

Abstract
Nitrite is now recognized as a storage pool of bioactive nitric oxide (NO). Hemoglobin (Hb) and myoglobin (Mb) convert, under certain conditions, nitrite to NO. This newly discovered nitrite reductase activity of Hb and Mb provides an attractive alternative to mammalian NO synthesis from the NO synthase pathway that requires dioxygen. We recently reported the X-ray crystal structure of the nitrite adduct of ferric horse heart Mb, and showed that the nitrite ligand binds in an unprecedented O-binding (nitrito) mode to the d5 ferric center in MbIII(ONO) (D. M. Copeland, A. Soares, A. H. West, G. B. Richter-Addo, J. Inorg. Biochem. 100 (2006) 1413-1425). We also showed that the distal pocket in Mb allows for different conformations of the NO ligand (120° and 144°) in MbIINO depending on the mode of preparation of the compound. In this article, we report the crystal structures of the nitrite and NO adducts of manganese-substituted hh Mb (a d4 system) and of the nitrite adduct of cobalt-substituted hh Mb (a d6 system). We show that the distal His64 residue directs the nitrite ligand towards the rare nitrito O-binding mode in MnIIIMb and CoIIIMb. We also report that the distal pocket residues allow a stabilization of an unprecendented bent MnNO moiety in MnIIMbNO. These crystal structural data, when combined with the data for the aquo, methanol, and azide MnMb derivatives, provide information on the role of distal pocket residues in the observed binding modes of nitrite and NO ligands to wild-type and metal-substituted Mb.
Keywords: Nitrite, Nitric oxide, X-ray, Heme, Manganese, Cobalt, Myoglobin
1. Introduction
Nitrite (NO2−) is a simple oxyanion of nitrogen that has long been thought of as being a “dead end” product of nitric oxide (NO) metabolism. Exogenous nitrite can be harmful. A well-known consequence of nitrite poisoning is methemoglobinemia [1]. Nitrite can oxidize ferrous hemoglobin to its ferric form (metHb), and the increased in vivo level of metHb relative to the oxygen-binding ferrous form results in the reduced capacity of the total Hb to efficiently transport dioxygen [1, 2]. Common symptoms of methemoglobinemia are greyish cyanotic skin color (10-20% metHb), lethargy and headaches (20-50% metHb), seizures (50% metHb) and death (>70% metHb), although at least one patient with 68% or higher metHb due to nitrite poisoning survived [2].
The physiological role of nitrite is only now being fully recognized [3, 4]. For example, infusion of nitrite into the forearm brachial artery results in increased blood flow in the forearm before and during exercise, suggesting a unique role of nitrite in vasodilation [5]. Pulmonary vasodilation by nitrite has been reported [6]. It has been known for some time that deoxyHb will convert nitrite to NO and ferric heme under anaerobic conditions [5, 7, 8]. This “enzymatic nitrite reductase” function of Hb, at first glance, is quite unexpected since most of the NO generated is then captured by deoxyHb to form a ferrous HbNO product. Gladwin and coworkers have recently reported a careful study of this nitrite reductase reaction using anaerobic and partially oxygenated solutions to demonstrate that the NO produced can be released as free NO under normal and hypoxic physiological conditions [9]. This nitrite reducase activity of Hb to produce NO under conditions of hypoxia thus essentially bypasses the dioxygen-dependent nitric oxide synthase pathway [3, 10]. Further, the related reaction of deoxyMb with nitrite to produce NO has been shown to regulate mitochondrial respiration [11].
Manganese is an essential trace element that has one less electron than iron. Manganese-substituted derivatives of a number of heme proteins have been prepared and studied. In many cases, these Mn-substituted derivatives were investigated in order to provide insight on the role of heme iron in the reactions of the native proteins [12]. Manganese-substituted derivatives of heme proteins that have been reported to date include those of Hb [13, 14], myoglobin (Mb) [15-17], cytochrome P450 [18], sGC [19], cytochrome c [20], nitric oxide synthase [21], horseradish peroxidase [14, 16, 22], cytochrome b5 [23], CcP [16, 24, 25], and PGHS-1 [26].
It is interesting to note that nearly fifty years ago, Borg and Cotzias discovered that injection of radiolabeled 54MnCl2 into patients in vivo resulted in the incorporation of MnII into red cell fractions, and that the incorporated MnII was non-exchangeable and non-dialyzable [27]. Based on their data, they suggested that the MnII was most likely incorporated into heme, thus providing some circumstantial evidence for the production of a natural manganese heme in humans [28]. Mahoney and Sargent confirmed this finding, and showed that between 0.5-9.0% of administered radiolabeled 54Mn was incorporated into red blood cells, and that 60-70% of the radioactivity was recovered in the resulting crystalline hemin preparation [29]. Hancock and Fritze later showed that, indeed, injection of 54MnCl2 into rats resulted in the formation of a 54Mn-containing species that had identical gel chromatographic elution behavior as hemoglobin, bis-pyridine hemochrome, and hematin [30]; this provided further evidence for the in vivo formation of a manganese porphyrin, confirming the earlier results of Borg and Cotzias. These authors noted, however, that added MnII was not able to displace iron from heme by a simple metal displacement reaction, suggesting the incorporation of MnII at the heme biosynthesis stage. In a separate study, Wibowo et al. determined that increased levels of protoporphyrin in erythrocytes (induced by exposure to Pb) resulted in incorporation of in vivo manganese by the porphyrin [31]. In metal ion accumulation studies using Norway lobsters, it was determined that exposure of the lobsters to MnII resulted in accumulation of the metal ion in nerve tissue, and in the hemolymph where it was bound mainly to the respiratory protein hemocyanin (the authors noted that MnII was not able to displace copper in the protein) [32].
Unlike the native iron analogues, MnII-substituted Hb (MnIIHb) and –myoglobin (MnIIMb) do not bind dioxygen or carbon monoxide [16], although the MnIII-derivatives bind azide [16, 33-35]. Mitra and coworkers have shown that MnIIIMb binds cyanide and thiocyanate at a location >6Å from the metal center [36].
Interestingly, Mn-substituted Hb (MnHb) exhibits allosteric effects in its binding of NO [34, 37-39]. The resulting adduct, written simply as MnIIHbNO, has been prepared and characterized by spectroscopy [34, 40, 41]. The NO adduct of Mn-substituted Mb is also known [34, 40], as are the NO adducts of Mn-substituted sGC [19], cytochrome c [20], CcP [40], cytochrome P450 [18], and that of a monomeric MnHb from the insect Chironomus thummi thummi [42]. Lan and coworkers have examined the ability of sol-gel encapsulated MnIIMb to act as an NO sensor under physiological conditions (since it binds NO but not O2) [43]. We, and others, have prepared and characterized several synthetic manganese porphyrins ((por)Mn) that bind NO [44-49]. In some cases, synthetic (por)Mn compounds have been shown to electrocatalyze the reduction of NO to hydroxylamine and ammonia [50].
Clearly, Mn porphyrins are becoming increasingly recognized for their potential applications. Despite numerous reports on the spectroscopy of Mn-substituted heme proteins, there are only a few reports of crystal structures of these complexes. Moffat and coworkers reported the structure of MnHb using X-ray difference Fourier techniques [51, 52], and demonstrated the similarity of the structure of this complex to that of the native iron compound. Arnone later reported the 3.0 Å resolution structure of the heterometallic complex Hb(α-FeIICO)(β-MnII) [53]. Very recently, Loll and coworkers reported the 2.0 Å resolution crystal structure of Mn-PGHS [26]. To the best of our knowledge, despite the increased attention given to the potential biological activities of Mn-hemes, these are the only reported crystal structures of Mn-substituted heme proteins.
We are interested in the structures of the heme pockets in Mb derivatives of nitrogen oxides (NOx; x = 1, 2) and how the distal amino acid residues might affect the binding preferences of these NOx ligands. In this manuscript, we describe, for the first time, the crystal structures of Mn-substituted Mb and its NOx-liganded derivatives, and the crystal structure of the analogous cobalt-substituted Mb-nitrite compound. Cobalt-substituted heme proteins have also provided valuable information about the native systems. For example, Makino and coworkers showed that cobalt-substituted sGC is a functional enzyme that is responsive to NO [54]. The crystal structures reported here help explain some of the contradictory results on the spectroscopy previously reported in the literature.
2. Experimental
Manganese(III) protoporphyrin IX chloride ((PPIX)MnIIICl), sodium dithionite (85%), and 2-butanone were purchased from Aldrich Chemical Company. Tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl, molecular biology grade), and ammonium sulfate (99.5%) were obtained from Fluka. Sodium nitrite (98.0%) was obtained from Baker and Adamson. Cobalt(III) protoporphyrin IX chloride ((PPIX)CoIIICl) was purchased from Frontier Scientific Company. Nitric oxide (98%, Matheson Gas) was passed through KOH pellets and two cold traps (dry ice/acetone, −78 °C) to remove higher nitrogen oxides.
Horse heart met-myoglobin (hh metMb) was purchased from Sigma. The heme group was removed from metMb (30 mg/ml) using the method of Yonetani [24] and Teale [55]. (PPIX)MnIII was reconstituted into apoMb following the method of Yonetani [16], and the product characterized by UV-visible (UV-vis) spectroscopy (λmax 378 and 471 nm) [16, 17]. The CoIIIMb complex was similarly prepared and characterized (λmax 425 nm) [56]. The purity of the metal-reconstituted myoglobins were established by SDS PAGE.
2.1 Crystallization and complex formation
All eight Mn- and Co-substituted myoglobin crystalline complexes were prepared and structurally characterized in (at least) duplicate.
2.1.1. MnIIIMb and CoIIIMb
Crystals of MnIIIMb and CoIIIMb were grown using identical procedures. The crystals were grown at room temperature (∼23 °C) using the hanging-drop vapor diffusion method. A hanging drop (10 μL) containing 0.45 mM protein (8 mg/mL) and 0.8 M (NH4)2SO4 in 10 mM Tris-HCl buffer at pH 7.4, was suspended over a well containing 500 μL of 3.4 M (NH4)2SO4 in 10 mM Tris-HCl buffer at pH 7.4. The crystals grew in clusters of plates and reached a suitable size in 3-5 days. A suitable crystal was harvested with a cryoloop, transferred to 10 μL of artificial mother liquor containing 10% glycerol as a cryoprotectant, and flash frozen in liquid nitrogen.
2.1.2. MnIIIMb(ONO−)
The nitrito-liganded complex, MnIIIMb(ONO−), was obtained by soaking crystals of MnIIIMb in a 10 μL droplet of the buffer (10 mM Tris-HCl, pH 7.4) under anaerobic conditions by submerging the droplet in mineral oil. The droplet contained (NH4)2SO4 (3.4 M), 7.5% glycerol as cryoprotectant, sodium dithionite (187 mM). Sodium nitrite (to give 100 mM) was then added to the droplet, and the crystal soaked for 2 min in this mixture. The crystals were then harvested with cryoloops and flash frozen in liquid nitrogen. We note that in our hands, the MnIIIMb(ONO−) complex could not be obtained either by soaking the MnIIIMb crystals in a droplet free of sodium dithionite (but containing the other components), or from co-crystallization of MnIIIMb in presence of excess NaNO2.
2.1.3. CoIIIMb(ONO−)
The CoIIIMb(ONO−) complex was prepared by mixing a solution of CoIIIMb (10 mg/mL) with a solution of NaNO2 (0.5 M) in a 1:1 v/v ratio and keeping the mixture at 4 °C for 30 min. The product was then crystallized using the vapor diffusion method using the same conditions as used for CoIIIMb(H2O), and suitable size crystals grew in 3-5 days. Suitable crystals were harvested with cryoloops and flash frozen in liquid nitrogen. The complex was also obtained by soaking CoIIIMb crystals in a droplet of Tris-HCl buffer (10 mM, pH 7.4) containing (NH4)2SO4 (3.4 M), 10% glycerol as cryoprotectant, and NaNO2 (500 mM) for 30 min. However, this latter method resulted in a product containing nitrite at only ∼60 % occupancy as determined by crystallography.
2.1.4. MnIIMb
The reduced MnIIMb crystal was obtained by soaking a crystal of MnIIIMb anaerobically in a 20 μL droplet of the buffer (10 mM Tris-HCl, pH 7.4) containing (NH4)2SO4 (3.4 M), 10% glycerol as cryoprotectant, and sodium dithionite (114 mM) for at least 20 min. The crystal and the droplet were submerged in mineral oil under an atmosphere of nitrogen. During the soaking of the crystal, it cracked into several pieces and its color changed from purple-red to orange-red indicative of the formation of the reduced MnIIMb [16, 57]. A suitable crystal was harvested with a cryoloop and flash frozen in liquid nitrogen.
2.1.5. MnIIMbNO
This complex was obtained by transferring a crystal of MnIIMb into a 10 μL droplet of the buffer (10 mM Tris-HCl, pH 7.4) under anaerobic conditions and submerged in mineral oil. The droplet contained (NH4)2SO4 (3.4 M), 10% glycerol as cryoprotectant, and NO (∼2 mM). The crystal was soaked in this solution for ∼2 min, and the color changed from orange-red to pink suggesting formation of the nitrosyl adduct MnIIMbNO. The crystal was harvested with a cryoloop and flash frozen in liquid nitrogen.
2.1.6. MnIIIMb(MeOH)
The MnIIIMb(MeOH) complex was prepared by adding 20 μL MeOH to a MnIIIMb solution (180 μL, 0.9 mM) in 10 mM Tris-HCl buffer at pH 7.4. The mixture was kept at 4°C for 10 min before setting up for crystallization using the hanging drop vapor diffusion method employing the same conditions as described above for MnIIIMb. A suitable crystal was harvested and soaked for 10 min in a droplet of 10 mM Tris-HCl buffer (pH 7.4) containing (NH4)2SO4 (3.4 M), 10% glycerol as cryoprotectant, and MeOH (2% v/v). The crystal was then flash frozen in liquid nitrogen.
2.1.7. MbIIIMb(N3−)
The azide complex was prepared as reported previously [35, 58] by adding 20 μL of a sodium azide solution (200 mM) directly into a solution of MnIIIMb (180 μL, 0.9 mM) in 10 mM Tris-HCl buffer at pH 7.4. The mixture was left at 4 °C for 10 min, and the resulting azide complex crystallized using the hanging-drop vapor diffusion method employing the same conditions as described above for MnIIIMb. Crystals grew in 3-5 days. A suitable crystal was harvested and soaked in a 10 μL droplet of 10 mM Tris-HCl buffer (pH 7.4) containing (NH4)2SO4 (3.4 M), 10% glycerol as cryoprotectant, and NaN3 (200 mM) for 10 min, and flash frozen in liquid nitrogen. The excess NaN3 added to the cryoprotectant was to maximize the occupancy of the azide ligand in the complex.
2.2. X-ray diffraction data collection and processing
Diffraction data sets were collected at 100 K by using CuKα radiation (λ = 1.5418 Å) produced from a RigakuMSC RU-H3R X-ray generator operated at 50 kV/100 mA. Diffracted X-rays were detected using an R-AXIS IV++ dual image plate detector system. The crystal-to-detector distance was set at 100 mm, and 220 frames of data were collected for each crystal, with 1° oscillations and a 5 min exposure time per frame. X-ray intensity data were indexed and processed with the stand-alone d*TREK program (Macintosh v.2D) [59] available from Molecular Structure Corporation.
2.3. Structure Solution and Refinement
The CCP4 Suite of programs [60] was used for structure solution and the subsequent refinement of all structures reported here. Phase information was obtained using molecular replacement as implemented in CCP4 (MOLREP) [61]. The search model was the 1.3 Å resolution structure of MbIINO (PDB access code 2FRJ) [62] with all the solvent molecules, sulfate anions, and the NO ligand removed from the structure. After molecular replacement, the iron atom was replaced by manganese or cobalt, and restrained refinement was performed for all atoms. However, there were no restraints placed on the axial metal-ligand or metal-N(His93) bond distances or angles. In all the structures, no electron density was observed for the C-terminal residue Gly153, thus it was not included in the models. In all cases, ARP/wARP was used to add water molecules to the structure during refinement. Lys47 was refined in two positions at 50% occupancy each. Also, two sulfate groups (three in case of reduced MnIIMb) were added to the models based on the initial Fo-Fc map. After completion of the refinement of the individual structures, the interactive macromolecular structure validation tool MolPROBITY (available online from the Richardson Lab at Duke University at http://kinemage.biochem.duke.edu/molprobity/) [63, 64] was utilized to assign the final rotamer orientations of Asn, Gln, and His side chains, and to test for any unusual side chain contacts.
In general, the structure solution and refinement procedures were similar for all eight structures reported here. The statistics of the data collection and refinement are summarized in Table 1. Selected geometrical data, calculated using SHELXTL v5.1, are presented in Table 2. The Fo-Fc difference electron density maps shown in Figs. 1 and 2 were generated using CNS [65], and Fcs were calculated using the final model but by omitting the ligand from the structure. All figures were drawn using PyMOL (Delano Scientific, 2002; http//www.pymol.org) and labels added using Adobe® Photoshop.
Table 1.
Statistics of X-Ray data collection and refinement
| MnIIIMb(H2O) | MnIIIMb(ONO) | CoIIIMb(H2O) | CoIIIMb(ONO) | MnIIMb | MnIIMb(NO) | MnIIIMb(MeOH) | MnIIIMb(N3) | |
|---|---|---|---|---|---|---|---|---|
| PDB code | 2O58 | 2O5O | 2O5T | 2O5S | 2O5B | 2O5Q | 2O5L | 2O5M |
| Data collection a | ||||||||
| Space group | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
| Cell Dimen. | ||||||||
| a (Å) | 35.41 | 35.43 | 35.31 | 35.17 | 35.21 | 35.47 | 35.41 | 35.48 |
| b (Å) | 28.65 | 28.67 | 28.75 | 28.68 | 28.74 | 28.64 | 28.59 | 28.75 |
| c (Å) | 62.94 | 62.99 | 63.03 | 63.36 | 64.20 | 63.19 | 63.20 | 62.91 |
| β (°) | 106.0 | 105.5 | 106.5 | 106.1 | 105.45 | 105.7 | 105.74 | 105.9 |
| Resolution (Å) | 1.65 | 1.60 | 1.60 | 1.60 | 2.00 | 1.90 | 1.70 | 1.65 |
| Mean I/σ(I) | 15.2 (4.4) | 14.5 (4.0) | 10.1 (2.9) | 20.4 (4.7) | 9 (2.7) | 9.0 (2.5) | 12.5 (3.9) | 17.0 (4.6) |
| No. Reflections | ||||||||
| Observed | 63842 | 68845 | 64903 | 65732 | 25281 | 35301 | 58562 | 64306 |
| Unique | 14874 | 16177 | 15721 | 15758 | 8510 | 9669 | 13275 | 14301 |
| Comp. (%) | 99.6 (98.2) | 98.5 (95.9) | 96.2 (87.2) | 96.5 (86.6) | 98.7 (96.0) | 97.8 (99.9) | 96.8 (93.7) | 95.3 (91.0) |
| Rmerge (%)b | 5.6 (26.4) | 5.3 (31.6) | 7.7 (34.8) | 4.4 (16.8) | 7.6 (31.8) | 9.5 (23.6) | 6.8 (36.4) | 5.1 (27.2) |
| Refinement | ||||||||
| Res. Range (Å) | 19.47-1.65 | 25.92-1.60 | 26.51-1.60 | 26.58-1.60 | 20.63-2.00 | 26.57-1.90 | 21.91-1.70 | 26.75-1.65 |
| R-factor (%)c | 17.9 | 18.6 | 19.3 | 17.4 | 19.4 | 17.7 | 17.4 | 18.4 |
| Rfree (%)d | 22.3 | 22.4 | 24.8 | 21.5 | 23.9 | 21.8 | 23.3 | 22.1 |
| rmsd bond dist. | 0.015 | 0.013 | 0.017 | 0.012 | 0.023 | 0.021 | 0.014 | 0.015 |
| rmsd angles (°) | 1.57 | 2.26 | 2.47 | 2.28 | 2.05 | 2.49 | 1.53 | 2.29 |
| B factor (Å2) | ||||||||
| Mean | 17.6 | 16.88 | 21.59 | 15.36 | 32.12 | 22.02 | 18.17 | 16.2 |
| rmsd main | 0.82 | 0.81 | 0.85 | 0.77 | 1.15 | 1.05 | 0.86 | 0.81 |
| rmsd side | 2.4 | 2.27 | 2.49 | 2.37 | 3.06 | 2.80 | 2.48 | 2.34 |
Values in parentheses correspond to the highest resolution shells for MnIIIMb(H2O) (1.71–1.65 Å), MnIIIMb(ONO) (1.66–1.60 Å), CoIIIMb(H2O) (1.66–1.60 Å), CoIIIMb(ONO) (1.66–1.60 Å), MnIIMb (2.07-2.00), MnIIMb(NO) (1.97–1.90 Å), MnIIIMb(MeOH) (1.76–1.70 Å), and MnIIIMb(N3-) (1.71–1.65 Å).
Rmerge =Σ|I - <I>|/Σ(I) where I is the individual intensity observation and <I> is the mean of all measurements of I.
R-factor = Σ‖Fo| - |Fc‖/Σ|Fo| where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree is calculated using randomly selected reflections comprising 5% of the data not used throughout refinement.
Table 2.
Selected geometrical data for the Mn- and Co-substituted myoglobin complexes.
| Compound | M–N(por) (Å) | M–X(axial)a (Å) | M-X-Y (°) | α (°)b | M–N(His93) | (M)X⋯N(His64)c (Å) | ΔMd (Å) |
|---|---|---|---|---|---|---|---|
| MnIIIMb(H2O) | 2.04-2.08 | 2.52e | – | 15.3e | 2.22 | 2.62e | −0.18 |
| MnIIIMb(ONO−) | 2.02-2.10 | 2.33 | 112 | 9.7 | 2.34 | 2.57 | −0.07 |
| CoIIIMb(H2O) | 2.02-2.09 | 2.46 | – | 11.7 | 2.15 | 2.54 | −0.04 |
| CoIIIMb(ONO−) | 2.06-2.07 | 2.14 | 105 | 8.8 | 2.02 | 2.71 | −0.04 |
| MnIIMb | 2.03-2.10 | – | – | – | 2.27 | – | −0.34 |
| MnIIMb(NO)f | 2.06-2.11 | 2.53 f | 130 | 12.5 | 2.20 f | 2.60 | −0.20f |
| MnIIIMb(MeOH) | 2.02-2.07 | 2.48 | 119 | 8.2 | 2.29 | 2.56 | −0.07 |
| MnIIMb(N3−) | 2.04-2.06 | 2.37 | 121 | 8.9 | 2.50 | 2.64 | −0.08 |
Distance between the metal and the coordinating atom of the ligand in the distal pocket.
Tilt of the coordinating atom X (of the distal ligand) from the normal to the heme 4N plane.
Distance between the coordinating atom X (of the distal ligand) and the nearest N-atom of the distal His64 residue.
Apical displacement of the central metal atom from the heme 4N plane towards the distal ligand.
These data are for the component with coordinated H2O (70% occupancy).
The NO ligand refined to 70% occupancy. Hence, these data likely represent an average of the MnIIMb (30%) and MnIIMb(NO) (70%) components (see text). The hydrogen bonding distance to the distal His64 residue is for the major His64 component.
Fig. 1.
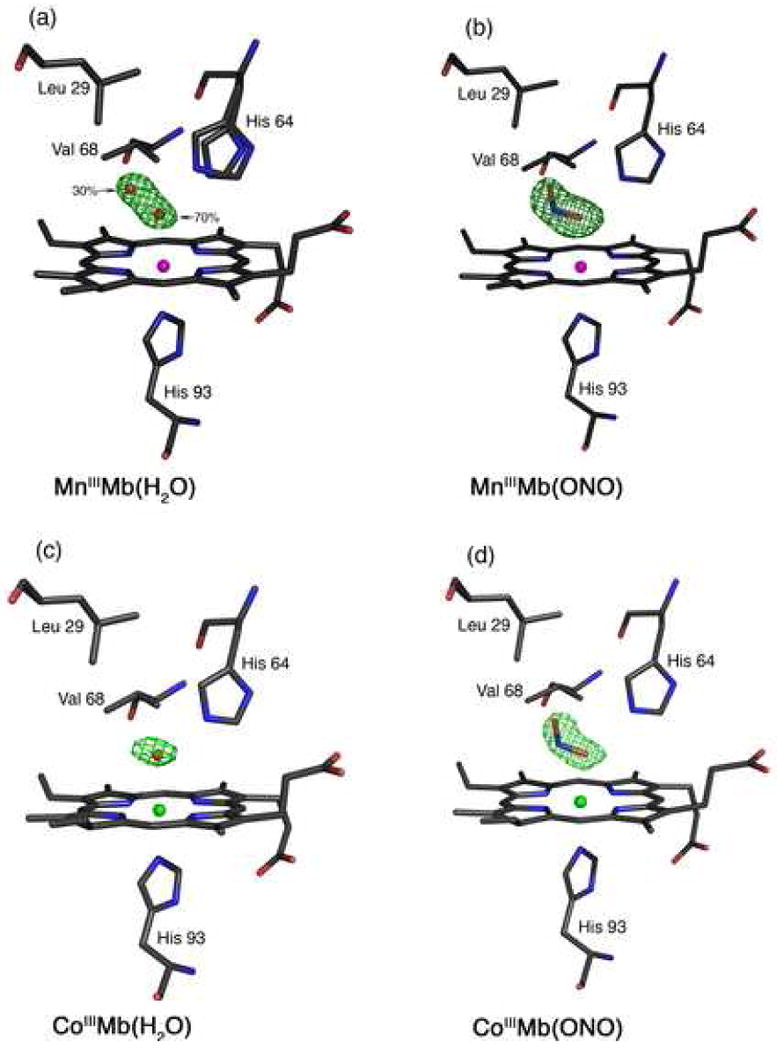
Final models and final Fo-Fc difference electron density maps showing side views of hh MnMb and CoMb derivatives. Carbon, oxygen, nitrogen, Mn and Co atoms are colored grey, red, blue, magenta and green, respectively. The bonds to Mn and Co are not shown for clarity. (a) MnIIIMb(H2O): The difference electron density map contoured at 3σ shows a single water molecule in two different positions. (b) MnIIIMb(ONO): The difference electron density map contoured at 2.5σ. (c) CoIIMb(H2O): The difference electron density map contoured at 2.5σ. (d) CoIIIMb(ONO): The difference electron density map contoured at 2.5σ.
Fig. 2.
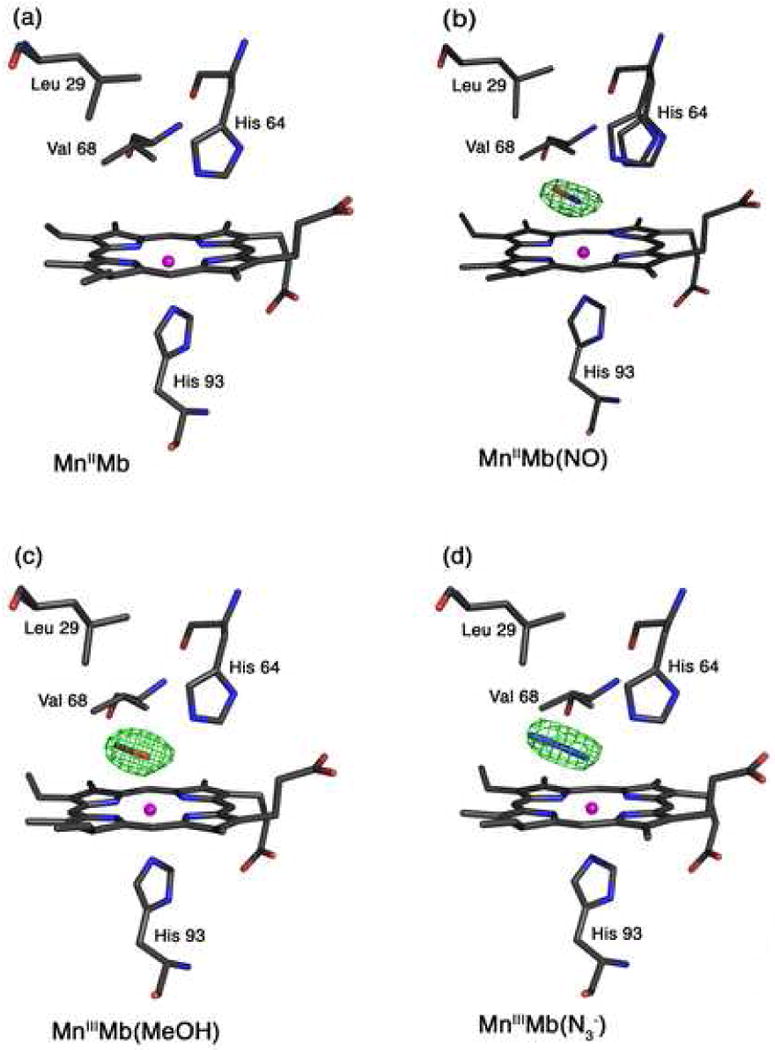
Final models and final Fo-Fc difference electron density maps showing side views of hh MnMb and CoMb derivatives. Carbon, oxygen, nitrogen, Mn, and Co atoms are colored grey, red, blue, magenta, and green, respectively. The bonds to Mn and Co are not shown for clarity. (a) The reduced MnIIMb showing the absence of electron density for a coordinated water molecule in the active site. (b) MnIIMb(NO): The difference electron density map contoured at 3σ shows an NO molecule that refined to 70% occupancy. (c) MnIIIMb(ONO): The difference electron density map is contoured at 3σ. (d) MnIIIMb(N3−): The difference electron density map is contoured at 3σ.
2.3.1. MnIIIMb(H2O)
After molecular replacement, the R-factor was 30.98%. After 10 cycles of restrained refinement, the R-factor was 24.86% and initial electron density maps were generated. At this stage, the initial Fo-Fc difference electron density map revealed the presence of cone-shaped electron density in the distal Mn-heme pocket. We initially tried to model this as a methanol molecule (see Results Section). This cone-shaped electron density was finally refined as a single water molecule in two disordered positions with 70% and 30% occupancies. Two conformations of the distal His64 residue were modeled into the electron density, and the occupancies refined to 70% and 30%. Solvent water molecules were added, followed by an additional 15 cycles of refinement with B-factors refined anisotropically, and the R-factor dropped to 17.9%.
2.3.2. MnIIIMb(ONO−)
At the start of refinement, the R-factor was 32.11%. After 15 cycles of restrained refinement, the R-factor dropped to 24.36%. The Fo-Fc difference electron density map showed clear density for the nitrite ligand in the Mn-heme distal pocket, and nitrite was modeled into the density. After the addition of solvent molecules and further cycles of refinement, the R-factor dropped to 18.6%.
2.3.3. CoIIIMb(H2O)
After molecular replacement, the R-factor was 33.50%. After 10 cycles of restrained refinement, the R-factor dropped to 24.38%. The Fo-Fc difference electron density map showed the presence of electron density consistent with the presence of a bound water molecule (bound to the heme cobalt atom). After the addition of solvent molecules and further cycles of refinement, the R-factor dropped to 19.3%.
2.3.4. CoIIIMb(ONO−)
After molecular replacement, the R-factor was 30.52%. After 10 cycles of restrained refinement, the R-factor dropped to 24.49%. The Fo-Fc difference electron density map showed clear density for the nitrite ligand in the Co-heme distal pocket, and nitrite was modeled into the density. After the addition of solvent molecules and further cycles of refinement, the final R-factor dropped to 17.4%.
2.3.5. MnIIMb
After molecular replacement, the R-factor was 33.09%. After 10 cycles of restrained refinement the R-factor dropped to 22.09% and the initial Fo-Fc difference electron density map showed the absence of electron density for any ligand in the Mn-heme distal pocket. After the addition of the water molecules and further cycles of refinement, the R-factor dropped to 19.4%.
2.3.6. MnIIMb(NO)
At the start of refinement, the R-factor was 30.77%. After 10 cycles of restrained refinement, the R-factor dropped to 22.23%. The Fo-Fc difference electron density map showed clear density for the nitrosyl ligand in the Mn-heme distal pocket, and NO was modeled into the density. After the addition of solvent molecules and further cycles of refinement, the final R-factor dropped to 17.7%. The NO ligand refined to 70% occupancy. Two conformations of the distal His64 were modeled into the electron density associated with this residue, and were refined with 70% and 30% occupancies.
2.3.7. MnIIIMb(MeOH)
At the start of refinement, the R-factor was 28.79%. After 10 cycles of restrained refinement, the R-factor dropped to 22.74% and initial electron density maps were generated. The Fo-Fc difference electron density map showed clear electron density for the methanol ligand in the Mn-heme distal pocket, and methanol was modeled into this density. After the addition of solvent molecules and further cycles of refinement, the R-factor dropped to 17.4%. The methanol ligand refined to full occupancy with thermal factors of 14.25 and 11.69 Å2 for its O and C atoms, respectively.
2.3.8. MnIIIMb(N3−)
After molecular replacement, the R-factor was 31.52%. Ten cycles of restrained refinement resulted in the lowering of the R-factor to 24.93%. The Fo-Fc difference electron density map showed clear density for the azide ligand in the Mn-heme distal pocket, and azide was modeled into the density. Solvent molecules were added, and further cycles of refinement dropped the R-factor to 18.4 %. The azide ligand refined to near-full (94%) occupancy, with thermal factors of 17.95, 20.28 and 22.17 Å2 for its N1, N2, and N3 atoms, respectively.
2.4. Protein Data Bank accession numbers
Atomic coordinates and structure factor amplitudes have been deposited with the RCSB Protein Data Bank. The accession codes are listed in Table 1.
3. Results
The crystal structures of the Mn- and Co-substituted myoglobin derivatives reported here display the normal fold expected for the native horse heart myoglobin. The differences lie in the nature of the heme axial ligand and the resulting distal pocket structure. Hence, we focus on the heme environment in these complexes, and selected structural data are shown in Table 2.
The crystal structure of the parent Mn-substituted myoglobin had not been reported prior to this study. Further, the crystal structures of the derivatives reported here were obtained by derivatizing crystals of the parent Mn-substituted myoglobin. Hence, it was important that we determined its crystal structure.
3.1. MnIIIMb(H2O)
The MnIII-reconstituted myoglobin (MnIIIMb) was prepared from the reaction of MnIII-protoporphyrin IX chloride and apo-Mb in solution, and its formation confirmed by UV-vis spectroscopy. The compound in solution displays a characteristic split Soret absorption at 378 and 471 nm as reported by Yonetani et al. [24]. We determined the crystal structure of this complex to 1.65 Å resolution. The MnIII-heme environment is shown in Fig. 1a. We also show the final Fo-Fc difference electron density map, contoured at 3σ, that reveals the presence of the axial water ligand. This axial water is best refined as a disordered molecule in two distinct positions, with 70% (coordinated) and 30% (uncoordinated) occupancies. We located two positions for the distal His64 residue, and the occupancies correlate with the two water molecule positions. The 2Fo-Fc map showing the electron density associated with the two His64 conformations is shown in Fig. S1 in the Supplementary Information. The distance between the Nε atoms of the two His64 conformations is 0.74 Å, with the minor conformation closer to the interior of the protein and closer to the uncoordinated water molecule.
The major disordered position of the water ligand is close to the MnIII center, with a Mn–O distance of 2.52 Å. This MnIII–O(H2O) distance is longer than those determined for the six-coordinate model compound [(TPP)MnIII(H2O)2]ClO4 (2.271(2) Å) [66] and the five-coordinate compounds [(TPP)MnIII(H2O)]SbF6 (2.145(5) Å) [66], [(TPP)MnIII(H2O)]OTf (2.105(4) Å) [67], and [(OEP)MnIII(H2O)]ClO4 (2.149(3) Å) [68]. The O-atom of the water ligand in MnIIIMb(H2O) is 2.62 Å from the Nε atom of the major conformation of the distal His64 residue, indicative of hydrogen-bonding between these moieties. In addition, the O-atom of the coordinated water ligand is tilted 15° from the normal to the heme four-nitrogen (4N) plane towards to the interior of the protein.
The minor (30%) disordered position of the distal water molecule is 3.96 Å from the MnIII center, and is 2.84 Å from the Nε atom of the corresponding minor conformation of the His64 residue; this distance between the water molecule and this distal His64 residue is consistent with hydrogen-bond stabilization of this non-coordinated water in the distal pocket. The Leu29 and Val68 residues are the closest distal pocket moieties to this water molecule; the distance between the water O-atom and the nearest C-atoms of Leu29 and Val68 are 3.14 and 3.32 Å, respectively. The distance between the O-atom positions of the coordinated and uncoordinated water molecules is 1.70 Å.
We attempted to refine the Mn atom at two positions to correlate with the presence of coordinated and uncoordinated water, but we were not successful. Thus, it is likely that the Mn atom position represents an average between two close-by positions. In the final refined structure, the Mn atom is apically displaced by 0.18 Å from the heme 4N plane towards the proximal His93 residue, with a MnIII–N(His93) distance of 2.22 Å. The displacement of the Mn atom towards the proximal His93 residue is common feature for all the structures reported here (see Table 2), although variations in the extent of the apical displacement are observed depending on the compound being examined.
3.2. MnIIIMb(ONO−)
We were not able to crystallize this complex from the addition of nitrite to MnIIIMb(H2O); either by co-crystallization or by soaking of the MnIIIMb(H2O) crystals with nitrite. Further, we did not observe any spectral change in the Soret region of the UV-vis spectrum upon addition of nitrite to a solution of MnIIIMb(H2O), consistent with the results from a previous study by Lan et al. [43]. However, addition of nitrite to reduced MnIIMb results in a spectral shift in the UV-vis spectrum from 442 nm to a new “split” Soret peak at 471 and 378 nm indicative of the formation of a new MnIIIMb species. Indeed, we were successful in obtaining crystals of MnIIIMb(ONO−) by this method, by adding nitrite to crystals of reduced MnIIMb.
As demonstrated in Fig. 1b, the nitrito (i.e., O-bonded) mode of coordination of the nitrite ligand is present in the MnIIIMb(ONO) complex. The conformation of the Mn–O–N–O moiety is trans. The Mn–O(nitrite) bond length is 2.33 Å, and the coordinating O-atom of the nitrite ligand is tilted 10° from the normal to the Mn-heme 4N plane. The Mn–O–N and O–N–O angles are 112° and 119°, respectively. The coordinating atom of the nitrite ligand is 2.57 Å from the Nε atom of the distal His64 residue, indicative of a strong hydrogen-bonding interaction. Further, the nitrite N-atom is 3.3 Å from the His64 Nε atom and the Val68 (nearest C-atom) residues. The terminal O-atom of the nitrite is 3.26 Å from the His64 Nε atom, but is only 3.07 Å from the Leu29 (nearest C-atom) residue. The bond length between the Mn atom and the proximal His93 residue is 2.34 Å, and the Mn atom is displaced by 0.07 Å from the Mn-heme 4N plane towards His93.
3.3. CoIIIMb(H2O)
The Co-heme environment in the crystal structure of CoIIIMb(H2O) is shown in Fig. 1c. Selected structural data are shown in Table 2. We note that the 1.65 Å resolution structure for sperm whale CoIIIMb(H2O), (Co–O = 2.19 Å; Co–N(His93) = 2.06 Å; (Co)O⋯N(His64) = 2.87 Å) has been reported previously [69]. The superposition of the heme environments of horse heart (this work) and sperm whale CoIIIMb(H2O) is shown in Fig. S2 in the Supplementary Information.
3.4. CoIIIMb(ONO−)
Addition of excess nitrite to a solution of CoIIIMb(H2O) results in a 6 nm shift of λmax from 425 nm to 431 nm. This shift is not immediately noticeable (e.g, even after 1 min), but is clearly evident several minutes after mixing of the nitrite and CoIIIMb(H2O) solutions. This 6 nm red-shift is larger than the 3 nm red-shift observed in the reaction of nitrite ion with native aquometMb (λmax 409 nm) to form Mb(ONO) (λmax 412 nm) [62, 70].
Co-crystallization of CoIIIMb(H2O) in the presence of excess sodium nitrite gives crystals of the desired complex with full occupancy of the nitrite ion ligand as determined by crystallography. Soaking crystals of CoIIIMb(H2O) in a solution containing a high concentration of nitrite for at least 30 min gives the same CoIIIMb(ONO) product, but with only ∼60% occupancy of the nitrite ligand.
The Co-heme environment of the CoIIIMb(ONO) complex obtained from the co-crystallization experiment (with full occupancy of the nitrite ligand) is shown in Fig. 1d. As with the MnIIIMb(ONO) complex described earlier (Fig. 1b), the nitrite ligand displays the nitrito (O-bound) coordination mode with respect to the metal center, and the Co–O–N–O moiety is in a trans conformation. The axial Co–O(nitrite) distance is 2.14 Å, and the Co–O–N(nitrite) and O–N–O angles are 105° and 117°, respectively. The coordinating atom of the nitrite ligand is within hydrogen-bonding distance to the Nε atom of the distal His64 residue (at a distance of 2.71 Å), and is tilted by 9° from the normal to the Co-heme 4N plane; the axial (His93)N–Co–O1 bond angle is 173°. The terminal O-atom of the nitrite ligand is situated 3.32 Å from the Nε atom of the distal His64 residue, and is 3.09 and 3.27 Å from the distal Leu29 and Val68 residues, respectively.
3.5. The reduced MnIIMb
Crystals of the reduced MnIIMb complex were prepared by soaking crystals of MnIIIMb in buffer containing dithionite. The structure of the Mn-heme environment in the resulting MnIIMb is shown in Fig. 2a. The absence of electron density in the Fo-Fc map was consistent with the absence of an axial water ligand in the distal pocket. Notable features of this structure include a 0.34 Å apical displacement of the Mn atom from the heme 4N plane towards the proximal His93 residue in this five-coordinate compound, with a MnII–N(His93) distance of 2.27 Å. This is a larger apical displacement of the Mn atom than that observed for the six-coordinate MnIIIMb(H2O) compound (0.18 Å). The MnII–N(His93) distance is, however, similar to that in the oxidized MnIIIMb(H2O) analogue (i.e., only a 0.05 Å difference).
The structure of the heme environment of MnIIMb reported here has some similarities with the structure of the model five-coordinate compound (TPP)MnII(1-MeIm) [71]. Both MnIIMb and (TPP)MnII(1-MeIm) have a vacant sixth coordination site, with the MnII centers displaced from the porphyrin 4N planes towards the imidazole groups. In the model compound (TPP)MnII(1-MeIm), the axial Mn–N(MeIm) bond length is 2.192(2) Å, with the Mn atom apically displaced by 0.51 Å from the four-nitrogen plane of the porphyrin towards the imidazole ligand [71]. Thus, the Mn-N(imidazole) bond length is ∼0.08 Å longer in MnIIMb compared with the related distance in the model complex, and the apical displacement of the Mn atom is 0.16 Å shorter in MnIIMb.
3.6. MnIIMb(NO)
The interaction of NO with reduced MnIIMb in solution results in a blue shift of the Soret absorption band of MnIIMb at 442 nm to 429 nm (with β and α bands at 544 and 583 nm, respectively) indicative of the formation of MnIIMb(NO) [40]. Related absorption bands at (i) 424, 538 and 580 nm,[43] and (ii) 433, 538 and 580 nm [40, 72] have been reported for MnIIMb(NO). For convenience, we will use this oxidation state formalism to describe the adduct formed when NO binds to MnIIMb (i.e., to distinguish it from the adduct between NO and MnIIIMb). We discuss the oxidation state formalism later in the Discussion.
We were not able to obtain, from solution, crystals of the preformed MnIIMb(NO) complex. Fortunately, soaking crystals of MnIIMb (obtained by reacting crystals of MnIIIMb with dithionite) with NO also produced the desired nitrosyl product. We determined the 1.9 Å resolution structure of this product, with the NO ligand refining to 70% occupancy. We also located two conformations of the distal His64 residue that refined to 70% and 30% occupancies, with the major conformation assigned to MnIIMb(NO), and the minor conformation assigned to MnIIMb. Fig. 2b shows the Mn-heme environment of the crystalline MnIIMb(NO) product.
The Mn-NO moiety is distinctly bent with a Mn–N–O angle of 130°. The nitrosyl N-atom is tilted by 12° from the normal to the Mn-heme 4N plane, and the NO group is oriented away from the distal His64 residue in the direction of Leu29. The nitrosyl N and O atoms are, respectively, 2.60 and 3.28 Å from the Nε atom of the major conformation of the distal His64 residue, consistent with hydrogen-bonding stabilization of the NO group in the distal pocket. The next closest distance between the NO group and the distal pocket residues is with a carbon atom of Val68 (3.37 Å from the nitrosyl O-atom).
In the structure of this MnIIMb(NO) complex, the NO ligand refined to 70% occupancy. The apical displacement of Mn by 0.20 Å from the Mn-heme 4N plane towards the proximal His93 residue suggests that the structure shown in Fig. 2b is likely a mixture of MnIIMb(NO) and MnIIMb (note that ΔMn for MnIIMb is 0.34 Å). We were not able to refine the two separate Mn positions, and the rather long Mn–N(O) distance of 2.53 Å may thus not correctly represent this bond length. For comparison, the Mn–N(O) bond length in the model compound (TPP)Mn(NO)(1-MeIm) is 1.641(1) Å [48].
3.7. MnIIIMb(MeOH)
As part of a control experiment (see Discussion), we prepared the methanol adduct of MnIIIMb to determine the binding geometry of this ligand. The structure of the MnIII-heme environment is shown in Fig. 2c. The Mn–O(MeOH) bond length is 2.48 Å, which is longer than the analogous Mn–O distance in the d4 high-spin model non-protein complexes (TPP)MnIII(N3)(MeOH) (2.329(7) Å) [73], [(TPP)MnIII(MeOH)2]ClO4 (2.252(2), 2.270(2) Å) [74], and for the five-coordinate [(OEP)Mn(EtOH)]ClO4 (2.145(2) Å) [75].
The major differences between the structure of MnIIIMb(MeOH) and MnIIIMb(H2O) are (i) only one position of MeOH was found, at full occupancy, (ii) only one His64 conformation was found in MnIIIMb(MeOH), and (iii) the MnIII atom was displaced only 0.07 Å from the heme 4N plane towards the proximal His93 residue. The methyl group of the MeOH ligand is oriented towards the interior of the protein in the direction of the Leu29 and Ile107 amino acid residues; the distance between the C-atom of the MeOH ligand and the nearest carbon atom of the closest residue Val68 is 3.51 Å. The MnIII–O–C(Me) angle of 119° is slightly more acute than that observed in the model compound [(TPP)MnIII(MeOH)2]ClO4 (124.6(3), 124.0(3)°) [74], perhaps due to the presence of the hydrogen-bonding interaction present in MnIIIMb(MeOH) which might also be responsible for the slightly longer Mn-O bond in the protein.
3.8. MnIIIMb(N3−)
The azide anion is a known inhibitor of cellular respiration, and it also inhibits the catalytic activities of catalase, cytochrome oxidase, and peroxidases [33]. The azide adduct of Mn-substituted Mb has been characterized by optical and resonance Raman spectroscopy [16, 58, 76]. Hoffman and Gibson reported anomalous binding of azide to MnIIIMb, and determined that the reaction of azide with MnIIIMb proceeded in two kinetically separable steps to eventually generate MnIIIMb(N3−) [35]. In contrast, azide binding to iron-containing metMb is consistent with a single binding equilibrium [77]. The 2.8 Å resolution crystal structure of the azide complex of the iron-containing horse metHb is known [78, 79]. The related crystal structure of the azide adduct of metMb was first reported by Stryer et al. in 1964 [80]. A comparison of the crystal structures of hh MbIII(N3−) (at 2.0 Å resolution) and its His64Thr mutant (at 1.8 Å resolution) was reported by Maurus et al. in 1998 [81]. In the wild type adduct, the Fe–N–N(N) bond was 119°, and the Fe–N(azide) bond was 2.11 Å. In the His64Thr mutant structure, two conformations of the azide ligand were present; the major (∼90%) conformation oriented the azide ligand towards the interior of the protein (as seen in the wild type structure), and the minor conformation oriented the azide ligand towards to exterior of the protein towards the solvent. The latter minor conformation is reminiscent of the 1.9 Å resolution crystal structure of the azide adduct of ferric myoglobin isolated from the buccal muscles of the Mediterranean mollusc Aplysia limacine, where the azide ligand is oriented towards the solvent [82]; this protein lacks a distal His64(E11) residue.
Given the interest in the conformational preferences of the azide ligand in myglobin, we determined the 1.65 Å resolution crystal structure of hh MnIIIMb(N3−). The heme environment is shown in Fig. 2d, and is similar to that of wild type MbIII(N3−) [81], in which only one conformation of the azide ligand was observed. In both compounds, the azide ligand is oriented towards the interior of the protein. The MnIII–N(azide) distance of 2.37 Å is longer than those observed in the model complexes (TPP)MnIII(N3−)(MeOH) (2.176(9) Å) [73], (TPP)MnIII(N3−) (2.045 Å) [83], and (Schiff base)MnIII(N3−) (2.221(4) Å) [84]. The coordinating atom of the azide ligand is 2.64 Å away from the Nε atom of the distal His64 residue, indicative of a hydrogen-bonding stabilization of the azide ligand in the distal pocket. The next closest nonbonding distance between the azide ligand and distal pocket residues is that between the terminal N3 atom of the azide with the side-chain of Leu29, with an (azide)N-to-Leu(C-atom) distance of 3.46 Å.
As is the case of the iron-containing MbIII(N3−), the azide ligand is oriented towards the distal Ile107 residue; other distances between the azide terminal N-atom and non-H atoms of distal pocket residues are with Ile107 (3.55 Å) and Val68 (3.97 Å). The Mn atom in MnIIIMb(N3−) is almost in the heme 4N plane, with an apical displacement of only 0.08 Å. The near in-plane position of Mn is consistent with the six-coordination of the metal center and near-complete (94%) occupancy of the azide ligand. The proximal Mn–N(His93) bond length in MnIIIMb(N3−) of 2.50 Å is, however, longer than that observed in the iron-containing MbIII(N3−) (2.06 Å), demonstrating the effect that the MnIIId4 center has on this axial bond length when compared with the ferric d5 center. Ferric MbIII(N3−) has been shown to exhibit a ground-state low-spin electronic configuration and an observable low-spin/high-spin equilibrium at room temperature [77]. In contrast, the d4 complex MnIIIMb(N3−) is known to be high-spin [35], and this likely accounts for its longer proximal metal–N(His93) bond length.
4. Discussion
The preparation and ligand binding properties of Mn-substituted Mb have been studied in detail by spectroscopic methods. Both MnIIIMb and MnIIMb are high-spin species [12, 14, 57, 85]. The overall structure of Mn-substituted horse heart Mb is very similar to that of the iron-containing analogue, and the protein retains the typical Mb fold. Maurus et al. compared the crystal structures of natural and recombinant wild-type horse heart myoglobin, and showed that small differences existed in the structures [86]. When the final MnIIIMb model is superimposed on the 1.7 Å resolution crystal structure of recombinant horse heart Mb reported by Maurus et al. (pdb access code 1WLA) [86], the root-mean square deviations in the backbone and side-chain atomic positions are 0.32 Å and 1.01 Å, respectively. When compared with the structure of natural Mb (pdb access code 1YMB) [87], these values are 0.32 and 1.12 Å, respectively. We find that the structures of the heme environments in our Mn-substituted horse heart myglobins more closely match those of the recombinant iron analogues, particularly with the Leu29 and Val68 side-chain orientations. The superposition of the heme environment structures of MnIIIMb(H2O) and the two aquometMb structures are shown in Fig. 3. It is interesting to note that Atassi reported earlier that MnIIIMb and wild-type Mb exhibit identical immunological properties [15].
Fig. 3.
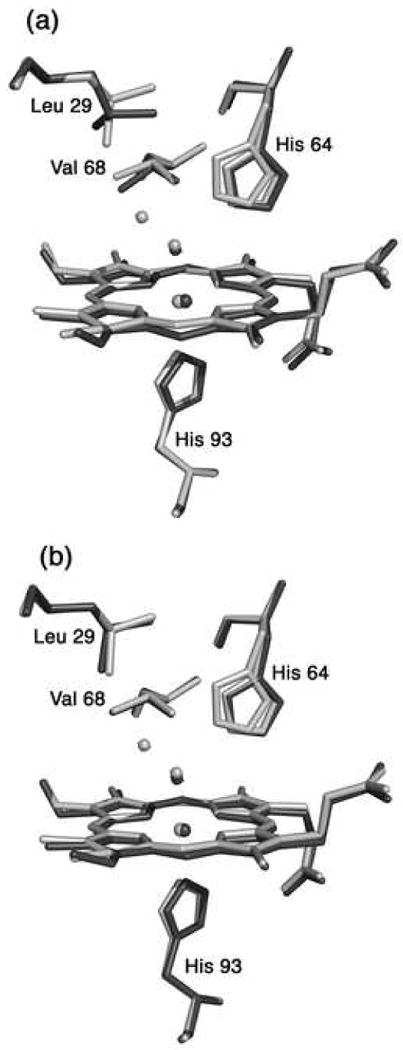
A superposition of the heme environments in the crystal structures of hh MnIIIMb(H2O) (this work, shown in light grey) and (a) natural hh FeIIIMb(H2O) (pdb code 1YMB [80], shown in dark grey), and (b) recombinant hh FeIIIMb(H2O) (pdb code 1WLA [79], shown in dark grey), using a global Cα structural alignment. The major conformation of His64 in hh MnIIIMb(H2O) is similar to the conformation in FeIIIMb(H2O). The Leu29 and Val68 conformations of the Mn- and Fe-proteins overlay better in the structures shown in Fig. 3b.
In this manuscript, we report the crystal structures of the nitrite adducts of Mn- and Co-substituted myoglobin. In addition, we report the structures of the reduced MnIIMb compound, the nitrosyl product MnIIMb(NO), and the methanol and azide derivatives of MnIIIMb. Crystals of the parent MnIIIMb were needed for this study, hence we begin by describing the structure of this compound.
4.1. The nature of the heme site in MnIII-substituted myoglobin
Manganese-substituted myoglobin (MnMb) has been prepared and studied in solution for decades. However, the coordination environment around the metal center in MnMb derivatives has been a subject of considerable debate. MnIIIMb displays a characteristic “split” Soret band pattern at λmax 378 and 471 nm, whereas MnIIMb displays a single Soret peak at λmax 438 nm [12, 24, 57]. Mitra and coworkers used multinuclear NMR and optical spectroscopy to determine that the sixth axial position in MnIIIMb was unlikely to be occupied by a water molecule [36]. In the latter study, they did not observe a change in the optical spectrum of MbIIIMb in the pH 4–11 range, suggesting the absence of an aquo↔hydroxo equilibrium for a hypothetical axial water ligand. This result differed somewhat from that reported by Spiro and coworkers who studied the resonance Raman spectra of MnIIIMb and found evidence for a weakly interacting axial water ligand in the sixth position [88]. As stated in the Introduction, there is a noticeable paucity of crystal structures of Mn-substituted heme proteins in the literature. Using X-ray difference Fourier techniques, Moffat and coworkers determined that for MnIIIHb, the α hemes contained six-coordinate MnIII centers (with axial water) whereas the β hemes were five-coordinate (i.e., no axial water) [51].
Farmer and coworkers investigated the electrochemical properties of MnIIIMb in dimethyldidodecylammonium bromide surfactant films [89]. They discovered a dynamic exchange between two MnIII/MnII redox couples (E1 = −0.25 V vs SCE; E2 = −0.41 V vs SCE), and they attributed this to the presence of two forms of MnIIIMb that probably differed in heme site geometry. Interestingly, Hoffman and Gibson reported that MnIIIMb reacted with azide in two kinetically separable steps, and that this was likely due to the presence of two forms of the Mn-porphyrin that differed in the extent of metal displacement from the porphyrin plane [35].
The crystal structure of MnIIIMb(H2O) reported here sheds new light on this debate regarding the axial coordination geometry of the metal site in this protein. Three independent preparations, crystallizations, and structure solutions at 1.65 Å resolution yielded identical MnIIIMb(H2O) crystal structures. In the structure, there is a mixture of a six-coordinate species (with axial water; 70% occupancy) and a five-coordinate species (without axial water; a non-bonded water refined to 30% in the distal pocket). We also found two conformations for the distal His64 residue, and these conformations refined to the corresponding 70% and 30% occupancies (Figs. 1a and S1).
During the initial stages of refinement of the structure, we were unsure if the electron density (attributed to an axial ligand) observed in the Fo-Fc difference map was due to an adventitious solvent molecule such as methanol. To test this, we performed the following additional experiments. (i) We effected reduction of the purple-red MnIIIMb crystal with dithionite, and obtained the structure of the resulting orange-red five-coordinate reduced compound MnIIMb (see later). We then reoxidized this MnIIMb crystal with ferricyanide and determined its 1.9 Å resolution structure; it was identical to that obtained for MnIIIMb(H2O) as shown in Fig. 1a. This result reaffirmed to us that the electron density in the distal pocket was not due to an adventitious exogenous ligand such as methanol. (ii) In addition, we prepared the previously unknown methanol adduct by cocrystallizing MnIIIMb in the presence of methanol, and determined the structure of the resulting MnIIIMb(MeOH) product (Fig. 2c, and Results section).
The presence of distal-pocket uncoordinated water in the structure of MnIIIMb(H2O) is, while initially unexpected, not without precedent in crystal structures of native and mutant iron myoglobins. For example, a similar distal-pocket (non-coordinated) water has been located in the 1.15 Å resolution crystal structures of wild-type ferrous deoxyMb (horse heart and sperm whale), and this water is stabilized by hydrogen-bonding with the distal His64 residue (HOH⋯N(His) distance of 2.8 Å) [90, 91]. In the case of hh deoxyMb, this distal pocket water was located 3.53(5) Å from the Fe center (c.f. 2.13 Å for liganded water in aquometMb) [90]. Further, distal pocket water has been observed in other reported structures of wild-type [92-94] and mutant (V68A [93], V68F [93], V68N (molecule B) [95]) deoxyMb's.
The crystal structure of the Val68Thr mutant of pig deoxyMb provides an interesting comparison with the structure of hh MnIIIMb(H2O) reported here. In molecule A of the Val68Thr deoxyMb, a single water molecule is distributed between two positions; one position is within direct bonding distance to Fe (Fe–O = 1.9 Å; 30% occupancy of H2O), and the other is positioned 3.5 Å from the Fe (70% occupancy of H2O) yet within hydrogen-bonding distance of the distal His64 residue (HOH⋯ Nε(His) = 2.6 Å) [96]. In molecule B, a single position of the distal water located 3Å from Fe is observed, and this is also within hydrogen-bonding distance to the His64 residue (HOH⋯NεHis) = 2.6 Å) [96].
The distance between the two distal pocket water molecule positions in MnIIIMb(H2O) is 1.70 Å. This distance is inconsistent with the presence of a dioxygen ligand (c.f., the O–O distance of 1.24 Å in oxyMb) [90], as are the refined occupancies of the O atoms. Further, this 1.70 Å distance is too short for a HOH⋯OH2 hydrogen-bonding interaction, and rules out the presence of both coordinated and unligated H2O in the same molecule (such presence of coordinated and uncoordinated H2O has been observed in the 1.9 Å resolution structure of native sw aquometMb) [92].
The location of the distal pocket (non-coordinated) water molecule in MnIIIMb(H2O) is reminiscent of the distal docking site for photodissociated CO from MbCO as determined by X-ray crystallography [97-99]. For example, the crystal structure of photolyzed hh MbCO shows the CO ligand in a position that is 3.6 Å from the Fe atom and atop a carbon pyrrole [97]. This site is in the general area as, but not the same as, the distal pocket Xe binding site (i.e., the Xe4 site) [100].
Importantly, the displacement of the distal pocket water in deoxyMb has been linked to the kinetic barriers for CO binding to ferrous Mb [92, 101]. It is thus likely that the presence of two types of distal H2O in MnIIIMb(H2O) (i.e., coordinated and uncoordinated) helps explain the presence of two MnIII/MnII redox couples for the electrochemical reduction of MnIIIMb [89] and the two kinetically separable steps for the reaction of azide with MnIIIMb [35]. Finally, we note that Moffat and coworkers have utilized X-ray difference Fourier techniques to determine that the Mn center in the α-heme of MnIIIHb binds H2O as a sixth ligand, whereas the β-heme does not and is five-coordinate [51].
4.2. The nitrite complexes of MnIIIMb and CoIIIMb
There is renewed interest in the nature of binding of the nitrite anion to heme proteins [102-106]. It has long been appreciated that the direct heme-nitrite interaction is an essential one insofar as nitrite reduction by the heme-containing nitrite reductases (NiRs) is concerned. Nitrite is reduced to NO by the NiRs [107-109], and this process is believed to involve initial binding of nitrite directly to the iron center in the heme-containing NiRs.
To date, the reported crystal structures of the nitrite adducts of heme-containing NiRs and related compounds all display N-binding of the nitrite ligand. For example, the crystal structure of the nitrite adduct of cytochrome cd1 NiR from P. Pantotrophus reveals the N-binding (i.e., nitro) mode of nitrite to the iron center [110].
This nitrite adduct was obtained by soaking crystals of ferrous NiR with nitrite, it is reasonable to assume that such an N-binding mode would facilitate protonation of a nitrite terminal O-atom to result in eventual release of NO and a water molecule. The crystal structure of the nitrite adduct of ferric cytochrome c NiR from Wolinella succinogenes also shows N-binding of the nitrite anion to the iron center, and density functional calculations on the related model compound (por)FeIII(NH3)(NO2) reveals that the N-binding mode is more stable than the nitrito O-binding mode by >10 kcal/mol [111]. The crystal structure of the nitrite adduct of the sulfite reductase hemoprotein from E. coli similarly reveals an N-binding mode of the nitrite anion to the iron center [112].
Interestingly, DFT calculations on the nitrite adduct of cytochrome cd1 NiR provide intriguing possibilities for nitrite coordination to the heme center in this enzyme [113]. Although the nitrito O-binding mode was determined to be 4.5 kcal/mol higher in energy than the corresponding nitro N-binding mode in the ferric form (6 kcal/mol in the ferrous form), the nitrito form was considered to also be a viable intermediate in NiR catalysis.
The nitro N-binding mode has been determined for all crystallographically characterized synthetic iron porphyrins containing the nitrite ligand, regardless of whether the iron center is formally in the ferric or ferrous state [114]. The only exception to this is that for the nitrite in the anionic complex [(TpivPP)Fe(NO)(NO2)]− which displays both N-binding (nitro) and O-binding (nitrito) of the disordered nitrite group [115]. Results of DFT calculations on a model iron porphyrin with NO and nitrite ligands show that the nitrito linkage isomer (porphine)Fe(NO)(ONO) is only 4.3 kcal/mol higher in energy than the related nitro isomer (porphine)Fe(NO)(NO2) [116].
We recently reported the 1.3 Å resolution X-ray crystal structure of the nitrite adduct of ferric horse heart Mb, and determined that the nitrite anion was bound to the ferric center via the nitrito O-binding mode [62]. Importantly, this structure was the first to demonstrate that a stable O-binding mode of nitrite is possible in heme proteins. Given this rather unexpected nitrito binding mode for the nitrite ligand in the iron-containing d5 compound MbIII(ONO−) and the role of the distal pocket in stabilizing this (to date) unusual binding mode, we sought to investigate the nitrite binding preferences within the distal pockets of the d4 MnIII and d6 CoIII analogues.
To the best of our knowledge, there is only one report of a crystal structure of a synthetic manganese porphyrin containing nitrite, namely that of (TPP)MnIII(ONO−) [117]. This compound clearly reveals a nitrito O-binding mode of the nitrite anion. Interestingly, although continuous photolysis of this compound shows the formation of (TPP)Mn(O) and NO [117, 118], flash photolysis studies on this compound reveal dissociation of NO2 from the metal center [119]. Recombination of NO2 with the (TPP)MnII photoproduct then proceeds via a nitro (TPP)Mn(NO2) intermediate that isomerizes to the more stable (TPP)Mn(ONO) compound [119]. Not surprisingly, therefore, the crystal structure of MnIIIMb(ONO−) also shows that the nitrite anion binds to the manganese center via the nitrito O-binding mode (Fig. 2b).
Our crystallography results for the nitrite adduct of cobalt-substituted myoglobin was unexpected, however. To date, all crystallographically characterized nitrite adducts of synthetic cobalt porphyrins demonstrate that the nitrite anion is bound to the cobalt centers via the nitro N-binding mode [120-123]. To the best of our knowledge, there is no exception to this observation for model cobalt porphyrin compounds. Indeed, it was demonstrated that during the recombination of NO2 with (TPP)CoII (generated from flash photolysis of (TPP)CoIII(NO2)), an intermediate forms which decays to (TPP)CoIII(NO2); the intermediate was assigned as the nitrito linkage isomer (TPP)CoIII(ONO) [124].
To the best of our knowledge, no nitrite adducts of cobalt-substituted heme proteins or related cobalt biomolecules have been characterized. Thus, the structure of CoIIIMb(ONO−) represents the first report of a cobalt nitrite heme protein, and is the first stable nitrito binding to cobalt porphyrins to be established. Clearly, it is evident that although information from crystal structures of cobalt nitrite compounds of model porphyrins have helped in understanding the mechanisms of action of these compounds as catalysts for chemical transformation such as O-atom transfers in oxidation reactions (generating (por)Co(NO) intermediates) [123], the structures may not correlate very well with the geometries present in cobalt-substituted heme proteins. The ability of the distal pocket in CoIIIMb to effect the nitrito O-binding preference is significant, and suggests that the hydrogen-bonding provided by the distal His64 residue plays a primary role in stabilizing this binding mode. Since such a moderate-to-strong hydrogen bonding capability is absent in most synthetic cobalt porphyrins with nitrite ligands, it is thus not surprising that this O-binding mode has yet to be observed in these model compounds. It is somewhat surprising, however, that even in the picket fence porphyrin derivatives such as (TpivPP)Co(NO2)(1-MeIm) which provide electrostatic interactions between “distal” NH groups and bound nitrite, that this stabilization is not sufficient to induce O-binding of the nitrite group [121], perhaps due to the artificial symmetrical nature of the distal pockets.
In summary, the X-ray crystal structures reported to date of synthetic Mn, Fe, and Co porphryins with nitrite ligands reveal a preference for nitrito O-binding for the Mn compound, and nitro N-binding for the Fe and Co compounds. In the Mb derivatives, however, all three metal-nitrite derivatives (MnIII, FeIII, and CoIII) reveal the nitrito O-binding mode, suggesting that the distal pocket of Mb is the major determinant in this structural preference for these Mb nitrite derivatives. This observation provides evidence that the O-binding mode of nitrite is not exempt from involvement in nitrite reduction to NO. This is further demonstrated by the successful reduction of the MnIIIMb(ONO−) (this work) and MbIII(ONO−) [62] by dithionite to their NO-derivatives.
In the next two sections, we discuss the crystal structures of the dithionite-reduced compound MnIIMb and its NO derivative.
4.3. The structure of the reduced MnIIMb complex
The reduction of MnIIIMb to MnIIMb in solution is a well-studied reaction, and the reduction can be effected using dithionite [16, 57, 76], radiolysis [57], or electrochemical methods employing surfactant films [89] or employing the mediator oxazine-170 perchlorate [125]. Hori et al. employed powder and single-crystal multifrequency EPR spectroscopy to determine that the metal center in MnIIMb is high-spin 3d5 (S = 5/2) [85], and this is consistent with a singly-occupied strongly antibonding dx2-y2 orbital [12]. Arnone and coworkers reported the 3.0 Å resolution crystal structure of the metallohybrid hemoglobin Hb(α-FeIICO)(β-MnII) that crystallizes in the deoxyHb T-state [53]. They determined that the β-MnII subunit is structurally isomorphous with the normal deoxy β-Fe subunit.
The crystal structure of MnIIMb reported here reveals that the distal pocket water molecules of MnIIIMb(H2O) are displaced upon dithionite reduction. Thus, unlike the case of MnIIIMb(H2O), no water molecules were located in the vicinity of the MnII center. This contrasts with several reported crystal structures of iron-containing deoxyMb where a non-coordinated water molecule has been located in the distal pocket [90-94, 126].
The apical displacement of the Mn atom in MnIIMb (ΔMn = 0.34 Å) from the heme 4N plane is larger than that observed for MnIIIMb(H2O) (ΔMn = 0.18 Å). This increase in apical displacement upon reduction of the protein is similar to that seen in the high-resolution structures of reduced Mb (ΔFe = 0.363(11) Å) vs. aquometMb (ΔFe = 0.106(7) Å) [90]. The heme environments of MnIIIMb(H2O) and MnIIMb, obtained from the overlay of the protein Cα backbones, are shown in Fig. 4. Differences are evident in the water ligation and distal His64 placement in these proteins. The His64 residue in the reduced MnIIMb more closely overlaps the minor conformation of the His64 residue in MnIIMb(H2O). Fig. S3 shows an overlay of the heme sites in the reduced MnIIMb and deoxyMb (FeIIMb, Fig. S3). In the reduced FeIIMb case, one of the two conformations of the distal His64 residue reveals an inward movement that supposedly helps stabilize the distal pocket water molecule through hydrogen-bonding, whereas in the reduced MnIIMb case such a hydrogen-bond stabilization is not present due to the lack of distal pocket water.
Fig. 4.
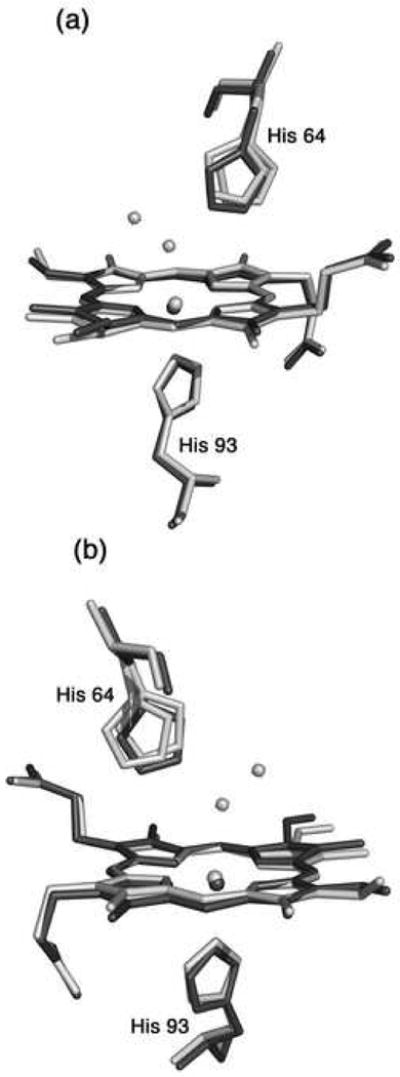
A superposition of the heme environments in the crystal structures of hh MnIIIMb(H2O) (this work, shown in light grey) and hh MnIIMb (this work, shown in dark grey) using a global Cα structural alignment, and shown from two different views. The Nε atoms of the His64 and His93 residues of MnIIMb are shifted from their major/minor positions in MnIIIMb(H2O) by 0.95/0.21 and 0.37 Å, respectively. Also, the MnII center is shifted 0.25 Å from its MnIII position. A meso carbon and its connecting pyrrole ring A are shifted towards the proximal side in MnIIMb relative to those in MnIIIMb(H2O).
4.4. The nitric oxide complex
For convenience, the nitrosyl adduct of MnIIMb is represented as MnIIMb(NO), although it is perhaps better represented as {MnNO}6 (see later). In order to place the NO ligand conformation observed in MnIIMb(NO) in proper context, some discussion of formalism is warranted [127, 128]. For example, the oxidation state formalism has often been used to describe metal nitrosyl linkages. In this formalism, NO binding to a metal center (M) to generate a linear M-NO unit results in a complex formulated as M−(NO+), where a formal electron transfer to the metal has occurred. Conversely, when a bent M-NO moiety is generated, the complex is formulated as M+(NO−). Clearly, this formalism is overly simplistic. Although it does take into account that NO is a redox active ligand, it does not account for cases where the unpaired electron density on NO essentially still resides on the ligand in its metal complex. Enemark and Feltham developed a notation that treats the metal-NO fragment as a single unit, and the use of the resulting formalism avoids the oxidation state simplification of the metal-NO group [129-131]. In this {MNO}n formalism, n denotes the number of assigned electrons to the metal d orbitals (without consideration of the NO ligand(s)) plus an electron for each of the attached NO ligands. Thus, the (por)Mn(NO) complex belongs to the {MnNO}6 class, and the (por)Fe(NO) belongs to the {MNO}7 class. According to this formalism as applied to nitrosyl metallopophyrins, {MNO}6 compounds have preferred linear NO geometries, and {MNO}7 compounds have bent (∼145°) NO geometries. In synthetic six-coordinate iron nitrosyl porphyrins of the {MNO}7 class, the FeNO angles are in the tight 137–140° range (avg. ∼138.5°) [114, 132]. A much wider range of FeNO bond angles (112°–160°) is observed in nitrosyl heme proteins, however [62]. We reported the 1.3 Å resolution crystal structures of hh MbNO, and showed that two reproducible FeNO angles (∼144° and 120°) are obtained depending on the method of preparation of the complex [62]. The variability of FeNO angle in the heme pocket of MbNO had been demonstrated previously by Hori et al. using single crystal EPR spectroscopy; the FeNO angle was 153 ± 5 at 293K, but was 109 ± 5 at 77 K [133]. Chien and Dickenson showed, using single crystal EPR spectroscopy, that the FeNO angles in HbKNO were 167° in the α-subunits and 105° in the β-subunits [41]. It is thus likely that subtle distal pocket effects influence the metal-NO bond geometry in nitrosyl adducts of heme proteins. We now consider the case of MnIIMb(NO) and compare the observed geometry to that expected from synthetic manganese nitrosyl porphyrins.
To date, all synthetic (por)Mn(NO)-containing compounds of the {MnNO}6 class (i.e., MnIINO) that have been characterized by X-ray crystallography display linear Mn-NO groups [48, 49, 134, 135]. The expected and observed linearity of the MnNO bonds is supported by the spectroscopic characterization of several low-spin (por)Mn(NO)-containing compounds [44, 46, 128, 136]. Yu et al. employed resonance Raman spectroscopy to demonstrate that steric constraints within the distal face of a synthetic “strapped” (por)Mn(NO) complex enhances the Mn–N–O bending mode, and they attributed this observation to a tilting of the trans Mn–N(N-base) bond [136].
As stated in the Introduction, NO adducts of Mn-substituted heme proteins have been prepared and characterized by spectroscopy. MnIIIMb does not bind NO, however MnIIMb does [40]. Interestingly, Gersonde and coworkers demonstrated, using resonance Raman spectroscopy, that the monomeric MnIIIHb compound from the insect Chironomus thummi thummi binds NO to give the MnIIINO adduct, but this undergoes autoreduction to the MnIINO derivative [42].
To the best of our knowledge, the crystal structure of MnIIMb(NO) reported in this work (Fig. 2b) represents the first distinctly bent MnNO moiety in a natural or synthetic manganese heme complex. The NO ligand refines to 70% occupancy, but the MnNO bend is clearly evident in the structure. The nitrosyl N atom is 2.6 Å away from the Nε atom of the major conformation of the distal His64 residue, suggesting strong hydrogen-bond stabilization of the NO molecule in the distal pocket. As mentioned in the Results section, the observed long Mn–NO distance of 2.53 Å is likely an average distance that has contributions from the unligated five-coordinate MnIIMb complex and the nitrosyl adduct. Assuming that the Mn atom were in the heme 4N plane in MnIIMb(NO), the resulting Mn–NO distance would still be rather long. Thus, we propose that the geometry of the MnNO moiety observed here represents that of a loosely bound NO molecule to the metal center that is held in place by strong hydrogen-bonding to the distal His64 residue. This geometry is also likely determined by the method of preparation of the complex, namely the nitrosylation of the preformed distal pocket in the MnIIMb crystals. Such a procedure may disfavor linear (strong) binding of the NO ligand without significant movement of the distal His64 residue away from its position in MnIIMb. We are continuing attempts to crystallize pre-formed MnIIMb(NO) to test this hypothesis.
Supplementary Material
Acknowledgments
The authors are grateful to the National Institutes of Health (GM 64476; GBR-A) for funding for this research. ZNZ thanks the Government of Egypt for a Ph.D. scholarship.
5. Abbreviations
- CcP
cytochrome c peroxidase
- DFT
density functional theory
- Hbk
hemoglobin Kansas
- hh
horse heart
- 1-MeIm
1-methylimidazole
- Mb
myoglobin
- NiR
nitrite reductase
- OEP
dianion of 2,3,7,8,12,13,17,18-octaethylporphyrin
- OTf
triflate anion
- PGHS
prostaglandin H2 synthase
- por
porphyrinato dianion
- PPIX
dianion of protoporphyrin IX
- SCE
saturated calomel electrode
- sGC
soluble guanyly cyclase
- sw
sperm whale
- TPP
dianion of 5,10,15,20-tetraphenylporphyrin
Footnotes
Supplementary data: Figs. S1-S3 accompany this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Percy MJ, McFerran NV, Lappin TRJ. Blood Reviews. 2005;19:61–68. doi: 10.1016/j.blre.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Chui JS, Poon WT, Chan KC, Chan AY, Buckley TA. Anaesthesia. 2005;60:496–500. doi: 10.1111/j.1365-2044.2004.04076.x. [DOI] [PubMed] [Google Scholar]
- 3.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, III, Kelm M, Wink DA, Espey MG, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR, Jr, Feelisch M, Lundberg JO. Nat Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. and references therein. [DOI] [PubMed] [Google Scholar]; 2006;2(2):110. Erratum: [Google Scholar]
- 4.Bryan NS. Free Rad Biol Med. 2006;41:691–701. doi: 10.1016/j.freeradbiomed.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 5.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields M, Kim-Shapiro DB, Schechter AN, Cannon RO, III, Gladwin MT. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 6.Hunter CJ, Dejam A, Blood AB, Shields H, Kim-Shapiro DB, Machado RF, Tarekegn S, Mulla N, Hopper AO, Schecter AN, Power GG, Gladwin MT. Nat Med. 2004;10:1122–1127. doi: 10.1038/nm1109. [DOI] [PubMed] [Google Scholar]
- 7.Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D. J Biol Chem. 1981;256:12393–12398. [PubMed] [Google Scholar]
- 8.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. J Biol Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 9.Grubina R, Huang A, Shiva S, Joshi MS, Azarov I, Basu S, Ringwood LA, Jiang A, Hogg N, Kim-Shapiro DB, Gladwin MT. J Biol Chem. 2007;282:12916–12927. doi: 10.1074/jbc.M700546200. [DOI] [PubMed] [Google Scholar]
- 10.Samouilov A, Kuppusamy P, Zweier JL. Arch Biochem Biophys. 1998;357:1–7. doi: 10.1006/abbi.1998.0785. [DOI] [PubMed] [Google Scholar]
- 11.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Cir Res. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- 12.Boucher LJ. Coord Chem Rev. 1972;7:289–329. [Google Scholar]
- 13.Fabry TL, Simo C, Javaherian K. Biochim Biophys Acta. 1968;160:118–122. doi: 10.1016/0005-2795(68)90072-x. [DOI] [PubMed] [Google Scholar]
- 14.Yonetani T, Drott HR, Leigh JS, Jr, Reed GH, Waterman MR, Asakura T. J Biol Chem. 1970;245:2998–3003. [PubMed] [Google Scholar]
- 15.Atassi MZ. Biochem J. 1967;103:29–35. doi: 10.1042/bj1030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yonetani T, Asakura T. J Biol Chem. 1969;244:4580–4588. [PubMed] [Google Scholar]
- 17.Breslow E. J Biol Chem. 1964;239:486–496. [PubMed] [Google Scholar]
- 18.Gelb MH, Toscano WA, Jr, Sligar SG. Proc Natl Acad Sci USA. 1982;79:5758–5762. doi: 10.1073/pnas.79.19.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dierks EA, Hu S, Vogel KM, Yu AE, Spiro TG, Burstyn JN. J Am Chem Soc. 1997;119:7316–7323. doi: 10.1007/s007750050354. [DOI] [PubMed] [Google Scholar]
- 20.Dickinson LC, Chien JCW. J Biol Chem. 1977;252:6156–6162. [PubMed] [Google Scholar]
- 21.Hemmens B, Gorren ACF, Schmidt K, Werner ER, Mayer B. Biochem J. 1998;332:337–342. doi: 10.1042/bj3320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theorell H. Nature (London) 1945;156:474. [Google Scholar]
- 23.Ozols J, Strittmatter P. J Biol Chem. 1964;239:1018–1023. [PubMed] [Google Scholar]
- 24.Yonetani T. J Biol Chem. 1967;242:5008–5013. [PubMed] [Google Scholar]
- 25.Yonetani T, Asakura T. J Biol Chem. 1968;243:3996–3998. [PubMed] [Google Scholar]
- 26.Gupta K, Selinsky BS, Loll PJ. Acta Cryst. 2006;D62:151–156. doi: 10.1107/S0907444905036309. [DOI] [PubMed] [Google Scholar]
- 27.Borg DC, Cotzias GC. Nature. 1958;182:1677–1678. doi: 10.1038/1821677a0. [DOI] [PubMed] [Google Scholar]
- 28.Borg DC, Cotzias GC. Fed Proc. 1959;18:470. [Google Scholar]
- 29.Mahoney JP, Sargent K. J Clin Invest. 1967;46:1090. [Google Scholar]
- 30.Hancock RGV, Fritze K. Bioinorg Chem. 1973;3:77–87. [Google Scholar]
- 31.Wibowo AAE, Salle HJA, del Castiho P, Zielhuis RL. Int Arch Occup Environ Health. 1979;43:177–182. doi: 10.1007/BF00381189. [DOI] [PubMed] [Google Scholar]
- 32.Baden SP, Neil DM. Comp Biochem Physiol A: Mol Integr Physiol. 1998;119A:351–359. doi: 10.1016/s1095-6433(97)00437-6. [DOI] [PubMed] [Google Scholar]
- 33.Keilin D. Proc Roy Soc B. 1936;121:165–173. [Google Scholar]
- 34.Hoffman BM, Gibson QH, Bull C, Crepeau RH, Edelstein SJ, Fisher RG, McDonald MJ. Ann N Y Acad Sci. 1975;244:174–186. doi: 10.1111/j.1749-6632.1975.tb41530.x. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman BM, Gibson QH. Biochemistry. 1976;15:3405–3410. doi: 10.1021/bi00661a002. [DOI] [PubMed] [Google Scholar]
- 36.Mondal MS, Mazumdar S, Mitra S. Inorg Chem. 1993;32:5362–5367. [Google Scholar]
- 37.Gibson QH, Hoffman BM, Crepeau RH, Edelstein SJ, Bull C. Biochem Biophys Res Commun. 1974;59:146–151. doi: 10.1016/s0006-291x(74)80186-5. [DOI] [PubMed] [Google Scholar]
- 38.Bull C, Fisher RG, Hoffman BM. Biochem Biophys Res Commun. 1974;59:140–145. doi: 10.1016/s0006-291x(74)80185-3. [DOI] [PubMed] [Google Scholar]
- 39.Gibson QH, Hoffman BM. J Biol Chem. 1979;254:4691–4697. [PubMed] [Google Scholar]
- 40.Yonetani T, Yamamoto H, Erman JE, Leigh JJS, Reed GH. J Biol Chem. 1972;247:2447–2455. [PubMed] [Google Scholar]
- 41.Chien JCW, Dickinson LC. J Biol Chem. 1977;252:1331–1335. [PubMed] [Google Scholar]
- 42.Lin SH, Yu NT, Gersonde K. FEBS Lett. 1988;229:367–371. doi: 10.1016/0014-5793(88)81158-x. [DOI] [PubMed] [Google Scholar]
- 43.Lan EH, Dave BC, Fukuto JM, Dunn B, Zink JI, Valentine JS. J Mater Chem. 1999;9:45–53. [Google Scholar]
- 44.Wayland BB, Olson LW. Inorg Chim Acta. 1974;11:L23–L24. [Google Scholar]
- 45.Wayland BB, Olson LW, Siddiqui ZU. J Am Chem Soc. 1976;98:94–98. [Google Scholar]
- 46.Piciulo PL, Scheidt WR. Inorg Nucl Chem Lett. 1975;11:309–311. [Google Scholar]
- 47.Kelly S, Lançon D, Kadish KM. Inorg Chem. 1984;23:1451–1458. [Google Scholar]
- 48.Zahran ZN, Shaw MJ, Khan MA, Richter-Addo GB. Inorg Chem. 2006;45:2661–2668. doi: 10.1021/ic051190n. [DOI] [PubMed] [Google Scholar]
- 49.Zahran ZN, Lee J, Alguindigue SS, Khan MA, Richter-Addo GB. Dalton Trans. 2004:44–50. doi: 10.1039/b308143p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu CH, Su YO. J Electroanal Chem. 1994;368:323–327. [Google Scholar]
- 51.Moffat K, Loe RS, Hoffman BM. J Mol Biol. 1976;104:669–685. doi: 10.1016/0022-2836(76)90128-5. [DOI] [PubMed] [Google Scholar]
- 52.Moffat K, Loe RS, Hoffman BM. J Am Chem Soc. 1974;96:5259–5261. doi: 10.1021/ja00823a046. [DOI] [PubMed] [Google Scholar]
- 53.Arnone A, Rogers P, Blough NV, McGourty JL, Hoffman BM. J Mol Biol. 1986;188:693–706. doi: 10.1016/s0022-2836(86)80015-8. [DOI] [PubMed] [Google Scholar]
- 54.Makino R, Matsuda H, Obayashi E, Shiro Y, Iizuka T, Hori H. J Biol Chem. 1999;274:7714–7723. doi: 10.1074/jbc.274.12.7714. [DOI] [PubMed] [Google Scholar]
- 55.Teale FWJ. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 56.Yonetani T, Yamamoto H, Woodrow GV., III J Biol Chem. 1974;249:682–690. [PubMed] [Google Scholar]
- 57.Langley R, Hambright P, Alston K. Inorg Chem. 1986;25:114–117. [Google Scholar]
- 58.Yu NT, Tsubaki M. Biochemistry. 1980;19:4647–4653. doi: 10.1021/bi00561a017. [DOI] [PubMed] [Google Scholar]
- 59.Pflugrath J. Acta Crystallogr. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 60.Project C. Acta Crystallogr. 1994;D50:760–763. [Google Scholar]
- 61.Vagin A, Teplyakov A. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 62.Copeland DM, Soares A, West AH, Richter-Addo GB. J Inorg Biochem. 2006;100:1413–1425. doi: 10.1016/j.jinorgbio.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 63.Arendall WB, III, Tempel W, Richardson JS, Zhou W, Wang S, Davis IW, Liu ZJ, Rose JP, Carson WM, Luo M, Richardson DC, Wang BC. J Struct Funct Genomics. 2005;6:1–11. doi: 10.1007/s10969-005-3138-4. [DOI] [PubMed] [Google Scholar]
- 64.Davis IW, Murray LW, Richardson JS, Richardson DC. Nucl Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. Web Server. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Reed RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 66.Williamson MM, Hill CL. Inorg Chem. 1987;26:4155–4160. [Google Scholar]
- 67.Williamson MM, Hill CL. Inorg Chem. 1986;25:4668–4671. [Google Scholar]
- 68.Cheng B, Cukiernik F, Fries PH, Marchon JC, Scheidt WR. Inorg Chem. 1995;34:4627–4639. [Google Scholar]
- 69.Brucker EA, Olson JS, Phillips GN, Jr, Dou Y, Ikeda-Saito M. J Biol Chem. 1996;271:25419–25422. doi: 10.1074/jbc.271.41.25419. [DOI] [PubMed] [Google Scholar]
- 70.Sono M, Dawson JH. J Biol Chem. 1982;257:5496–5502. and references therein. [PubMed] [Google Scholar]
- 71.Kirner JF, Reed CA, Scheidt WR. J Am Chem Soc. 1977;99:2557–2563. doi: 10.1021/ja00450a025. [DOI] [PubMed] [Google Scholar]
- 72.Parthasarathi N, Spiro TG. Inorg Chem. 1987;26:2280–2282. [Google Scholar]
- 73.Day VW, Stults BR, Tasset EL, Day RO, Marianelli RS. J Am Chem Soc. 1974;96:2650–2652. doi: 10.1021/ja00815a075. [DOI] [PubMed] [Google Scholar]
- 74.Hatano K, Anzai K, Iitaka Y. Bull Chem Soc Jpn. 1983;56:422–427. [Google Scholar]
- 75.Cheng B, Scheidt WR. Acta Crystallogr. 1996;C52:585–588. doi: 10.1107/s0108270195010705. [DOI] [PubMed] [Google Scholar]
- 76.Cox RP, Hollaway MR. Eur J Biochem. 1977;74:575–587. doi: 10.1111/j.1432-1033.1977.tb11427.x. [DOI] [PubMed] [Google Scholar]
- 77.Bogumil R, Hunter CL, Maurus R, Tang HL, Lee H, Lloyd E, Brayer GD, Smith M, Mauk AG. Biochemistry. 1994;33:7600–7608. doi: 10.1021/bi00190a013. [DOI] [PubMed] [Google Scholar]
- 78.Perutz MF, Mathews FS. J Mol Biol. 1966;21:199–202. doi: 10.1016/0022-2836(66)90088-x. [DOI] [PubMed] [Google Scholar]
- 79.Deatherage JF, Obendorf SK, Moffat K. J Mol Biol. 1979;134:419–429. doi: 10.1016/0022-2836(79)90361-9. [DOI] [PubMed] [Google Scholar]
- 80.Stryer L, Kendrew JC, Watson HC. J Mol Biol. 1964;8:96–104. doi: 10.1016/s0022-2836(64)80152-2. [DOI] [PubMed] [Google Scholar]
- 81.Maurus R, Bogumi B, Nguyen NT, Mauk AG, Brayer G. Biochem J. 1998;322:67–74. doi: 10.1042/bj3320067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mattevi A, Gatti G, Coda A, Rizzi M, Ascenzi P, Brunori M, Bolognesi M. J Mol Recogn. 1991;4:1–6. doi: 10.1002/jmr.300040102. [DOI] [PubMed] [Google Scholar]
- 83.Day VW, Stults BR, Tasset EL, Marianelli RS. Inorg Nucl Chem Lett. 1975;11:505–509. [Google Scholar]
- 84.Brooker S, McKee V. Acta Cryst. 1993;C49:441–445. [Google Scholar]
- 85.Hori H, Ikeda-Saito M, Reed GH, Yonetani T. J Magn Reson. 1984;58:177–185. [Google Scholar]
- 86.Maurus R, Overall CM, Bogumi R, Luo Y, Mauk AG, Smith M, Brayer GD. Biochem Biophys Acta. 1997;1341:1–13. doi: 10.1016/s0167-4838(97)00064-2. [DOI] [PubMed] [Google Scholar]
- 87.Evans SV, Brayer GD. J Mol Biol. 1990;213:885–897. doi: 10.1016/S0022-2836(05)80270-0. [DOI] [PubMed] [Google Scholar]
- 88.Parthasarathi N, Spiro TG. Inorg Chem. 1987;26:3792–3796. [Google Scholar]
- 89.Lin R, Immoos CE, Farmer PJ. J Biol Inorg Chem. 2000;5:738–747. doi: 10.1007/s007750000163. [DOI] [PubMed] [Google Scholar]
- 90.Vojtechovsky J, Chu K, Berendzen J, Sweet RM, Schlichting I. Biophys J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kachalova GS, Popov AN, Bartunik HD. Science. 1999;284:473–476. doi: 10.1126/science.284.5413.473. [DOI] [PubMed] [Google Scholar]
- 92.Quillin ML, Arduini RM, Olson JS, Philips GN., Jr J Mol Biol. 1993;234:140–155. doi: 10.1006/jmbi.1993.1569. [DOI] [PubMed] [Google Scholar]
- 93.Quillin ML, Li T, Olson JS, Phillips GN, Jr, Y D, Ikeda-Saito M, Regan R, Carlson M, Gibson QH, Li H, Elber R. J Mol Biol. 1995;245:416–436. doi: 10.1006/jmbi.1994.0034. [DOI] [PubMed] [Google Scholar]
- 94.Takano T. J Mol Biol. 1977;110:569–584. doi: 10.1016/s0022-2836(77)80112-5. [DOI] [PubMed] [Google Scholar]
- 95.Krzywda S, Murshudov GN, Brzozowksi AM, Jaskolski M, Scott EE, Klizas SA, Gibson QH, Olson JS, Wilkinson AJ. Biochemistry. 1998;37:15896–15907. doi: 10.1021/bi9812470. [DOI] [PubMed] [Google Scholar]
- 96.Cameron AD, Smerdon SJ, Wilkinson AJ, Habash J, Helliwell JR, Li T, Olson JS. Biochemistry. 1993;32:13061–13070. doi: 10.1021/bi00211a016. [DOI] [PubMed] [Google Scholar]
- 97.Chu K, Vojtchovsky J, McMahon BH, Sweet RM, Berendzen J, Schlichting I. Nature. 2000;403:921–923. doi: 10.1038/35002641. [DOI] [PubMed] [Google Scholar]
- 98.Schotte F, Soman J, Olson JS, Wulff M, Anfinrud PA. J Struct Biol. 2004;147:235–246. doi: 10.1016/j.jsb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 99.Nienhaus GU, Nienhaus K. J Biol Phys. 2002;28:163–172. doi: 10.1023/A:1019990522433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brunori M, Bourgeois D, Vallone B. J Struct Biol. 2004;147:223–234. doi: 10.1016/j.jsb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 101.Springer BA, Sligar SG, Olson JS, Phillips GN., Jr Chem Rev. 1994;94:699–714. [Google Scholar]
- 102.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gladwin MT. Nat Chem Biol. 2005;1:245–246. doi: 10.1038/nchembio1005-245. [DOI] [PubMed] [Google Scholar]
- 104.Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. J Inorg Biochem. 2005;99:237–246. doi: 10.1016/j.jinorgbio.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 105.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 106.Duranski MR, Greer JJM, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Averill BA. Chem Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 108.Hollocher TC. The Enzymology and Occurrence of Nitric Oxide in the Biological Nitrogen Cycle. In: Lancaster J, editor. Nitric Oxide Principles and Applications. Academic Press; San Diego: 1996. pp. 289–344. [Google Scholar]
- 109.Eady RR, Hasnain SS. Denitrification. In: Que L Jr, Tolman WB, editors. Comprehensive Coordination Chemistry II. Elsevier; San Diego, CA: 2004. pp. 759–786. [Google Scholar]
- 110.Williams PA, Fulop V, Garman EF, Saunders NFW, Ferguson SJ, Hajdu J. Nature. 1997;389:406–412. doi: 10.1038/38775. [DOI] [PubMed] [Google Scholar]
- 111.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. J Am Chem Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 112.Crane BR, Siegel LM, Getzoff ED. Biochemistry. 1997;36:12120–12137. doi: 10.1021/bi971066i. [DOI] [PubMed] [Google Scholar]
- 113.Silaghi-Dumitrescu R. Inorg Chem. 2004;43:3715–3718. doi: 10.1021/ic035403p. [DOI] [PubMed] [Google Scholar]
- 114.Wyllie GRA, Scheidt WR. Chem Rev. 2002;102:1067–1089. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 115.Nasri H, Ellison MK, Chen S, Huynh BH, Scheidt WR. J Am Chem Soc. 1997;119:6274–6283. [Google Scholar]
- 116.Novozhilova IV, Coppens P, Lee J, Richter-Addo GB, Bagley KA. J Am Chem Soc. 2006;128:2093–2104. doi: 10.1021/ja0567891. [DOI] [PubMed] [Google Scholar]
- 117.Suslick KS, Watson RA. Inorg Chem. 1991;30:912–919. [Google Scholar]
- 118.Suslick KS, Watson RA. New J Chem. 1992;16:633–642. [Google Scholar]
- 119.Hoshino M, Nagashima Y, Seki H, Leo MD, Ford PC. Inorg Chem. 1998;37:2464–2469. [Google Scholar]
- 120.Yamamoto K, Iitaka Y. Chem Lett. 1989:697–698. [Google Scholar]
- 121.Jene PG, Ibers JA. Inorg Chem. 2000;39:3823–3827. doi: 10.1021/ic000127p. [DOI] [PubMed] [Google Scholar]
- 122.Adachi H, Suzuki H, Miyazaki Y, Iimura Y, Hoshino M. Inorg Chem. 2002;41:2518–2524. doi: 10.1021/ic0108686. [DOI] [PubMed] [Google Scholar]
- 123.Goodwin J, Kurtikyan T, Standard J, Walsh R, Zheng B, Parmley D, Howard J, Green S, Mardyukov A, Przybla DE. Inorg Chem. 2005;44:2215–2223. doi: 10.1021/ic048701a. [DOI] [PubMed] [Google Scholar]
- 124.Seki H, Okada K, Iimura Y, Hoshino M. J Phys Chem A. 1997;101:8174–8178. [Google Scholar]
- 125.Taniguchi I, Li CZ, Ishida M, Yao Q. J Electroanal Chem. 1999;460:245–250. [Google Scholar]
- 126.Nienhaus K, Ostermann A, Nienhaus GU, Parak FG, Schmidt M. Biochemistry. 2005;44:5095–5105. doi: 10.1021/bi047513t. [DOI] [PubMed] [Google Scholar]
- 127.Richter-Addo GB, Legzdins P. Metal Nitrosyls. Oxford University Press; New York: 1992. [Google Scholar]
- 128.Cheng L, Richter-Addo GB. Binding and Activation of Nitric Oxide by Metalloporphyrins and Heme. In: Guilard R, Smith K, Kadish KM, editors. The Porphyrin Handbook. Vol. 4. Academic Press; New York: 2000. pp. 219–291. [Google Scholar]
- 129.Feltham RD, Enemark JH. Top Stereochem. 1981;12:155–215. [Google Scholar]
- 130.Westcott BL, Enemark JH. Transition Metal Nitrosyls. In: Lever ABP, Solomon EI, editors. Inorganic Electronic Structure and Spectroscopy. Chapter 7 Wiley and Sons; New York: 1999. [Google Scholar]
- 131.Enemark JH, Feltham RD. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 132.Wyllie GRA, Schultz CE, Scheidt WR. Inorg Chem. 2003;42:5722–5734. doi: 10.1021/ic034473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hori H, Ikeda-Saito M, Yonetani T. J Biol Chem. 1981;256:7849–7855. [PubMed] [Google Scholar]
- 134.Scheidt WR, Hatano K, Rupprecht GA, Piciulo PL. Inorg Chem. 1979;18:292–299. [Google Scholar]
- 135.Piciulo PL, Rupprecht G, Scheidt WR. J Am Chem Soc. 1974;96:5293–5295. [Google Scholar]
- 136.Yu NT, Lin SH, Chang CK, Gersonde K. Biophys J. 1989;55:1137–1144. doi: 10.1016/S0006-3495(89)82910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.