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- [44].Kuzmanovic DA, Elashvili I, Wick C, O’Connell C, Krueger S. Structure. 2003;11:1339. doi: 10.1016/j.str.2003.09.021. [DOI] [PubMed] [Google Scholar]
- [45].Liu C-M, Jin Q, Sutton A, Chen L. Bioconjugate Chem. 2005;16:1054. doi: 10.1021/bc050108+. [DOI] [PubMed] [Google Scholar]
- [46].Irving MB, Pan O, Scott JK. Curr. Opin. Chem. Biol. 2001;5:314. doi: 10.1016/S1367-5931(00)00208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cao B, Mao CB. Biomacromolecules. 2009;10:555. doi: 10.1021/bm801224q. [DOI] [PubMed] [Google Scholar]
- [48].Lee SW, Mao CB, Flynn CE, Belcher AM. Science. 2002;296:892. doi: 10.1126/science.1068054. [DOI] [PubMed] [Google Scholar]
- [49].Petrenko VA, Smith GP. In: Phage Display in Biotechnology and Drug Discovery. Sidhu S, editor. CRC, Taylor & Francis; Boca Raton: 2005. p. 714. [Google Scholar]
- [50].Modali S, Abbineni G, Jayanna P, Petrenko V, Mao CB. Nanotechnology 2008: Life Sciences, Medicine & Bio Materials. Vol. 2. CRC; Boca Raton: 2008. p. 465. [Google Scholar]
- [51].Petrenko VA, Smith GP, Gong X, Quinn T. Protein Eng. 1996;9:797. doi: 10.1093/protein/9.9.797. [DOI] [PubMed] [Google Scholar]
- [52].Flynn CE, Mao CB, Hayhurst A, Williams J, Georgiou G, Iverson B, Belcher AM. J. Mater. Chem. 2003;13:2414. [Google Scholar]
- [53].Kuzmicheva GA, Jayanna PK, Sorokulova IB, Petrenko VA. Protein Eng. Des. Sel. 2009;22:9. doi: 10.1093/protein/gzn060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Katz BA. Annu. Rev. Biophys. Biomol. Struct. 1997;26:27. doi: 10.1146/annurev.biophys.26.1.27. [DOI] [PubMed] [Google Scholar]
- [55].Kehoe JW, Kay BK. Chem. Rev. 2005;105:4056. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- [56].Fernandez-Gacio A, Uguen M, Fastrez J. Trends Biotechnol. 2003;21:408. doi: 10.1016/S0167-7799(03)00194-X. [DOI] [PubMed] [Google Scholar]
- [57].Barrientos LG, Louis JM, Gronenborn AM. J. Magn. Reson. 2001;149:154. doi: 10.1006/jmre.2000.2276. [DOI] [PubMed] [Google Scholar]
- [58].Dogic Z, Fraden S. Phys. Rev. Lett. 1997;78:2417. [Google Scholar]
- [59].Petrenko VA, Vodyanoy VJ. J. Microbiol. Methods. 2003;53:253. doi: 10.1016/s0167-7012(03)00029-0. [DOI] [PubMed] [Google Scholar]
- [60].Larocca D, Burg MA, Jensen-Pergakes K, Ravey EP, Gonzalez AM, Baird A. Curr. Pharm. Biotechnol. 2002;3:45. doi: 10.2174/1389201023378490. [DOI] [PubMed] [Google Scholar]
- [61].Brigati JR, Petrenko VA. Anal. Bioanal. Chem. 2005;382:1346. doi: 10.1007/s00216-005-3289-y. [DOI] [PubMed] [Google Scholar]
- [62].Fukusaki E, Nakanishi T, Ogawa K, Kogayashi A. Electrochemistry. 2001;69:966. [Google Scholar]
- [63].Olofsson L, Ankarloo J, Andersson PO, Nicholls IA. Chem. Biol. 2001;8:661. doi: 10.1016/s1074-5521(01)00041-2. [DOI] [PubMed] [Google Scholar]
- [64].Dultsev FN, Speight RE, Fiorini MT, Blackburn JM, Abell C, Ostanin VP, Klenerman D. Anal. Chem. 2001;73:3935. doi: 10.1021/ac0100897. [DOI] [PubMed] [Google Scholar]
- [65].Uttenthaler E, Schraml M, Mandel J, Drost S. Biosens. Bioelectron. 2001;16:735. doi: 10.1016/s0956-5663(01)00220-2. [DOI] [PubMed] [Google Scholar]
- [66].Olsen EV, Sorokulova IB, Petrenko VA, Chen I-H, Barbaree JM, Vodyanoy VJ. Biosens. Bioelectron. 2006;21:1434. doi: 10.1016/j.bios.2005.06.004. [DOI] [PubMed] [Google Scholar]
- [67].Nanduri V, Sorokulova IB, Samoylov AM, Simonian AL, Petrenko VA, Vodyanoy V. Biosens. Bioelectron. 2007;22:986. doi: 10.1016/j.bios.2006.03.025. [DOI] [PubMed] [Google Scholar]
- [68].Olsen EV, Sykora J, Sorokulova I, Neely W, Petrenko V, Chen I-H, Barbaree J, Vodyanoy V. ECS Trans. 2007;2:9. [Google Scholar]
- [69].Decker J, Weinberger K, Prohaska E, Hauck S, Kosslinger C, Wolf H, Hengerer A. J. Immunol. Methods. 2000;233:159. doi: 10.1016/s0022-1759(99)00187-8. [DOI] [PubMed] [Google Scholar]
- [70].Sauerbrey GZ. Physics. 1959;155:206. [Google Scholar]
- [71].Hutchinson AM. Mol. Biotechnol. 1995;3:47. doi: 10.1007/BF02821334. [DOI] [PubMed] [Google Scholar]
- [72].Ngeh-Ngwainbi J, Suleiman AA, Guilbault GG. Biosens. Bioelectron. 1990;5:13. doi: 10.1016/0956-5663(90)80023-7. [DOI] [PubMed] [Google Scholar]
- [73].Schmitt N, Tessier L, Watier H, Patat F. Sens. Actuators B. 1997;43:217. [Google Scholar]
- [74].Muramatsu H, Kajiwara K, Tamiya E, Karube I. Anal. Chim. Acta. 1986;188:257. [Google Scholar]
- [75].Plomer M, Guilbault GG, Hock B. Enzyme Microb. Technol. 1992;14:230. doi: 10.1016/0141-0229(92)90071-u. [DOI] [PubMed] [Google Scholar]
- [76].Ivnitski D, Abdel-Hamid I, Atanasov P, Wilkins E. Biosens. Bioelectron. 1999;14:599. doi: 10.1016/s0956-5663(99)00004-4. [DOI] [PubMed] [Google Scholar]
- [77].Pathirana ST, Barbaree J, Chin BA, Hartell MG, Neely WC, Vodyanoy V. Biosens. Bioelectron. 2000;15:135. doi: 10.1016/s0956-5663(00)00067-1. [DOI] [PubMed] [Google Scholar]
- [78].Fung YS, Wong YY. Anal. Chem. 2001;73:5302. doi: 10.1021/ac010655y. [DOI] [PubMed] [Google Scholar]
- [79].Vaughan RD, O’Sullivan CK, Guilbault GG. Enzyme Microb. Technol. 2001;29:635. [Google Scholar]
- [80].Brigati J, Williams DD, Sorokulova IB, Nanduri V, Chen I-H, Turnbough CL, Jr., Petrenko VA. Clin. Chem. 2004;50:1899. doi: 10.1373/clinchem.2004.038018. [DOI] [PubMed] [Google Scholar]
- [81].Ben-Dov I, Willner I. Anal. Chem. 1997;69:3506. doi: 10.1021/ac970216s. [DOI] [PubMed] [Google Scholar]
- [82].Koenig B, Graetzel M. Anal. Lett. 1993;26:1567. [Google Scholar]
- [83].Shen Z, Huang M, Xiao C, Zhang Y, Zeng X, Wang PG. Anal. Chem. 2007;79:2312. doi: 10.1021/ac061986j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Susmel S, O’Sullivan CK, Guilbault GG. Enzyme Microb. Technol. 2000;27:639. doi: 10.1016/s0141-0229(00)00196-4. [DOI] [PubMed] [Google Scholar]
- [85].Abad JM, Pariente F, Hernández L, Lorenzo E. Anal. Chim. Acta. 1998;368:183. [Google Scholar]
- [86].Eun AJ-C, Huang L, Chew F-T, Li SF-Y, Wong S-M. Phytopathology. 2002;92:654. doi: 10.1094/PHYTO.2002.92.6.654. [DOI] [PubMed] [Google Scholar]
- [87].Tamarin O, Déjous C, Rebière D, Pistré J, Comeau S, Moynet D, Bezian J. Sens. Actuators B. 2003;91:275. doi: 10.1016/s0956-5663(03)00022-8. [DOI] [PubMed] [Google Scholar]
- [88].Petrenko VA, Smith GP. Protein Eng. 2000;13:589. doi: 10.1093/protein/13.8.589. [DOI] [PubMed] [Google Scholar]
- [89].Houshmand H, Fröman G, Magnusson G. Anal. Biochem. 1999;268:363. doi: 10.1006/abio.1998.3076. [DOI] [PubMed] [Google Scholar]
- [90].Tan WS, Tan GH, Yusoff K, Seow HF. J. Clin. Virol. 2005;34:35. doi: 10.1016/j.jcv.2005.01.007. [DOI] [PubMed] [Google Scholar]
- [91].Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindqvist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD. Proc. Natl. Acad. Sci. USA. 1998;95:6157. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Charlton K, Harris WJ, Porter AJ. Biosens. Bioelectron. 2001;16:639. doi: 10.1016/s0956-5663(01)00192-0. [DOI] [PubMed] [Google Scholar]
- [93].Samoylov AM, Samoylova TI, Pathirana ST, Globa LP, Vodyanoy VJ. J. Mol. Recognit. 2002;15:197. doi: 10.1002/jmr.574. [DOI] [PubMed] [Google Scholar]
- [94].Shriver-Lake LC, Taitt CR, Ligler FS. J. AOAC Int. 2004;87:1498. [PubMed] [Google Scholar]
- [95].Paoli GC. ACS Symp. Ser. 2006;931:41. [Google Scholar]
- [96].Hengerer A, Kosslinger C, Decker J, Hauck S, Queitsch I, Wolf H, Dubel S. BioTechniques. 1999;26:956. doi: 10.2144/99265rr05. [DOI] [PubMed] [Google Scholar]
- [97].Nanduri V, Samoylova AM, Petrenko V, Vodyanoy V, Simonian AL. 206th Meeting of The Electrochemical Society; Honolulu (USA). 2004. [Google Scholar]
- [98].Marks KM, Rosinov M, Nolan GP. Chem. Biol. 2004;11:347. doi: 10.1016/j.chembiol.2004.03.004. [DOI] [PubMed] [Google Scholar]
- [99].Goodridge L, Chen J, Griffiths M. Int. J. Food Microbiol. 1999;47:43. doi: 10.1016/s0168-1605(99)00010-0. [DOI] [PubMed] [Google Scholar]
- [100].Goodridge L, Chen J, Griffiths M. Appl. Environ. Microbiol. 1999;65:1397. doi: 10.1128/aem.65.4.1397-1404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Loessner M, Rudolf M, Scherer S. Appl. Environ. Microbiol. 1997;63:2961. doi: 10.1128/aem.63.8.2961-2965.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Goldman ER, Pazirandeh MP, Charles PT, Balighian ED, Anderson GP. Anal. Chim. Acta. 2002;457:13. [Google Scholar]
- [103].Goldman ER, Hayhurst A, Lingerfelt BM, Iverson BL, Georgiou G, Anderson GP. J. Environ. Monit. 2003;5:380. doi: 10.1039/b302012f. [DOI] [PubMed] [Google Scholar]
- [104].Goldman ER, Pazirandeh MP, Mauro JM, King KD, Frey JC, Anderson GP. J. Mol. Recognit. 2000;13:382. doi: 10.1002/1099-1352(200011/12)13:6<382::AID-JMR511>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [105].Lu X, Weiss P, Block T. J. Virol. Methods. 2004;119:51. doi: 10.1016/j.jviromet.2004.02.014. [DOI] [PubMed] [Google Scholar]
- [106].Zebedee SL, Barbas CF, Hom Y, Coathien RH, Graff R, DeGraw J, Pyati J, LaPolla R, Burton DR, Lerner RA, Thorton GB. Proc. Natl. Acad. Sci. USA. 1992;89:3175. doi: 10.1073/pnas.89.8.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Block T, Miller R, Korngold R, Jungkind D. Biotechniques. 1989;7:756. [PubMed] [Google Scholar]
- [108].Pulli T, Hoyhtya M, Soderlund H, Takkinen K. Anal. Chem. 2005;77:2637. doi: 10.1021/ac048379l. [DOI] [PubMed] [Google Scholar]
- [109].Torrance L, Ziegler A, Pittman H, Paterson M, Toth R, Eggleston I. J. Virol. Methods. 2006;134:164. doi: 10.1016/j.jviromet.2005.12.012. [DOI] [PubMed] [Google Scholar]
- [110].Nanduri V, Balasubramanian S, Sista S, Vodyanoy VJ, Simonian AL. Anal. Chim. Acta. 2007;589:166. doi: 10.1016/j.aca.2007.02.071. [DOI] [PubMed] [Google Scholar]
- [111].Nanduri V, Bhunia AK, Tu S-I, Paoli GC, Brewster JD. Biosens. Bioelectron. 2007;23:248. doi: 10.1016/j.bios.2007.04.007. [DOI] [PubMed] [Google Scholar]
- [112].Balasubramanian S, Sorokulova IB, Vodyanoy VJ, Simonian AL. Biosens. Bioelectron. 2007;22:948. doi: 10.1016/j.bios.2006.04.003. [DOI] [PubMed] [Google Scholar]
- [113].Daly S, Dillon P, Manning B, Dunne L, Killard A, O’Kennedy R. Food Agric. Immunol. 2002;14:255. [Google Scholar]
- [114].Wikman M, Rowcliffe E, Friedman M, Henning P, Lindholm L, Olofsson S, Ståhl S. Biotechnol. Appl. Biochem. 2006;45:93. doi: 10.1042/BA20060016. [DOI] [PubMed] [Google Scholar]
- [115].Wu Y, Brovko L, Griffiths MW. Lett. Appl. Microbiol. 2001;33:311. doi: 10.1046/j.1472-765x.2001.01002.x. [DOI] [PubMed] [Google Scholar]
- [116].Ripp S, Jegier P, Birmele M, Johnson CM, Daumer KA, Garland JL, Sayler GS. J. Appl. Microbiol. 2006;100:488. doi: 10.1111/j.1365-2672.2005.02828.x. [DOI] [PubMed] [Google Scholar]
- [117].Petrenko VA, Brigati JR. In: Immunoassay and other Bioanalytical Techniques. Emon JMV, editor. CRC, Taylor & Francis Group; Boca Raton: 2007. p. 187. [Google Scholar]
- [118].Ramanujam P, Tan WS, Nathan S, Yusoff K. Bio Techniques. 2004;36:296. doi: 10.2144/04362RR04. [DOI] [PubMed] [Google Scholar]
- [119].Blasco R, Murphy MJ, Sanders MF, Squirrell DJ. J. Appl. Microbiol. 1998;84:661. doi: 10.1046/j.1365-2672.1998.00393.x. [DOI] [PubMed] [Google Scholar]
- [120].Lakshmanan RS, Hu J, Guntupalli R, Wan J, Huang S, Yang H, Petrenko VA, Barbaree JM, Chin BA. SPIE-The International Society for Optical Engineering. Bellingham; Washington: 2006. p. 6218. [Google Scholar]
- [121].Rowe CA, Tender LM, Feldstein MJ, Golden JP, Scruggs SB, MacCraith BD, Cras JJ, Ligler FS. Anal. Chem. 1999;71:3846. doi: 10.1021/ac981425v. [DOI] [PubMed] [Google Scholar]
- [122].Jaworski JW, Raorane D, Huh JH, Majumdar A, Lee SW. Langmuir. 2008;24:4938. doi: 10.1021/la7035289. [DOI] [PubMed] [Google Scholar]
- [123].Stryer L, Haugland RP. Proc. Natl. Acad. Sci. USA. 1967;58:719. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Selvin PR. Nat. Struct. Biol. 2000;7:730. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- [125].Brockman JM, Nelson BP, Corn RM. Annu. Rev. Phys. Chem. 2000;51:41. doi: 10.1146/annurev.physchem.51.1.41. [DOI] [PubMed] [Google Scholar]
- [126].Haes AJ, Stuart DA, Duyne RPV. In: Nanotechnology in Biology and Medicine: Methods, Devices, and Applications. Vo-Dinh T, editor. CRC; Boca Raton: 2007. p. 20/1. [Google Scholar]
- [127].Nanduri V, Bhunia AK, Tu S-I, Paoli GC, Brewster JD. Biosens. Bioelectron. 2007;23:248. doi: 10.1016/j.bios.2007.04.007. [DOI] [PubMed] [Google Scholar]
- [128].Mu Y, Song D, Li Y, Zhang H-Q, Li W, Luo G-M, Jin Q-H. Talanta. 2005;66:181. doi: 10.1016/j.talanta.2004.11.014. [DOI] [PubMed] [Google Scholar]
- [129].Åström E, Ohlson S. J. Mol. Recognit. 2006;19:282. doi: 10.1002/jmr.786. [DOI] [PubMed] [Google Scholar]
- [130].Neufeld T, Schwartz-Mittelmann A, Biran D, Ron EZ, Rishpon J. Anal. Chem. 2003;75:580. doi: 10.1021/ac026083e. [DOI] [PubMed] [Google Scholar]
- [131].Neufeld T, Mittelman AS, Buchner V. J. Rishpon, Anal. Chem. 2005;77:652. doi: 10.1021/ac0488053. [DOI] [PubMed] [Google Scholar]
- [132].Thomas JH, Kim SK, Hesketh PJ, Halsall HB, Heineman WR. Anal. Chem. 2004;76:2700. doi: 10.1021/ac035503c. [DOI] [PubMed] [Google Scholar]
- [133].Kuramitz H, Dziewatkoski M, Barnett B, Halsall HB, Heineman WR. Anal. Chim. Acta. 2006;561:69. [Google Scholar]
- [134].Benhar I, Eshkenazi I, Neufeld T, Opatowsky J, Shaky S, Rishpon J. Talanta. 2001;55:899. doi: 10.1016/s0039-9140(01)00497-0. [DOI] [PubMed] [Google Scholar]
- [135].Jia Y, Qin M, Zhang H, Niu W, Li X, Wang L, Li X, Bai Y, Cao Y, Feng X. Biosens. Bioelectron. 2007;22:3261. doi: 10.1016/j.bios.2007.01.018. [DOI] [PubMed] [Google Scholar]
- [136].Yang L-MC, Tam PY, Murray BJ, Mclntire TM, Overstreet CM, Weiss GA, Penner RM. Anal. Chem. 2006;78:3265. doi: 10.1021/ac052287u. [DOI] [PubMed] [Google Scholar]
- [137].Yang L-MC, Diaz JE, McIntire TM, Weiss GA, Penner RM. Anal. Chem. 2008;80:5695. doi: 10.1021/ac8008109. [DOI] [PubMed] [Google Scholar]
- [138].Yang L-MC, Diaz JE, McIntire TM, Weiss GA, Penner RM. Anal. Chem. 2008;80:933. doi: 10.1021/ac071470f. [DOI] [PubMed] [Google Scholar]
- [139].Dill K, Montgomery DD, Ghindilis AL, Schwarzkopf KR, Ragsdale SR, Oleinikov AV. Biosens. Bioelectron. 2004;20:736. doi: 10.1016/j.bios.2004.06.049. [DOI] [PubMed] [Google Scholar]
- [140].Gabig-Ciminska M, Los M, Holmgren A, Albers J, Czyz A, Hintsche R, Wegrzyn G, Enfors S-O. Anal. Biochem. 2004;324:84. doi: 10.1016/j.ab.2003.09.020. [DOI] [PubMed] [Google Scholar]
- [141].Yemini M, Levi Y, Yagil E, Rishpon J. Bioelectromagnetics. 2007;70:180. doi: 10.1016/j.bioelechem.2006.03.014. [DOI] [PubMed] [Google Scholar]
- [142].Liu A, Honma I, Zhou HS. Biosens. Bioelectron. 2005;21:809. doi: 10.1016/j.bios.2005.03.005. [DOI] [PubMed] [Google Scholar]
- [143].Liu A, Watanabe T, Honma I, Wang J, Zhou H. Biosens. Bioelectron. 2006;22:694. doi: 10.1016/j.bios.2006.02.006. [DOI] [PubMed] [Google Scholar]
- [144].Liu A, Honma I, Zhou H. Biosens. Bioelectron. 2007;23:74. doi: 10.1016/j.bios.2007.03.019. [DOI] [PubMed] [Google Scholar]
- [145].Liu A, Wei MD, Honma I, Zhou H. Adv. Funct. Mater. 2006;16:371. [Google Scholar]
- [146].Bard A, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. 2nd ed Wiley; New York: 2001. [Google Scholar]
- [147].Shah J, Wilkins E. Electroanalysis. 2003;15:157. [Google Scholar]
- [148].Liu A, Anzai J.-i. Anal. Chem. 2004;76:2975. doi: 10.1021/ac0303970. [DOI] [PubMed] [Google Scholar]
- [149].Bakker E, Qin Y. Anal. Chem. 2006;78:3965. doi: 10.1021/ac060637m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Kuramitz H, Dziewatkoski M, Barnett B, Halsall HB, Heineman WR. Anal. Chim. Acta. 2006;561:69. [Google Scholar]
- [151].Yoshinobu T, Iwasaki H, Ui Y, Furuichi K, Ermolenko Y, Mourzina Y, Wagner T, Näther N, Schöning MJ. Methods. 2005;37:94. doi: 10.1016/j.ymeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- [152].Diaz JE, Yang LM, Lamboy JA, Penner RM, Weiss GA. Methods Mol. Biol. 2009;504:255. doi: 10.1007/978-1-60327-569-9_16. [DOI] [PubMed] [Google Scholar]
- [153].Majumdar S, Hajduczki A, Mendez AS, Weiss GA. Bioorg. Med. Chem. Lett. 2008;18:5937. doi: 10.1016/j.bmcl.2008.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Weiss GA, Penner RM. Anal. Chem. 2008;80:3082. doi: 10.1021/ac086009h. [DOI] [PubMed] [Google Scholar]
- [155].Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- [156].Decher G. Science. 1997;277:1232. [Google Scholar]
- [157].Liu A, Kashiwagi Y, Anzai J.-i. Electroanalysis. 2003;15:1139. [Google Scholar]
- [158].Liu A, Anzai J.-i. Langmuir. 2003;19:4043. [Google Scholar]
- [159].Liu A, Anzai J.-i. Anal. Bioanal. Chem. 2004;380:98. doi: 10.1007/s00216-004-2728-5. [DOI] [PubMed] [Google Scholar]
- [160].Liu A, Abbineni G, Mao CB. Adv. Mater. 2009;21:1001. [Google Scholar]
- [161].Shenton W, Douglas T, Young M, Stubbs G, Mann S. Adv. Mater. 1999;11:253. [Google Scholar]
- [162].Mukherjee S, Pfeifer CM, Johnson JM, Liu J, Zlotnick A. J. Am. Chem. Soc. 2006;128:2538. doi: 10.1021/ja056656f. [DOI] [PubMed] [Google Scholar]
- [163].Loo L, Guenther RH, Basnayake VR, Lommel SA, Franzen S. J. Am. Chem. Soc. 2006;128:4502. doi: 10.1021/ja057332u. [DOI] [PubMed] [Google Scholar]
- [164].Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Nat. Med. 2006;12:354. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Srinivasan K, Cular S, Bhethanabotla VR, Lee SY, Harris MT, Culver JN. AIChE Annual Meeting; Cincinnati (USA). 2005. p. 74d/1. [Google Scholar]
- [166].Steinmetz NF, Lomonossoff GP, Evans DJ. Langmuir. 2006;22:3488. doi: 10.1021/la060078e. [DOI] [PubMed] [Google Scholar]
- [167].Niu Z, Liu J, Lee LA, Bruckman MA, Zhao D, Koley G, Wang Q. Nano Lett. 2007;7:3729. doi: 10.1021/nl072134h. [DOI] [PubMed] [Google Scholar]
- [168].Simões FR, Mattoso LHC, Vaz CMP. Sens. Lett. 2006;4:319. [Google Scholar]
- [169].Jung YS, Jung WC, Tuller HL, Ross CA. Nano Lett. 2008;8:3776. doi: 10.1021/nl802099k. [DOI] [PubMed] [Google Scholar]
- [170].Gerard M, Chaubey A, Malhotra BD. Biosens. Bioelectron. 2002;17:345. doi: 10.1016/s0956-5663(01)00312-8. [DOI] [PubMed] [Google Scholar]
- [171].West JL, Halas NJ. Annu. Rev. Biomed. Eng. 2003;5:285. doi: 10.1146/annurev.bioeng.5.011303.120723. [DOI] [PubMed] [Google Scholar]
- [172].Niemeyer CM. Angew. Chem. 2001;113:4254. [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:4128. [Google Scholar]
- [173].Pankhurst QA, Connolly J, Jones SK, Dobson J. J. Phys. D. 2003;36:R167. [Google Scholar]
- [174].Tartaj P, del Puerto Morales M, Veintemillas-Verdaguer S, González-Carreño T, Serna CJ. J. Phys. D. 2003;36:R182. [Google Scholar]
- [175].Berry CC, Curtis ASG. J. Phys. D. 2003;36:R198. [Google Scholar]
- [176].Liu A, Zhou H, Honma I, Ichihara M. Appl. Phys. Lett. 2007;90:253112. [Google Scholar]
- [177].Chen RJ, Bangsaruntip S, Drouvalakis KA, Kam NWS, Shim M, Li Y, Kim W, Utz PJ, Dai H. Proc. Natl. Acad. Sci. USA. 2003;100:4984. doi: 10.1073/pnas.0837064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Lu Y, Liu J. PCT Int. Appl. 2005:69. [Google Scholar]
- [179].Medintz IL, Sapsford KE, Konnert JH, Chatterji A, Lin T, Johnson JE, Mattoussi H. Langmuir. 2005;21:5501. doi: 10.1021/la0468287. [DOI] [PubMed] [Google Scholar]
- [180].Fischlechner M, Reibetanz U, Zaulig M, Enderlein D, Romanova J, Leporatti S, Moya S, Donath E. Nano Lett. 2007;7:3540. doi: 10.1021/nl0723580. [DOI] [PubMed] [Google Scholar]
- [181].Fischlechner M, Donath E. Angew. Chem. 2007;119:3246. doi: 10.1002/anie.200603445. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:3184. [Google Scholar]
- [182].Toellner L, Fischlechner M, Ferko B, Grabherr RM, Donath E. Clin. Chem. 2006;52:1575. doi: 10.1373/clinchem.2005.065789. [DOI] [PubMed] [Google Scholar]
- [183].Fischlechner M, Toellner L, Messner P, Grabherr R, Donath E. Angew. Chem. 2006;118:798. doi: 10.1002/anie.200502620. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:784. [Google Scholar]
- [184].Fischlechner M, Zschornig O, Hofmann J, Donath E. Angew. Chem. 2005;117:2952. doi: 10.1002/anie.200460763. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:2892. [Google Scholar]
- [185].Zhu H, White IM, Suter JD, Fan X. Biosens. Bioelectron. 2008;24:461. doi: 10.1016/j.bios.2008.04.028. [DOI] [PubMed] [Google Scholar]
- [186].Tseng RJ, Tsai C, Ma L, Ouyang J, Ozkan CS, Yang Y. Nat. Nanotechnol. 2006;1:72. doi: 10.1038/nnano.2006.55. [DOI] [PubMed] [Google Scholar]
- [187].Clarkson S. Nat. Rev. Microbiol. 2004;2:353. doi: 10.1038/nrmicro834. [DOI] [PubMed] [Google Scholar]
- [188].Chen C, Daniel MC, Quinkert ZT, De M, Stein B, Bowman VD, Chipman PR, Rotello VM, Kao CC, Dragnea B. Nano Lett. 2006;6:611. doi: 10.1021/nl0600878. [DOI] [PubMed] [Google Scholar]
- [189].Lim Y.-b., Lee E, Yoon Y-R, Lee MS, Lee M. Angew. Chem. 2008;120:4601. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:4525. [Google Scholar]
- [190].Lee ES, Kim D, Youn YS, Oh KT, Bae YH. Angew. Chem. 2008;120:2452. doi: 10.1002/anie.200704121. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:2418. [Google Scholar]
- [191].Haselhorst T, Garcia J-M, Islam T, Lai JCC, Rose FJ, Nicholls JM, Peiris JSM, von Itzstein M. Angew. Chem. 2008;120:1936. doi: 10.1002/anie.200704872. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1910. [Google Scholar]
- [192].Huang X, Bronstein LM, Retrum J, Dufort C, Tsvetkova I, Aniagyei S, Stein B, Stucky G, McKenna B, Remmes N, Baxter D, Kao CC, Dragnea B. Nano Lett. 2007;7:2407. doi: 10.1021/nl071083l. [DOI] [PubMed] [Google Scholar]
- [193].Jayanna PK, Torchilin VP, Petrenko VA. Nanomedicine. 2009;5:83. doi: 10.1016/j.nano.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lee S-W, Lee SK, Belcher AM. Adv. Mater. 2003;15:689. [Google Scholar]
- [195].Douglas T, Young M. Science. 2006;312:873. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- [196].Steinmetz NF, Findlay KC, Noel TR, Parker R, Lomonossoff GP, Evans DJ. ChemBioChem. 2008;9:1662. doi: 10.1002/cbic.200800070. [DOI] [PubMed] [Google Scholar]
- [197].Nam KT, Kim D-W, Yoo PJ, Chiang C-Y, Meethong N, Hammond PT, Chiang Y-M, Belcher AM. Science. 2006;312:885. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- [198].Takeda S, Ozaki H, Hattori S, Ishii A, Kida H, Mukasa K. J. Nanosci. Nanotechnol. 2007;7:752. doi: 10.1166/jnn.2007.512. [DOI] [PubMed] [Google Scholar]
- [199].Pantarotto D, Partidos CD, Hoebeke J, Brown F, Kramer E, Briand J-P, Muller S, Prato M, Bianco A. Chem. Biol. 2003;10:961. doi: 10.1016/j.chembiol.2003.09.011. [DOI] [PubMed] [Google Scholar]