

SUMMARY
The multifunctional, stress-inducible, molecular chaperone HSP70 has important roles in aiding protein folding and maintaining protein homeostasis. HSP70 expression is elevated in many cancers, contributing to tumor cell survival and resistance to therapy. We have determined that a small molecule called 2-Phenylethynesulfonamide (PES) interacts selectively with HSP70, and leads to a disruption of the association between HSP70 and several of its co-chaperones and substrate proteins. Treatment of cultured tumor cells with PES promotes cell death that is associated with protein aggregation, impaired autophagy, and inhibition of lysosomal function. Moreover, this small molecule is able to suppress tumor development and enhance survival in a mouse model of Myc-induced lymphomagenesis. The data demonstrate that PES disrupts actions of HSP70 in multiple cell signaling pathways offering an opportunity to better understand the diverse functions of this molecular chaperone, and also to aid in the development of new cancer therapies.
INTRODUCTION
Resistance to programmed cell death (apoptosis) is a characteristic of many cancer cells. Accordingly, efforts to develop new treatment strategies and therapeutic targets include attempts to identify and characterize proteins that regulate other survival or stress-response pathways. Heat shock proteins (HSPs) are encoded by evolutionarily conserved gene families and are required for cell survival following various forms of stress. HSPs generally are classified according to their approximate molecular size, and are structurally and functionally diverse; some are constitutively expressed, while others are stress-induced (Mayer and Bukau, 2005; Brodsky and Chiosis, 2006; Garrido et al., 2006; Schmitt et al., 2006; Powers and Workman, 2007). The stress-inducible protein HSP70 (also called HSP72, HSP70-1 or HSPA1A) is an approximately 70 kDa ATP-dependent molecular chaperone that is present at low or undetectable levels in most unstressed normal cells and tissues. Its abundance rapidly increases in response to a variety of metabolic or exogenous insults that, among other effects, can cause changes in protein conformation or stability. HSP70-inducing stresses include elevated temperatures, nutrient deprivation, heavy metals, oxidative stress and viral infections. The stress-inducible HSP70 is thought to help cells cope with these potentially deleterious conditions, in part by aiding with folding of nascent polypeptides or refolding of damaged proteins, preventing/reversing protein aggregation or self-association, promoting protein transport to intracellular locations for degradation, and aiding in the formation of protein complexes. HSP70 also is an important regulator of apoptotic signaling pathways, acting in part through direct interactions with substrate proteins that affect multiple steps in the process, including control of mitochondrial membrane integrity and caspase-activation (Mayer and Bukau, 2005; Brodsky and Chiosis, 2006; Garrido et al., 2006; Schmitt et al., 2006; Powers and Workman, 2007).
In contrast to its low abundance in unstressed normal cells, the inducible HSP70 protein is present at constitutively elevated levels in many human tumors of various origin. Such enhanced HSP70 expression correlates with resistance of the tumor cells to caspase-dependent and-independent cell death and is associated with poor patient prognosis (Brodsky and Chiosis, 2006; Garrido et al., 2006; Guzhova and Margulis, 2006; Schmitt et al., 2006). It is likely that the unfavorable conditions associated with the tumor microenvironment, such as hypoxia, nutrient deprivation, oxidative stress, oncogene activation, and exposure to chemotherapeutics lead to alterations in protein structure or processing, as well as upregulation of HSP70. The actions of this protein would be expected to help tumor cells tolerate, or adapt to, these conditions, and current evidence suggests that elevated HSP70 expression promotes tumorigenesis. Conversely, reducing HSP70 levels in some cultured tumor cells has been reported to induce cell death, and/or to sensitize them to cytotoxic agents, while having no obvious deleterious effects on non-tumor cells (Nylandsted et al., 2000, 2002; Rohde et al., 2005; Schmitt et al., 2006; Aghdassi et al., 2007; Powers et al., 2008).
In addition to its cytoprotective actions in promoting tumorigenesis, an altered expression or function of HSP70 also has been implicated in certain other human disorders that are associated with defects in protein conformation or folding. This includes disorders caused by the presence of mutant proteins, as well as some neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, and viral pathogenesis (McClellan et al., 2005; Muchowski and Wacker, 2005; Brodsky and Chiosis, 2006; Guzhova and Margulis, 2006; Morimoto, 2008). The identification of small molecules that specifically interact with, and modulate the activities of, HSP70 therefore has important implications for a number of human diseases. To date, however, only a limited number of compounds that specifically target HSP70 have been identified in chemical screens, and few are currently available to assess the physiologic impact of modulating HSP70 actions (Brodsky and Chiosis, 2006; Powers and Workman, 2007; Wisén and Gestwicki, 2008). Here we report that the small molecule 2-Phenylethynesulfonamide (PES), also called phenylacetylenylsulfonamide, or pifithrin-μ(PFTμ), interacts selectively with the stress-inducible HSP70 protein and inhibits its functions. Tumor cell lines treated with PES lose viability associated with evidence of protein aggregation and dysregulation of autophagic/lysosomal processes. Moreover, administration of PES inhibits Myc-induced lymphoma in a mouse model system.
RESULTS
PES Interacts with HSP70
PES was identified in a screen of the Chembridge DIVERSet library of drug-like small molecules for those exhibiting an ability to inhibit the direct mitochondrial pathway of p53-mediated apoptosis, and was referred to as PFTμ (Strom et al., 2006). Subsequently, we found that PES prevents the accumulation of p53 at mitochondria and inhibits caspase cleavage following cisplatin treatment of a human tumor cell line (Leu and George, 2007). In an attempt to better understand the molecular basis for these observations, we sought to identify intracellular targets of PES.
Based on the structure of PES (Figure S1A), we used a thiol-cleavable amine-reactive reagent to synthesize a biotinylated form of the molecule (biotin-PES), as described (see Methods). Several mammalian cell lines were treated with biotin-PES, cell lysates were prepared, and biotin-PES complexes were captured using NeutrAvidin Resins. PES-interacting proteins were eluted using 100 mM DTT, which cleaves the disulfide bond in the spacer arm of the biotin attached to PES. The associated proteins were resolved by SDS-PAGE in a 4%-20% gradient gel and visualized by Coomassie staining. Results obtained using two different mammalian cell lines were consistent, and revealed a major band of about 70 kDa (Figure 1A). The band was excised from the gel, subjected to trypsin digestion, and the resulting peptides were analyzed by liquid chromatography-tandem mass spectrometry. The results pointed to the presence of the stress-inducible HSP70 or the closely related constitutive HSC70 (“cognate” 70 kDa heat shock protein), which have peptides in common (see Figure S1B for peptide analysis).
Figure 1. PES Binds to HSP70.

(A) Whole cell extracts (WCE) prepared from A875 and BX-U2OS cells, treated with Biotin or Biotin-PES, were captured using NeutrAvidin agarose resins. HSP70-family proteins were identified as the major product in the excised band of ~ 70 kDa shown in both Coomassie gels. HSP70 and HSC70 peptide sequences are shown in Figure S1B.
(B and C) WCE were prepared from the cell lines indicated following 24 h treatment with 20μM biotin or B-PES and examined for the expression of proteins indicated (left panel of B and C) by western blot analysis; note that different exposure times were used to visualize these proteins. B-PES-containing complexes were captured by NeutrAvidin Resins, and eluted following 100 mM DTT treatment. Immunoprecipitation-western blot (IP-WB) analysis using the indicated antibodies reveals interaction of B-PES with endogenous HSP70, but there is no detectable interaction with endogenous BAK, BCL-xL, GRP78, p53, HSC70, or HSP90 even after longer exposure times.
(D) In vitro evidence for an interaction between B-PES and HSP70. Full-length human HSP70 and human HSP90 proteins were in vitro translated in the presence of 35S-methionine, mixed with B-PES coupled to NeutrAvidin resins, and eluted using 100 mM DTT. The resulting DTT eluates were separated by polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by autoradiography.
(E) B-PES interacts with the C-terminal region of HSP70. H1299 cells were transfected with the indicated hemagglutinin (HA)-tagged constructs and exposed to B-PES. B-PES containing complexes were captured by NeutrAvidin Resins, eluted following 100 mM DTT treatment, and immunblotted with anti-HA antibody following SDS-PAGE.
We next carried out an analysis of PES-treated cells using antibodies specific for different HSP proteins, including the stress-inducible HSP70, HSC70, the ER-localized HSP family member GRP78 (BiP), and the 90 kDa molecular chaperone HSP90. These analyses revealed that in several cell lines biotin-PES interacts with HSP70, but not with HSC70, GRP78, HSP90, or several other proteins (Figures 1B, 1C and S1C). Although HSC70 and HSP70 are highly homologous (Figure S1B), they do exhibit some functional differences, in part related to different interactions with some co-chaperones (Rohde et al., 2005; Tutar et al., 2006). To complement these analyses, we confirmed that biotin-PES interacts with in vitro translated HSP70, but not with HSP90 (Figure 1D). Based on deletion analysis, we determined that biotin-PES interacts with the carboxyl-terminal substrate-binding domain (amino acids 386-641), but not the amino-terminal ATPase domain (amino acids 2-385), of human HSP70 (Figure 1E). Interestingly, we also obtained evidence that PES interacts with DnaK, the bacterial orthologue of mammalian HSP70 (Figures S2A and S2B). Consistent with a disrupted function of DnaK resulting from an interaction with PES, we found that this small molecule adversely affects the response of the bacteria to elevated temperatures by producing slow growth, filamentation, and reduced viability (Figures S2C and S2D). Notably, these are identical to the phenotypes reported for some DnaK mutants (Bukau and Walker, 1989; McCarty and Walker, 1994).
PES Interferes with the Chaperone Function of HSP70
The cellular actions of HSP70 are mediated in large part by its physical association with a number of co-chaperones, including HSP40, HSP90, CHIP and BAG-1 (McDonough and Patterson, 2003; Fan et al., 2003; Wegele et al., 2004; Mayer and Bukau, 2005; Townsend et al., 2005; Kabbage and Dickman, 2008). Also, the cytoprotective role of HSP70 has been linked to its ability to modulate the conformation and actions of apoptosis-regulators like APAF1. Thus, we used immunoprecipitation-western blot analysis to determine if PES alters the interactions between HSP70 and these proteins. Several mammalian cell lines used for these analyses generated consistent results, and representative data are presented (Figures 2A and 2B). When compared to controls, PES-treated cells contain less HSP70 in association with CHIP, HSP40 and APAF1. In contrast, we detected no change in the abundance of HSP70/HSP90 complexes in the presence of PES; however, the functional integrity of the HSP70/HSP90 interaction has not yet been explored.
Figure 2. PES Interferes with HSP70 Actions.

(A) WCE from H1299 lung carcinoma cells, treated either with 20μM Biotin or B-PES for 24 h, were immunoprecipitated (IP) using anti-HSP70 antibody. Western blots assessed the relative abundance of the proteins indicated (left), and co-immunoprecipitation-western (IP-WB) analysis revealed a reduced degree of interaction between HSP70 and HSP40, CHIP, BAG-1M, and APAF1 in B-PES-treated cells (right).
(B) IP-WB analyses of WCE from PES-treated U2OS osteosarcoma cells reveal a lower abundance of HSP70 complexes containing BAG-1M or APAF1.
(C) WCE were prepared from A875 melanoma cells that were either untreated or pretreated with PES (20μM) for 1 h, followed by the addition of 50μM cisplatin for 8 h. Note evidence of caspase cleavage in cisplatin-treated cells, but not in the presence of PES. IP-WB analysis reveals the presence of p53/BAK and p53/HSP70 complexes in cisplatin-treated cells (right) that are reduced following exposure to PES.
(D) (left) H1299 cells were transfected with a NF-κB-dependent luciferase reporter. 24 h later, cells were either pretreated with DMSO or the indicated amount of PES for 1 h, followed by the addition of 20 ng/ml of TNFα for 5 h, as specified. Each graphical representation indicates the mean ± SD of at least three independent cultures relative to control (DMSO-treated) cells. (right) H1299 cells were treated with 20μM PES for 5.7 h, followed by treatment with 10 ng/ml TNF for 20 min. WCE were immunoblotted for the proteins indicated.
The co-chaperone BAG-1 has several isoforms (BAG-1L, BAG-1M, BAG-1S) that have been implicated in diverse cellular pathways, including the inhibition of stress-induced apoptosis (Townsend et al., 2005; Kabbage and Dickman, 2008). PES caused a clear decrease in the interaction between HSP70 and the BAG-1M isoform (also known as the receptor-associated protein, RAP-46). In contrast, there was no detectable change in the abundance of the HSP70/BAG-1S complex, while disruption of the HSP70/BAG-1L interaction occurred in H1299 cells, but not U2OS cells (Figures 2A and 2B). It should be pointed out, that the same results were obtained in our experiments whether or not PES was conjugated to biotin (Figures 2A, 2B, and data not shown).
It previously has been reported that wt p53 can be found in a complex with HSP70 under some stress conditions, such as following heat shock or UV-irradiation of cultured tumor cells (Matsumoto et al., 1994; Chen et al., 1999). In light of these observations and data that PES antagonizes p53 mitochondrial localization (Strom et al., 2006; Leu and George, 2007), we tested the hypothesis that PES alters the HSP70/p53 association. For these analyses, we treated A875 melanoma cells and HepG2 hepatoma cells with cisplatin. As previously demonstrated (Leu and George, 2007), cisplatin (50μM) promotes p53 mitochondrial localization, where it interacts with mitochondrial BAK, and promotes caspase cleavage, as revealed by analysis of caspase-3 and caspase-8 (Figures 2C and S3). Under these conditions, we detected a fraction of the stress-activated p53 in a complex with HSP70 (Figures 2C and S3). In contrast, protein complexes containing p53 and either BAK or HSP70 were not observed in cells treated with PES or with cisplatin and PES; for the latter, there was a concomitant reduction in the appearance of caspase cleavage products (Figures 2C and S3). In contrast to the altered HSP70/p53 interaction in response to PES, there was no detectable change in the association between HSP70 and HSP90 or between BAK and BCL-xL (Figures 2C and S3).
The nuclear transcription factor NF-κB is an important regulator of cellular responses to stress, and contributes to pathological processes such as inflammation and tumor cell survival. Through mechanisms that are not fully characterized, HSP70 has been implicated as interacting with regulatory components of NF-κB signaling pathways (Salminen et al., 2008). In initial studies, we have found that PES inhibited NF-κB activation along with the concomitant turnover of the NF-κB inhibitory protein IκBα, that is normally stimulated by tumor necrosis factor-alpha (TNFα). PES also inhibited unstimulated NF-κB activity in a dose-dependent manner (Figure 2D). These observations, together with data described below, support our working hypothesis that disruption of HSP70 functions by PES interferes with a number of cell survival and signaling pathways.
PES Induces Cell Death In the Absence of Caspase Activation
Caspases are critical cellular effectors of apoptosis. As shown, caspase activation was observed following treatment of the cells with a concentration of cisplatin (50μM) that produced about 50% cytotoxicity (Figures 2C, 3A and S3). Interestingly, while exposure to PES (20μM) caused a greater than 50% loss of cell viability in all of the tumor cell lines we examined (Figures 3A-3C), this was not accompanied by cleavage of caspase-3, caspase-8, or PARP, based on western blot analyses (Figures 2C, 3B and S3; Leu and George, 2007). In fact, co-treatment of cells with PES together with cisplatin effectively inhibited the appearance of these caspase-cleavage products (Figures 2C and S3; Leu and George, 2007). Moreover, although the broad-spectrum caspase inhibitor Z-VAD-FMK inhibited cisplatin- as well as camptothecin-mediated caspase cleavage, it did not alter PES-mediated loss of cell viability (Figure 3B). Together, these results indicate that PES leads to a loss of cell viability in a manner that is not dependent on caspase activation, and indeed that PES inhibits caspase activation. This is consistent with the identification of PES as a small molecule inhibitor of p53-mediated caspase-activation and apoptosis (Strom et al., 2006).
Figure 3. PES Reduces Viability of Tumor Cells and Induces Cytoplasmic Vacuolization.

(A) MTT assays of A875 cells treated with DMSO, 20μM PES, or 50μM cisplatin for 24 h. Each graphical representation indicates the mean ± SD of at least three independent cultures relative to control (DMSO-treated) cells.
(B) (Top) WCE were prepared from U2OS osteosarcoma cells that were either untreated or pretreated with Z-VAD-FMK (20μM) for 1 h, followed by the addition of 5μM camptothecin (Camp), 16μg/ml Cisplatin (Cis), or 20μM PES for 6 h. Note evidence of caspase cleavage in cisplatin- or camptothecin-treated cells, but not in the presence of PES or Z-VAD-FMK. (Bottom) The indicated cell lines were either untreated or pretreated with Z-VAD-FMK (20μM) for 1 h, followed by the addition of DMSO or 20μM PES for 24 h. Cell viability was determined by MTT assays. Results shown are the mean of at least three independent experiments.
(C) The indicated cell lines were treated with the indicated concentrations of PES for either 24 h (top) or for 48 h (middle and bottom). Representative MTT assays indicate cell viability in human cell lines, including non-transformed human WI38 fibroblasts, as well as several tumor cell lines with wt p53 (U2OS, BX-U2OS, MCF7, CaPan2), or with mutant/deleted p53 (SKBR3, MDA468, MDA231, CaPan1, MiaPaCa, and Panc1). Four independent cultures were assayed for each treatment and DMSO treated cells were used as internal controls. Values shown are normalized to the viability of the control (DMSO-treated) cells. Error bars represent standard deviation (SD).
We analyzed several tumor cell lines of different histologic type and found that PES treatment leads to a dose-dependent loss of cell viability for all of the tumor cell lines we examined (Figures 3A-3C). In contrast, non-transformed WI38 human fetal lung fibroblasts exhibit no decrease in cell viability (Figure 3C), except at the highest concentration of PES tested (20μM). These results suggest a differential sensitivity to PES between non-tumor cells and tumor cells. Tumor cells that lack a functioning p53 tumor suppressor protein or that overexpress anti-apoptotic members of the BCL2 family are known to exhibit resistance to many chemotherapeutic agents. It is of interest, therefore, that the loss of cell viability resulting from PES occurred in all of the tumor cell lines we examined, irrespective of the p53 status (Figure 3C). Additionally, the anti-apoptotic protein BCL-xL was unable to protect against a PES-mediated loss of cell viability, as evidenced by the similar dose-response curves of U2OS osteosarcoma cells and a U2OS-derivative (BX-U2OS) that overexpresses BCL-xL (Figures 3B and 3C). These data support the conclusion that PES-mediated cytotoxicity likely involves a mechanism distinct from the execution of classical apoptosis.
PES Leads to Dysfunctional Autophagy and Altered Lysosome Function
In some settings, an inhibition of caspase activation and apoptosis can induce autophagy (Debnath et al., 2005; Levine and Kroemer, 2008). Many of the PES-treated cells showed a progressive accumulation of intracytoplasmic vacuoles (Figure S4), similar to those often observed in cells undergoing autophagy. Autophagy is an evolutionarily conserved, lysosome-dependent, bulk protein degradation pathway activated in response to starvation or stress; it allows for sustained metabolism by virtue of the digestion and recycling of long-lived proteins and cellular organelles. Mammalian cells also utilize autophagy to promote the degradation of damaged proteins, or to mediate the large-scale clearance of misfolded proteins and aggregates (Debnath et al., 2005; Eskelinen, 2005; Levine and Kroemer, 2008; Mizushima et al., 2008). In eukaryotic cells, the process is characterized by the sequestration of portions of the cytoplasm and intracellular organelles within double-membrane autophagic vacuoles, or autophagosomes; sequestered material is targeted for degradation through fusion of autophagosomes with lysosomes or endosomes. Therefore we examined PES-treated cells for altered expression of autophagy markers, including the microtubule-associated protein-1 light-chain 3 (LC3), which is converted from the 18 kDa free form (LC3-I) to a proteolytically-processed smaller (16 kDa) form (LC3-II) during autophagy (Klionsky et al., 2008; Tasdemir et al., 2008). Based on western blot analysis, all of the PES-treated cell lines exhibited higher levels of LC3-II (Figure 4A), consistent with an impact of PES on autophagy.
Figure 4. PES-Treated Cells Exhibit Altered Autophagy.

(A) Western blot (WB) analysis reveals increased appearance of processed LC3-II in the indicated cells following 20μM PES treatment for the indicated times.
(B-H) Electron micrographs of H1299 cells with or without PES treatment (20μM) for 7 or 24 h. (D) Double membrane autophagic vacuoles (AV), and vacuoles within vacuoles are evident. (E and F) AVs of different sizes are evident, some containing recognizable cytoplasmic content. (G and H) Large AVs containing partially digested cytoplasmic material as well as amorphous, membranous, aggregated, or granular masses are shown.
(I) The average area of autophagic vacuoles (AV) calculated with ImageJ software per cell is indicated.
We next used a number of recommended criteria to evaluate autophagic flux in PES-treated cells (Klionsky et al., 2008; Tasdemir et al., 2008; Kaushik and Cuervo, 2009). As one approach, we used electron microscopy (EM) which confirmed that PES-treated cells exhibit a rapid and substantial increase in multiple double-membrane autophagosomes and single-membrane autophagolysosomes (Figures 4B-4I and Figure S5C-S5I). Additionally, we evaluated the bulk degradation of long-lived proteins using a well-established radiolabeled-amino-acid-based assay. This approach provides a reliable indicator of autophagic flux, as it assays the end-point of autophagy (Klionsky et al., 2008; Tasdemir et al., 2008). We found that the basal degradation of long-lived proteins was significantly reduced in PES-treated cells relative to controls (Figure 5A), consistent with an impairment of autophagic flux. The turnover of long-lived proteins is regulated in part by hydrolases present in lysosomes, hydrolytic bodies that functionally interact with the trans-Golgi network, endosomes, and autophagosomes. Lysosomes contain a number of enzymes, including cathepsin proteases, that help in the turnover of macromolecules and organelles during normal metabolism and during autophagy. To further explore the effects of PES on autophagic flux, we examined the expression of the lysosomal cysteine peptidase cathepsin L, which plays an important role in the degradation of lysosomal cargo. Like other members of this protein family, cathepsin L is synthesized as an inactive precursor that undergoes proteolytic processing to the mature, active form during transport to the acidic environment of the endosomal/lysosomal compartment through autoprocessing or cleavage by other cathepsins (Collette et al., 2004). We found that PES caused an accumulation of the precursor pro-cathepsin L and a markedly reduced abundance of the smaller, mature form of the enzyme (Figure 5B); this points to an impaired processing of this lysosomal enzyme and impaired lysosomal function.
Figure 5. PES Impairs Long-Lived Protein Degradation and Lysosomal Function.

(A) Decreased degradation of long-lived proteins in U2OS cells treated with PES (20μM) for the indicated time (black line), compared to vehicle treated cells (gray line). Data are reported in percent of protein degraded at each time point, and are the averaged data from two independent experiments done in duplicate; error bars represent standard deviation. The data were consistent in H1299 cells (data not shown).
(B) Western blot (WB) analysis indicating altered processing of cathepsin L from the larger precursor form to the smaller mature form in the indicated cells following 20μM PES treatment for 24 h.
(C) IP-WB analyses of WCE from untreated or PES-treated A875 melanoma cells (left) and MiaPaCa pancreatic cells (right) reveal a lower abundance of HSP70 complexes containing LAMP2 following 20μM PES exposure for 24 h (lower panels). The expression patterns of LAMP2 before and after 20μM PES exposure for 24 h are shown on the top panel.
LAMP2 is an abundant late endosomal/lysosomal protein marker (Eskelinen, 2006) which plays an important role in chaperone-mediated autophagy and cooperates with the HSC70 and HSP70 in transporting certain substrates from the cytoplasm into lysosomes (Bandyopadhyay et al., 2008; Ryhänen et al., 2008). Given the evidence for co-localization of LAMP2 with HSP70 in lysosomes, we investigated how treatment of cells with PES might affect an interaction between these proteins. The results of immunoprecipitation-western blot analysis demonstrate that PES significantly reduced the amount of LAMP2 in a complex with HSP70 (Figure 5C). These data, together with the evidence of altered cathepsin processing and reduced degradation of long-lived proteins in the presence of PES, further support a role for HSP70 in the optimal transport and degradation of macromolecules during macroautophagy and chaperone-mediated autophagy.
PES Promotes Oligomerization and Aggregation of p62/SQSTM1
We next examined another marker of autophagy, the adaptor/scaffold protein p62/SQSTM1 (sequestosome-1). This protein is up-regulated in response to various forms of stress, and it mediates diverse cellular functions including signal transduction, receptor internalization, nuclear gene transcription, and the shuttling of some poly-ubiquitylated protein aggregates to different intracellular locations for degradation (Wooten et al., 2006; Moscat et al., 2007). The steady-state level of p62/SQSTM1 is regulated by autophagy, and an accumulation or aggregation of p62/SQSTM1 is considered a marker for inhibition of autophagy or defective autophagic degradation (Bjørkøy et al., 2005; Komatsu et al., 2007; Pankiv et al., 2007; Ichimura et al., 2008; Shvets et al., 2008). Western blot analysis for p62/SQSTM1 revealed that PES promotes an accumulation and oligomerization of p62/SQSTM1 in a time- and dose-dependent manner (Figures 6A-6C). Similarly, immunofluorescence-staining of PES-treated cells revealed the presence of p62/SQSTM1 punctae and aggregates (Figure 6D), as have been seen in previous studies (Paine et al., 2005; Schvets et al., 2008; Bjørkøy et al., 2009).
Figure 6. PES Induces p62/SQSTM1 Oligomerization.

(A and B) Western blot (WB) analysis showing increased appearance of processed LC3-II and p62/SQSTM1 oligomerization in the indicated cells following 20μM PES treatment for the indicated time.
(C) MCF7 cells were treated with indicated amount of PES for 24 h, and examined for the indicated proteins.
(D) Immunostaining for p62/SQSTM1 in BX-U2OS cells, either before or following 20μM PES treatment for 24 h. Note the appearance of p62/SQSTM1 punctae and inclusion bodies.
(E) IP-WB analyses of WCE from vehicle or PES-treated MiaPaCa pancreatic cells (top panel) and A875 melanoma cells (lower panel). Note that LC3 binds to both the monomeric and oligomeric forms of p62/SQSTM1 following PES-exposure.
(F) WI38 or H1299 cells were transfected with a negative shRNA or with HSP70 shRNAs, and examined for the indicated proteins.
(G) WI38 cells were transfected with a negative shRNA or with HSP70 shRNAs. After 72 h, the cells were either untreated or treated with 10μM PES for 24 h, and examined for the indicated proteins.
(H) H1299 cells were transfected with a negative shRNA or with HSP70 shRNAs. After 48h, the cells were either treated with DMSO or 20μM PES for 24 h. Each graphical representation indicates the mean ± SD of at least three independent cultures relative to control cells transfected with a negative shRNA and treated with DMSO.
(I) MCF7 cells were treated either with 20μM PES or 100 nM Velcade for 24 h before harvesting cells in NP40-containing lysis buffer. Lysates were centrifuged to separate the clarified lysate (detergent-soluble) and NP40-insoluble (detergent-insoluble) fractions and assayed by western blot for the proteins indicated.
(J) BX-U2OS cells were treated with 20μM PES, 50μM chloroquine (CQ), or 15 nM 17-AAG for 24 h. Cells were harvested in 1% NP40-containing lysis buffer, fractionated into detergent-soluble and detergent-insoluble preparations, and assayed by western blot for the proteins indicated.
(K) A875 cells were either pre-treated with DMSO or 20μM PES for 1 h prior to the addition 50μg/ml of cycloheximide (CHX) for 5 h. Note the marked reduction in PES-induced p62/SQSTM1 oligomerization in the insoluble fraction, the significant inhibition of LC3-II processing following PES and CHX co-treatment, and the obvious loss of cathepsin L expression in the presence of CHX.
An interaction between LC3 and p62/SQSTM1 proteins, which targets p62/SQSTM1 to auotophagosomes and lysosomes for degradation, is important for autophagic degradation of p62/SQSTM1 (Bjørkøy et al., 2005, 2009; Komatsu et al., 2007; Pankiv et al., 2007; Ichimura et al., 2008; Shvets et al., 2008). Therefore, we were interested in determining if the interaction between these two proteins would be modified by PES. Immunoprecipitation-western blot analysis revealed that PES enhanced the interaction between LC3 and both monomeric and oligmeric forms of p62/SQSTM1 (Figure 6E). This observation is consistent with previous results suggesting that p62/SQSTM1 oligomerization is important for its interactions with LC3, and that p62/SQSTM1 oligomerization contributes to the formation of inclusion bodies that reside in the cytosol or within detergent-insoluble structures (Bjørkøy et al., 2005, 2009; Komatsu et al., 2007; Pankiv et al., 2007; Ichimura et al., 2008; Schvets et al., 2008). In contrast, PES attenuated the interaction between the monomeric form of p62/SQSTM1 and HSP70 as well as between the monomeric form of p62/SQSTM1 and TRAF6 (Figure S6), which are known to interact (Pridgeon et al., 2003; Chen et al., 2006; Duran et al, 2008).
To determine whether the cytotoxicity of PES involved its association with HSP70, we next chose to reduce HSP70 expression using shRNA and to measure the effects on cell viability and autophagy. This analysis revealed that silencing of HSP70, like PES, resulted in increased levels of p62/SQSTM1 and LC3-II (Figure 6F and 6G). Notably, silencing of HSP70 rescued PES-induced cell death (Figure 6H). These data support the premise that HSP70 is a critical target of PES-mediated cytotoxicity.
Accumulation of PES-Induced p62/SQSTM1 Oligomers in Soluble and Detergent-Insoluble Fractions
Endogenous p62/SQSTM1 has been found in detergent-insoluble preparations following inhibition of autophagic degradation (Bjørkøy et al., 2005; Shvets et al., 2008). Thus, we asked whether these oligomers are present in a detergent-insoluble fraction, indicating a change to a more aggregated conformation. For these analyses, cells were treated either with PES, the proteasome inhibitor Velcade (bortezomib), or the lysomotrophic drug chloroquine (CQ). CQ has been found to inhibit a late stage in autophagy and inhibit tumorigenesis by raising lysosomal pH, thereby impairing lysosomal protein degradation and autophagic vesicle clearance (Amaravadi et al., 2007; Maclean et al., 2008). PES-treated cells contain higher molecular weight forms of p62/SQSTM1 in both the soluble and detergent-insoluble fractions (Figures 6I and 6J). In contrast, CQ and Velcade promoted p62/SQSTM1 oligomerization primarily in the detergent-insoluble fraction (Figures 6I and 6J). Also, neither p62/SQSTM1 accumulation nor oligomerization was noted following treatment of cells with the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) (Figure 6J and data not shown), suggesting that the actions of PES are distinct from these agents, at least in some respects. The molecular chaperone HSP70 has been implicated in promoting proper folding of nascent polypeptides. To analyze nascent polypeptides, we co-treated cells with PES and the general protein translational inhibitor cycloheximide (CHX) for 6 h. This reduced the accumulation and oligomerization of the autophagy marker p62/SQSTM1 in the detergent-resistant fraction with a concomitant reduction of LC3-II levels (Figure 6K), and retarded the appearance of PES-induced cytoplasmic vacuolization (Figure S7A). These data are consistent with a PES-mediated inhibition of HSP70 function in protein quality control pathways.
PES Inhibits Myc-induced Lymphoma Development
Because the inducible HSP70 protein participates in multiple essential survival signaling pathways in tumor cells, we asked if PES could modify tumor development in vivo. For these studies we utilized a transgenic Eμ-Myc mouse model of lymphomagenesis. Beginning at 8 weeks of age, Eμ-Myc mice were treated intra-peritoneally (i.p.) once every five days with PES (40 mg/kg for 30 days), and the effects on lymphoma development and survival were determined. As shown in Figure 7, PES-treatment markedly impaired tumor development in this mouse model of human Burkitt lymphoma, significantly increasing the overall mean survival time in the PES-treated animals (p<0.02, Mantel Cox test). Taken together, these data suggest that PES may be an effective agent in the treatment of some forms of cancer.
Figure 7. PES Prevents Myc-Induced Lymphomagenesis and Prolongs the Survival of Eμ-Myc Transgenic Mice.
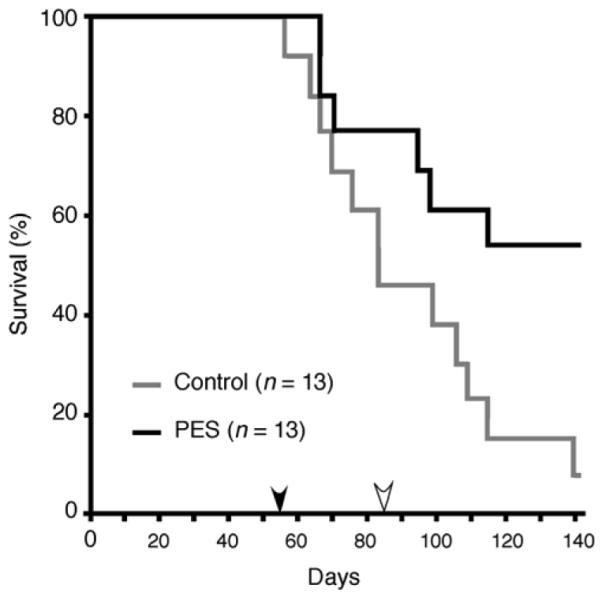
Beginning at 8 weeks of age, Eμ-Myc transgenic mice were treated either with vehicle or 40 mg/kg PES intra-peritoneally every 5 days for a total of 30 days (n = 13 for each group). The black arrow indicates the first day of treatment (day 56), and the open arrow indicates the final day of treatment (day 86). Note that PES-administration increased the overall mean survival time from about 80 days in vehicle treated animals to > 140 days in PES-treated cohorts.
DISCUSSION
The stress-inducible molecular chaperone HSP70 participates in numerous cellular pathways and interacts with a varied group of proteins, including key factors in signal transduction, transcription, cell cycle control and stress response (Mayer and Bukau, 2005; Brodsky and Chiosis, 2006; Garrido et al., 2006; Schmitt et al., 2006; Powers and Workman, 2007). HSP70 activities also have been implicated in the pathogenesis of several human diseases, including cancer. Thus, there is growing interest in the identification of HSP70 modulators to better understand the many cellular activities of this protein. In this study we provide evidence that the small molecule PES interacts with HSP70, alters its functions, and is cytotoxic to tumor cells. PES-induced tumor cell death is not dependent on caspase activation or p53 function, and is not inhibited by overexpression of the anti-apoptotic BCL-xL protein. Rather, loss of cell viability is associated with protein aggregation and an impairment of lysosomal functions resulting in a disruption of autophagic processes.
Autophagy is a catabolic process characterized by the self-digestion of cellular constituents in lysosomal compartments. This degradative process is an important mechanism for the disposal of altered cytoplasmic constituents, including aggregated proteins, and it can be activated in tumor cells by various stressors, including as a response to therapies, nutrient deprivation, or following an inhibition of apoptosis. The process generally serves to promote survival under adverse conditions, in part by helping to prevent the accumulation of damaged proteins and organelles, and by supporting the metabolic needs of the cell (Debnath et al., 2005; Eskelinen, 2005; Levine and Kroemer, 2008; Mizushima et al., 2008). Several lines of investigation point to HSP70 as a regulator of lysosomal activities and, thereby, of autophagy. For example, recent studies demonstrate that the stress-inducible HSP70 protein exhibits tumor-specific localization at membranes of the endosomal/lysosomal compartment, and that it contributes to tumor cell survival by inhibiting lysosomal permeabilization induced by diverse stimuli (Nylandsted et al., 2004; Daugard et al., 2007; Ryhänen et al., 2008). In addition, the interaction of HSP70 and the lysosomal marker LAMP2, which is disrupted by PES, has been implicated in the formation of complexes at lysosomes that are important for lysosomal activities such as the translocation of soluble substrates during chaperone-mediated autophagy. Previous work indicates that inhibiting lysosomal functions following activation of autophagy can result in cancer cell death (Amaravadi et al., 2007; Degtyarev et al., 2008; Maclean et al., 2008). As presented here, PES-treated cells exhibit a significant reduction in the degradation of long-lived proteins and an obvious defect in the processing of the precursor form of cathepsin L to the mature lysosomal form of this cysteine protease. Such observations support the conclusion that PES impairs autophagy in part by its inhibitory effects on lysosomal functions.
HSP70 also is a regulator of apoptosis that has been reported to associate with APAF1 and to either block, or promote, apoptosome formation, depending on experimental conditions (Beere et al., 2000; Saleh et al., 2000; Kim et al., 2008). Recent studies indicate that HSP70 interacts with the tumor suppressor protein PHAPI and the cellular apoptosis susceptibility protein CAS; together these three proteins play an important role in helping with the proper folding of APAF1 to prevent its aggregation and to stimulate apoptosome assembly and caspase activation (Kim et al., 2008). Consistent with such a model, our studies reveal that in the presence of PES, the interaction of HSP70 with its substrate APAF1 is diminished, and this correlates with a significant reduction in caspase activation following cisplatin treatment of tumor cell lines. PES also inhibits the appearance of the HSP70/p53 complex and the stress-induced localization of p53 to mitochondria, thus interfering with p53-mediated apoptosis.
HSP70 regulation and function is mediated by its interactions with co-factors or co-chaperones. PES disrupts several of these interactions, including the association of HSP70 with CHIP, BAG-1M, and HSP40. Both CHIP and BAG-1 help regulate the ATPase activity of HSP70. It has been suggested that, in binding to both HSP70 and substrates, co-factors like CHIP and BAG-1 also may serve as a direct physical link between the chaperone and the proteasome, perhaps aiding in the targeting or selection of substrates for degradation (Mayer and Bukau, 2005; Townsend et al., 2005; Kabbage and Dickman, 2008). The HSP40 co-factor also works with HSP70 to induce conformational changes of certain substrates, in part by promoting ATP hydrolysis and preventing protein aggregation (Fan et al., 2003; Vos et al., 2008). HSP-interacting proteins, including CHIP, HSP40, BAG-1 and the scaffold-adapter protein p62/SQSTM1, also associate with many other cellular proteins to mediate diverse biological processes and signaling pathways. Altering HSP70 function therefore also has consequences for the activities of these other regulatory proteins. For example, recent data show that an inability to eliminate p62 through autophagy can lead to a toxic increase in oxidative stress and DNA damage in some tissues, and that autophagy inhibition also can compromise the ubiquitin-proteasome system, leading to a potentially lethal accumulation of aggregation-prone or misfolded proteins (Korolchuk et al., 2008; Matthew et al., 2009). Considered together, these data are consistent with the idea that PES alters the activities of HSP70 in multiple cellular processes, disabling the normally cytoprotective role of this molecular chaperone. Interestingly, since PES selectively interacts with HSP70, the deleterious consequences of this compound may depend on the presence of this protein. Our investigations on the cell-death inducing properties of PES are supportive of this idea, in that cultured tumor cells which generally have greater levels of HSP70 are much more sensitive to the cell death effects of this small molecule than are non-transformed fibroblasts. Additionally, reducing HSP70 levels in tumor cells reduces the cytotoxic effects of PES exposure. In this regard, PES may cause HSP70 to adopt a potentially lethal “gain-of-function” activity along with a loss of its prosurvival role. Future studies, including structural analysis of the HSP70-PES interaction, should provide needed insight about this issue.
HSP70 has a key cytoprotective role in a broad range of activities that promote protein homeostasis, including the targeting of potentially toxic proteins for proteolysis. Cancer cells experience high levels of protein-modifying- and metabolic-stresses and seem to be particularly dependent on the various actions of HSP70 for survival. This phenomenon, referred to as “non-oncogene addiction”, suggests that it may be possible to target such critical survival proteins for the development of therapies aimed at the selective killing of neoplastic cells (Solomini et al., 2007). In support of this idea, our in vivo analysis indicate that administration of PES inhibits spontaneous tumor development and enhances survival in the Eμ-Myc model of lymphomagenesis. Thus PES represents a valuable new tool to advance basic investigations on the varied activities of the HSP70 protein, and also should have application in the development of effective therapies aimed at simultaneously disabling multiple cancer-critical biological processes.
Supplementary Material
ACKNOWLEDGMENTS
We thank G. Swain and the Penn Morphology Core, Center for Molecular Studies in Digestive and Liver Disease Morphology Core (Center Grant P30 DK50306) for assistance with immunostaining and photography. The authors would also like to thank Raymond Meade for technical assistance with electron microscopy and imaging. This work was supported by NIH grants RO1CA118761 (to D. L. G.), K01DK078025 (to J. I. L.), and RO1CA102184 (to M.M). The authors declare that they have no competing financial interests. We apologize to authors whose work was not cited in this paper because of space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–625. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Chiosis G. Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr Top Med Chem. 2006;6:1215–1225. doi: 10.2174/156802606777811997. [DOI] [PubMed] [Google Scholar]
- Bukau B, Walker GC. ΔdnaK52 Mutants of Escherichia coli have defects in chromosome segregation and plasmid maintenance at normal growth temperatures. J. Bacteriol. 1989;171:6030–6038. doi: 10.1128/jb.171.11.6030-6038.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Lin-Shiau SY, Lin JK. Involvement of heat-shock protein 70 and P53 proteins in attenuation of UVC-induced apoptosis by thermal stress in hepatocellular carcinoma cells. Photochem Photobiol. 1999;70:78–86. [PubMed] [Google Scholar]
- Chen H, Wu Y, Zhang Y, Jin L, Luo L, Xue B, Lu C, Zhang X, Yin Z. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006;580:3145–3152. doi: 10.1016/j.febslet.2006.04.066. [DOI] [PubMed] [Google Scholar]
- Collette J, Bocock JP, Ahn K, Chapman RL, Godbold G, Yeyeodu S, Erickson AH. Biosynthesis and alternate targeting of the lysosomal cysteine protease cathepsin L. Int Rev Cytol. 2004;241:1–51. doi: 10.1016/S0074-7696(04)41001-8. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nature Genet. 2003;33:357–365. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. doi: 10.1379/1466-1268(2003)008<0309:mfrohf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–2601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- Guzhova I, Margulis B. Hsp70 chaperone as a survival factor in cell pathology. Int Rev Cytol. 2006;254:101–149. doi: 10.1016/S0074-7696(06)54003-3. [DOI] [PubMed] [Google Scholar]
- Humbey O, Pimkina J, Zilfou JT, Jarnik M, Dominguez-Brauer C, Burgess DJ, Eischen CM, Murphy ME. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008;68:9608–9613. doi: 10.1158/0008-5472.CAN-08-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62/SQSTM1 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci. 2008;65:1390–1402. doi: 10.1007/s00018-008-7535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HE, Jiang X, Du F, Wang X. PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol Cell. 2008;30:239–247. doi: 10.1016/j.molcel.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes & Dev. 2007;21:3095–3109. doi: 10.1101/gad.1567107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Shimura M, Omatsu T, Okaichi K, Majima H, Ohnishi T. p53 proteins accumulated by heat stress associate with heat shock proteins HSP72/HSC73 in human glioblastoma cell lines. Cancer Lett. 1994;87:39–46. doi: 10.1016/0304-3835(94)90407-3. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JS, Walker GC. DnaK mutants defective in ATPase activity are defective in negative regulation of the heat shock response: expression of mutant DnaK proteins results in filamentation. J Bacteriol. 1994;176:764–780. doi: 10.1128/jb.176.3.764-780.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nature Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62/SQSTM1 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nature Rev. Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Nylandsted J, Brand K, Jäättelä M. Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci. 2000;926:122–125. doi: 10.1111/j.1749-6632.2000.tb05605.x. [DOI] [PubMed] [Google Scholar]
- Nylandsted J, Wick W, Hirt UA, Brand K, Rohde M, Leist M, Weller M, Jäättelä M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002;62:7139–42. [PubMed] [Google Scholar]
- Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen M, Weber E, Multhoff G, Rohde M, Jäättelä M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Paine MG, Babu JR, Seibenhener ML, Wooten MW. Evidence for p62/SQSTM1 aggregate formation: Role in cell survival. FEBS Lett. 2005;579:5029–5034. doi: 10.1016/j.febslet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME. ARF Induces Autophagy by Virtue of Interaction with Bcl-xl. J Biol Chem. 2009;284:2803–2810. doi: 10.1074/jbc.M804705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–3769. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–262. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Geetha T, Wooten MW. A Method to Identify p62’s UBA Domain Interacting Proteins. Biol Proced Online. 2003;5:228–237. doi: 10.1251/bpo66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jäättelä M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005;19:570–582. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryhänen T, Hyttinen JM, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, Viiri J, Holmberg CI, Immonen I, Meri S, et al. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Salminen A, Paimela T, Suuronen T, Kaarniranta K. Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett. 2008;117:9–15. doi: 10.1016/j.imlet.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, Solary E, Kroemer G, Garrido C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–4197. doi: 10.1158/0008-5472.CAN-05-3778. [DOI] [PubMed] [Google Scholar]
- Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1/SQSTM1 into autophagosomes. J Cell Sci. 2008;121:2685–2695. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–988. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2:474–479. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Stephanou A, Packham G, Latchman DS. BAG-1: a multi-functional pro-survival molecule. Int J Biochem Cell Biol. 2005;37:251–259. doi: 10.1016/j.biocel.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Tutar Y, Song Y, Masison DC. Primate chaperones Hsc70 (constitutive) and Hsp70 (induced) differ functionally in supporting growth and prion propagation in Saccharomyces cerevisiae. Genetics. 2006;172:851–861. doi: 10.1534/genetics.105.048926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- Wegele H, Müller L, Buchner J. Hsp70 and Hsp90--a relay team for protein folding. Rev Physiol Biochem Pharmacol. 2004;151:1–44. doi: 10.1007/s10254-003-0021-1. [DOI] [PubMed] [Google Scholar]
- Wisén S, Gestwicki JE. Identification of small molecules that modify the protein folding activity of heat shock protein 70. Anal Biochem. 2008;374:371–377. doi: 10.1016/j.ab.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62/SQSTM1’s role in neurodegenerative disease. J Biomed Biotechnol. 2006;2006:1–12. doi: 10.1155/JBB/2006/62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.