

Abstract
A hallmark of immune activation by certain RNA sequences is the generation of interferon responses. However, the study of immunostimulatory RNA (isRNA) has been hindered by costly and slow methods, particularly in vitro. We have developed a cell-based assay to detect human type I interferon (IFN) that reliably senses both IFN-α and IFN-β simultaneously. The human 293T cell line was stably transfected with a fusion gene of monomeric red fluorescent protein (mRFP) under the transcriptional control of an interferon-stimulated response element (ISRE). High levels of mRFP are expressed following activation of the type I IFN receptor (IFNAR). Using this method, detection limits for IFN similar to that of ELISA can be achieved but with a greater dynamic range and in a high-throughput manner. As a proof of concept, we utilized this method to screen a library of cationic lipid-like materials that for nanoparticle complexes with RNA for induction of innate immune responses in vitro. We expect the screening and detection methods described herein may provide a useful tool in elucidating mechanisms that govern the delivery of RNA molecules to effector cells and receptors of the innate immune system. We apply this tool to investigate isRNA drug delivery, but it may also find use in other applications for which high throughput detection of type 1 IFN is desired.
Keywords: RNA interference, siRNA, immunostimulatory RNA, drug delivery, high-throughput screening, innate immunity
Introduction
Clinically suitable intracellular delivery of RNA remains a major challenge in realizing the therapeutic potential of technologies such as RNA interference (RNAi). One approach has been to generate large libraries of potential delivery materials followed by high throughput screening (Goldberg et al. 2008). The screening of combinatorial materials is a powerful tool for the development of material structure-function relationships in systems too complex to be currently understood by first principles (Hubbell 2004; Kohn 2004) such as the immune system. One general workflow of combinatorial materials discovery is material generation followed by screening for biological readouts in vitro and finally the selection of candidate materials for in vivo study. Screening of large libraries typically requires in vitro assays that are rapid, automatable, and low cost. The complex immunologic responses to materials are often only considered once the level of in vivo work has been reached. However, materials may have physiologic effects, for example, by altering the biological responses to encapsulated drugs (Kabanov 2006). The development of in vitro screening protocols to investigate immunologic responses to drug delivery materials can provide additional data at an early stage in the process of material development.
RNA interference is mediated by small 21–23 nucleotide double-stranded short interfering RNAs (siRNA), and holds potential therapeutic utility by specifically repressing target genes involved in diseases such as infection, cancer, or a genetic pathology (Behlke 2006). Certain siRNA sequences have the potential to interact with the innate immune system, and can result in off-target effects and undesired immune activation (Eberle et al. 2008; Judge et al. 2006; Moss and Taylor 2003; Robbins et al. 2008). However, for certain disease states, it is possible that these immune responses could be harnessed for therapeutic benefit (Furset and Sioud 2007; Poeck et al. 2008). Small RNA molecules, including siRNAs, can stimulate the innate immune system (Moss and Taylor 2003) leading to antiviral properties (Stram and Kuzntzova 2006), anti-tumor activity (Scheel et al. 2006), and vaccine-adjuvant responses (Westwood et al. 2006).
RNA molecules that activate the immune response have been termed immunostimulatory (isRNA) (Hornung et al. 2005; Sioud 2006). These properties arise through interactions with pattern recognition receptors (PRR), such as the endosomal Toll-like receptors (TLR) and cytosolic RIG-I-like receptors (RLRs), which typically result in induction of type I interferon (IFN) (Hornung et al. 2005; Sioud 2006; Xiao 2008). In particular TLR7 and TLR8 seem to be the most physiologically important receptor for synthetic siRNA molecules in mice and humans for generating IFN responses (Hornung et al. 2005). The plasmacytoid dendritic cell (PDC) is a circulating cell of hematopoetic origin that is specialized for the detection of viral RNA genomes by expressing TLR7 and TLR9. The PDC responds to TLR engagement by producing IFN, specifically IFN-alpha (IFN-α), and other cytokines that activate anti-viral responses (Heil et al. 2004). A hallmark of the PDC is its ability to produce over 1000 times more IFN-α per cell than any other cell type (Hornung et al. 2005). Additionally, TLR8-mediated isRNA activity is observed in human myeloid dendritic cells, monocytes, and monocyte-derived dendritic cells (Hornung et al. 2002); TLR3 has also been implicated as an isRNA receptor (Kleinman et al. 2008).
The definition of isRNA is a functional one that depends on an RNA’s potential to interact with TLRs and other PRRs: RNA sequence (Hornung et al. 2005; Judge et al. 2005), structure and chemical modifications (Judge et al. 2006; Zamanian-Daryoush et al. 2008), and intracellular location (Sioud 2006) are all determinants of isRNA function. For example, TLR7 and TLR8 are segregated to the endosome and are reported to function only after endosomal acidification (Sioud 2005). By contrast, RLRs are located in the cytoplasm (Xiao 2008). Thus, immunostimulatory properties of a RNA molecule can be influenced by delivery to the proper intracellular compartment and cell type.
The power to harness the potentially therapeutic properties of RNAi or isRNA lies in the ability to deliver RNA in a clinically relevant manner. We recently developed a large library of lipid-like materials termed “lipidoids” (Akinc et al. 2008) for delivery of siRNA. Herein, we detail a systematic in vitro method of screening delivery agents for a specific type of innate immune response to RNA that results in PDC-mediated generation of type I IFN. A limiting factor in screening for IFN responses is the traditional reliance on low-throughput and costly ELISA assays to quantify IFN levels. Our high-throughput cell-based assay for detection of human IFN has a high dynamic range, robust sensitivity, and 96-well plate compatible workflow. As a further proof of concept, we demonstrate the utility of this method in screening a library of lipidoid materials for immunostimulatory siRNA drug delivery.
Materials and Methods
Recombinant protein standards, TLR agonists, and siRNAs
Carrier free recombinant human interferon-alpha A (IFN-α, specific activity 0.233 U/pg), human interferon-beta 1a (IFN-β, specific activity 0.3 U/pg), and human interferon-omega (IFN-ω, specific activity 0.1 U/pg) were purchased from PBL Laboratories (Piscataway, NJ), diluted to 1.0 x 106 U/mL in PBS with 0.1% BSA, and stored in 250 μL aliquots at −80°C until use. Infergen, a recombinant consensus human IFN-alpha, was from Three Rivers Pharmaceuticals (Cranberry Township, PA). All TLR agonists were purchased from Invivogen (San Diego, CA) and used at manufacturer’s recommended concentrations (see Supplemental Table 1 for details). Unmodified siRNA molecules with 3′UU overhangs were purchased from Dharmacon (Lafayette, CO) with the “A4” processing option. The siGFP (Ge et al. 2003), and siβgal (Judge et al. 2005) sequences were as previously reported (see supplemental Table 2 for sequence).
Stable transduction of a 293T cell line with reporter gene construct
The ISG54-mRFP-CAT cassette, which contains an IFN-stimulated response element (ISRE) promoting the transcription of a gene encoding a fusion protein consisting of the monomeric red fluorescent protein (mRFP) (Campbell et al. 2002) and chloramphenicol acetyltransferase (CAT), was previously described (Martinez-Sobrido et al. 2006). This cassette was cloned into a lentiviral vector plasmid at ClaI and SalI restriction endonuclease sites. To produce lentivirus particles, the lentiviral vector was cotransfected with a GAG-POL expressing plasmid and a VSV-G expressing plasmid in HEK293T (293T) cells. Supernatants containing lentivirus were then added to fresh 293T cells, and following transduction approximately 30 individual clones were isolated by limiting dilution. The most stable clone exhibiting the greatest mRFP expression (approaching 100% of all cells) following incubation with Infergen was selected for further studies. This cell line was cultured for multiple passages, treated with 10,000 U/mL of recombinant IFN-α for 24 hours, and then sorted by FACS with gating for mid-level RFP expression. This subset and subsequent generations (referred to as 293T-ISRE-RFP) were used in all experiments reported herein.
Flow cytometry analysis and quantification of type I IFN activity
For quantification of type I IFN activity in samples or standard curves, 293T-ISRE-RFP cells were seeded overnight at 10,000 cells per well in 150 μL of growth media (D-MEM, 10% FCS, 100 U/mL penicillin/streptomycin) in 96-well tissue culture plates. To generate standard curves, IFN-α or IFN-β was serially diluted in supplemented RPMI growth media (RPMI 1640 medium with 10% FCS, 1mM MEM sodium pyruvate, 10 mM HEPES, and 100 U/mL penicillin/streptomycin). In triplicate, 50 μL of standard or sample was added to wells for a final dilution of 1:4 followed by overnight incubation.
To prepare 293T-ISRE-RFP cells for flow cytometry, media was completely aspirated from each well and replaced with 25 μL of 0.25% Trypsin-EDTA (Invitrogen, Carlsbad, CA) for 10 minutes. An additional 50 μL of FACS buffer (PBS with 1% BSA and 2 mM EDTA) was added to each well and cells were resuspended by repeated pipetting. All 75+ μL was transferred to a round bottom 96-well analysis plate for high throughput flow cytometry. Fluorescent flow cytometry was performed on a Becton Dickson (Franklin Lakes, NJ) LSR II system equipped with the HTS plate reader option. Fluid handling settings were 50 μL sample per well at 2.5 μL/sec flow rate. Cells were excited with a 488 nm laser, mRFP signal was detected using the 610 nm with 20 nm bandpass filter set, and auto-fluorescence signal was detected using the 575 nm with 26 nm bandpass filter set. Treestar FlowJo software (Ashland, OR) was used for cytometry data analysis.
Preparation and activation of peripheral blood mononuclear cells
Donor-blind buffy coat packs were obtained from the Massachusetts General Hospital Blood Bank and diluted in PBS with 2mM EDTA. Peripheral blood mononuclear cells (PBMC) were obtained by density centrifugation at 400 RCF in Ficoll-Paque Plus (Amersham Biosciences) and washed twice in PBS/EDTA. Magnetic cell sorting was used to further separate PBMCs into PDCs and PDC-depleted PBMCs (PBMC(−)) using the MACS human Plasmacytoid Dendritic Cell Isolation Kit (Miltenyi Biotech, Auburn, CA).
Cells were resuspended in supplemented RPMI and plated in 96-well tissue culture plates at 5x105 cells/well in 175 μL, or 2x104 cells/well in 175 μL for PDCs. Cells were then activated with TLR agonists or transfected as described. Following 18 to 24 hours of incubation at 37°C, PBMC plates were centrifuged at 400 RCF to clear suspended cells. A 100 μL per well aliquot of supernatant was stored at −80°C for later quantification of type I IFN activity using the 293T-ISRE-RFP system as described above.
Screening for immunostimulatory delivery of siRNA with lipidoids
Lipidoids were synthesized as previously described (Akinc et al. 2008). These transfection materials were dissolved to 0.5 mg/mL in 25 mM sodium acetate (NaAc), pH 5.0; siRNA was diluted to 50 μg/mL in NaAc and arrayed into a 96-well round bottom assay plate to facilitate high throughput. Transfection materials were added to RNA at the indicated weight:weight ratios (15, 10, 5, 2.5: 1) and thoroughly mixed by pipetting. After 20 minutes of incubation at room temperature, stable complexes were diluted in supplemented RPMI and 25 μL of diluted complexes were added to PBMCs for a dose of 100 ng siRNA (~38 nM) per well. As a positive control, siRNAs were mixed with Lipofectamine 2000 (L2K) in Opti-MEM media (Invitrogen, Carlsbad, CA) for 20 minutes according to manufacturer’s protocols prior to adding 100 ng siRNA (~38 nM) per well to PBMCs. Mock transfection with uncomplexed siRNA diluted in NaAc and supplemented RPMI media was used as a negative control at 100 ng/well (~38 nM).
Results
Generation and characterization of reporter cell line
A stably-transfected cell line based on the human 293T cell line was created by retroviral insertion of a fusion gene construct of mRFP-CAT under control of interferon-stimulated response elements (ISRE) (Fig. 1a). Upon engagement of the type I IFN receptor (IFNAR), 293T-ISRE-RFP cells express mRFP (Fig. 1b). Expression of mRFP is IFN-dose dependent (Fig. 1c). Fluorescent signal can be detected by cell cytometry with excitation from a 488 nm laser and emission measured at 610 nm (Figs. 2a and 2b). Though mRFP has low excitation at 488 nm (peak excitation at 584 nm, peak emission at 607 nm (Campbell et al. 2002)), this careful gating allows distinction of red fluorescence signal even without a yellow excitation laser. We have not tested interferon-mediated induction of CAT activity, although this method may prove useful when flow cytometry is not available.
Figure 1.



A 293T cell line was stably transduced with the reporter construct depicted above. Expression of mRFP-CAT is induced by engagement of the type I interferon receptor (IFNAR) by IFN-β (blue triangle) or IFN-α (yellow triangle) (a). The 293T cell line was seeded in 24-well plates overnight followed by treatment with 1000 U/mL IFN-α or IFN-β (b) or 2-fold dilutions of IFN-β (c) for 30 hours. Expression of mRFP was observed by fluorescent light microscopy at 20X magnification.
Figure 2.
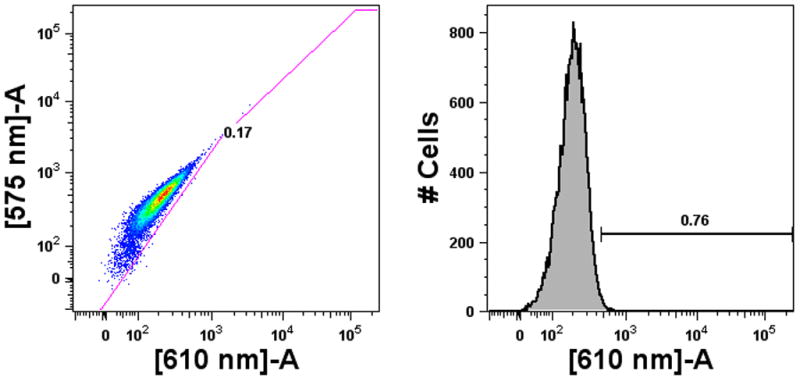
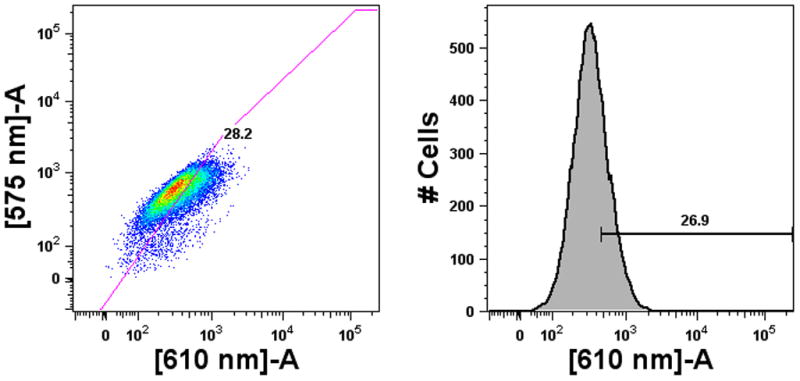
FACS analysis and gating of 293T-ISRE-RFP cells plated at 10,000 cells/well in a 96-well plate overnight prior to incubation for an additional 18–24 hours with media (a) or with 5000 Units/mL IFN-α standard (b). Cells were analyzed on a BD LSRII FACS analysis machine equipped with HTS plate handling. Two-dimensional gating (610 nm vs 575 nm, left) was found to be superior to one-dimensional (histogram of 610 nm, right) gating due to the ability to discern autofluorescence. mRFP has an emission peak at 607 nm.
Detection of type I interferons
While incubation with type I IFN induces mRFP expression, at low IFN concentrations only a few cells out of the entire population will express mRFP in large quantities while the vast majority of cells exhibit only background autofluorescence (Fig. 1c). Methods relying on a measurement of the fluorescence intensity averaged over the entire population, such as fluorescent microplate readers, may not detect increases in mRFP signal due to activation of only a few cells. Therefore, the most sensitive method for detection of IFN using the 293T-ISRE-RFP system will be methods such as flow cytometry that can distinguish an increase in single-cell fluorescence from the autofluorescence intensity of the entire population. The sensitivity for single-cell fluorescence is greatly increased by simultaneously controlling for cellular autofluorescence achieved by two-dimensional gating. Two-dimensional gating (Fig. 2, left) is crucial for distinguishing autofluorescent cells, as measured by 575 nm channel intensity, from RFP+ cells that would not be discernible from the background using single dimension gating (Fig. 2, right).
In the low range of IFN-α sample concentration, detection of RFP+ cells via two-dimensional gating for mRFP signal allows for quantification of as low as 2.44 U/mL of IFN-α, or approximately 10.5 pg/mL (Fig. 3a). The molecular weight of IFN-α is 19.4 kD, and 50 μL of this standard was diluted in 150 μL of media indicating a lower detection limit of 540 femtomolar IFN-α for the 293T-ISRE-RFP system. Dose concentration of IFN-α followed a log-log relationship with percentage of RFP+ cells that is highly linear (R2 > 0.99) up to at least 625 U/mL. Additionally, the 293T-ISRE-RFP cell line is dose-responsive to both IFN-α and IFN-β (Fig. 3c) as well as IFN-ω (Supplemental Fig. 1).
Figure 3.



Type 1 IFNs cause dose-dependent expression of mRFP. Standards were prepared by serial dilution of recombinant IFN-α. Pre-dilution concentrations are plotted on the x-axis. Low range and high range signals require different analysis for IFN quantification. (a) Low range linear standard curve (filled squares, left axis) between 2.44 U/mL and 625 U/mL IFN-α reveals a log-log correlation between percent RFP+cells and sample concentration by 2D-gating. The same data are plotted in log-linear transformation (open circles, right axis) for comparison. As low as 2.44 U/mL (~ 10.5 pg/mL) IFN-α can be detected. Error bars indicate standard deviation, n=2. (b) High range linear standard curve between 781 U/mL and 100,000 U/mL (~4.29x105 pg/mL) reveals a log-log correlation (filled squares, right axis) of mean fluorescence intensity (MFI) of 2D-gated cells and pre-diluted sample concentration. The same data are plotted in log-linear transformation (open circles, left ordinate) for comparison. Error bars indicate standard deviation, n=5. (c) Standard curves of recombinant IFN-α (black squares) and recombinant IFN-β (grey circles). Error bars indicate standard deviation, n=3.
As total IFN concentration increases, all cells become RFP-positive and produce increasing levels of mRFP. Expression of mRFP dynamically reflects IFN concentration such that simple on/off measurements of mRFP signal are no longer indicative of total IFN activity. At greater concentrations of IFN-α, the log of mean fluorescence intensity (MFI) of gated cells at the 610 nm emission channel correlates linearly to the log of IFN-α concentration (R2 > 0.99) (Fig. 3b). This analysis method, which measures an average of the entire RFP+ population, results in a detection range extending from 781 U/mL up to at least 100,000 U/mL (sample concentration), which is approximately 4.3x105 pg/mL f standard recombinant IFN-α. For comparison, the high-end limit of ELISA detection is approximately 5,000 pg/mL.
Activation of PBMCs to produce type-I interferon
Human PBMC’s were stimulated with agonists of TLRs 1 through 9 (Supplemental Table 1) for 24 hours and quantification of the type I IFN response was determined by 293T-ISRE-RFP assay. As expected, stimulation of TLR7 and TLR9 (receptors for small RNA and CpG DNA, respectively) that are highly expressed in PDCs (Hornung et al. 2002) induced large amounts of type I IFN (Fig. 4a). TLR1 and TLR3 agonists also induced expression of type I IFN, while agonists of TLR 2, 4, 5, 6, and 8 did not cause significant expression of type I IFN (less than 100 U/mL).
Figure 4.



Detection of isRNA activity in vitro in human PBMCs. (a) PBMCs were stimulated with agonists of TLRs 1 through 9 for 24 hours (a list of these agonists is included in Supplemental Table 1). TLR-stimulated secretion of type I IFN was detected by 293T-ISRE-RFP assay, using recombinant IFN-α as a standard. (b) PBMCs were transfected with 200 ng/well (~75 nM) siβgal or siGFP siRNAs complexed with Lipofectamine 2000 (L2K) or lipidoid 98N12(5)-1 (ND98-(5)1). (c) PBMCs were separated into PDCs and PDC-depleted PBMCS (PBMC(−)) and transfected with 200 ng/well (~75 nM) siβgal siRNAs complexed with Lipofectamine 2000 (L2K) or lipidoid ND98-(5)1. Secretion of type I IFNs was detected by 293T-ISRE-RFP assay, using recombinant IFN-α as a standard. Error bars indicate standard deviation, n=3.
We established standard positive and negative controls for comparing isRNA activity and the role of drug delivery. Delivery of siRNA to PBMCs induced type I IFN secretion depending upon both sequence and delivery agent. The siβgal sequence, a sequence with known immunostimulatory potential (Judge et al. 2005), is a much stronger isRNA sequence than the siGFP sequence (Fig. 4b); therefore the two sequences can be used as positive and negative controls, respectively. Further, immunostimulatory delivery of siβgal to PBMCs was increased with ND98-(5)1, a lipidoid delivery reagent (Akinc et al. 2008) (Supplemental Fig. 2), compared to delivery of siβgal with Lipofectamine 2000 (L2K), a commercially available cationic lipid transfection agent that can be used as a standard for comparison. Delivery of siGFP with ND98-(5)1 or L2K resulted in low stimulation of type I interferons.
To investigate the role of PDCs in the activity observed, PDCs were separated from non-PDCs by negative selection magnetic sorting. Whole PBMCs (5x105 cells/well), PDC-depleted PBMCs (5x105 cells/well), and untouched purified PDCs (2x104 cells/well) were then transfected with the siβgal immunostimulatory sequence. Both L2K and ND98-(5)1 delivered siβgal in a stimulatory fashion to PBMCs, while PDC-depleted PBMCs exhibited low secretion of type I IFN (Fig. 4c). PDCs alone were highly stimulated by ND98-(5)1-siβgal particles, although L2K-siβgal particles induced lower responses.
Screening of 10 lipidoids for isRNA delivery potential
The robust and simple nature of the PBMC model coupled with the 293T-ISRE-RFP assay enables high-throughput screening of materials for induction of type I IFN immune responses. We used a 3-step workflow, outlined in Fig. 5a, comprised of complex formation, transfection of PBMCS, and type I IFN detection. We have found that a single buffy coat (~35 – 45 mL) can provide enough cells for five to seven 96-well transfection plates. Additionally, the process can be accomplished in a piece-meal fashion providing further flexibility in workflow timing. PBMC supernatants can be frozen prior to 293T-ISRE-RFP assay, and PBMCs can be isolated a day in advance without loss of activity when stored overnight at 4°C in PBS with 2 mM EDTA (data not shown). Donor-to-donor variability in maximum secretion of type I IFNs is typically observed as evident when comparing the scale of Figs. 4a through 4c.
Figure 5.


HTS screening of isRNA delivery – (a) (1) Nanoparticles containing siRNA are formulated in a 96 well plate and incubated with PBMCs for 24 hours, (2) supernatants containing type I IFNs are transferred onto a 96-well plate containing 293T-ISRE-RFP cells, and (3) HTS FACS analysis for RFP expression levels are quantified after 24 hours of incubation. (b) PBMC were transfected with a set of 10 lipidoids by complexing at the indicated weight:weight ratios (lipidoid to RNA) with siβgal, a sequence with known isRNA activity, or siGFP, a sequence with low isRNA activity. Secretion of type I IFN was detected by 293T-ISRE-RFP assay, using recombinant IFN-α as a standard. Error bars indicate standard deviation, n=4.
As a proof-of-principle, we applied this screening method to a subset of the lipidoid material library synthesized in our lab for RNA delivery. The lipidoids are lipid-like materials generated by combinatorial chemistry that are cationic in nature and form nanoparticle-sized complexes with nucleic acids (Akinc et al. 2008). Candidate compounds (10 lipidoids) were pre-selected for screening based upon prior activity for delivery of siRNA molecules for RNA interference (Supplemental Fig. 2). We utilized the difference in isRNA potential between the siβgal and siGFP sequences (Fig. 4b) to standardize responses. Approximately 100 independent reactions and PBMC transfections were performed in quadruplicate in a 96-well plate format in less than one day, and measurement of type I IFN response was accomplished with minimal effort using the 293T-ISRE-RFP assay. In a 96-well plate format, samples were arrayed and complexed with siRNA at four different weight:weight ratios. The resulting particles were added to freshly isolated human PBMCs and incubated for 24 hours. Supernatants were collected and frozen for later analysis by the 293T-ISRE-RFP cell-based assay for type I IFN activity.
Of the 10 lipidoids tested, 4 compounds displayed significant induction of type I IFN above baseline levels. Lipidoids based on the 98-core chemistry, 3-tailed NA116, and 3-tailed NC100 all showed significant levels of activity with siβgal siRNA, (Fig. 5b). Only NC100-3 exhibit immunostimulatory effects with siGFP (Fig. 5b). As a control, naked RNA did not show isRNA properties. RNA concentration was held constant across all transfection conditions (100 ng/well or 38 nM), but higher ratios of lipidoid produced greater amounts of isRNA activity. Lipidoids alone were not investigated as these materials have significant positive charge when not complexed with anionic siRNA that may result in significant cytotoxicity.
One set of supernatants from PBMCs transfected with lipidoids complexed with siβgal or siGFP was assayed by ELISA for IFN-α and IFN-β levels to verify cell-based detection methods. Qualitatively, ELISA for (human multitype) IFN-α and cell-based analysis detected type I IFN in the same wells (Supplemental Fig. 3), but the absolute quantity detected by each method differed. Note that the 293T-ISRE-RFP assay is a functional assay and standardized to recombinant IFN-α reported in terms of interferon activity (activity units, or U/mL). ELISA results are standardized by quantity (mass, or pg/mL) independent of the given activity of the detected IFN protein. Specific activity of the recombinant IFN-α used as a standard for the 293T-ISRE-RFP assay was approximately 4.3 units per mg of protein, but the activity of IFN-α standards supplied with the ELISA kits used was unspecified. While significant IFN-α was detected by ELISA, IFN-β was not detected in any wells by a separate IFN-β ELISA (data not shown).
Discussion
Immune stimulation is a complex process involving multiple cell types and effector molecules that can be difficult to reconstitute completely in vitro. We report here a new method for high throughput screening of isRNA delivery utilizing cell-based in vitro assays for both innate immune activation and detection of responses. We have chosen to focus on measuring type I IFNs as secretion of these cytokines are a prominent component of isRNA-mediated innate immune stimulation through interaction with TLR7, TLR8 (in humans), and RLRs (Garcia-Sastre and Biron 2006; Hornung et al. 2005; Hornung et al. 2004; Marques and Williams 2005; Sioud 2006). A major component of siRNA-mediated immunostimulation is production of IFN-α by PDCs, which typically constitute 0.5% to 0.8% of the human PBMC population (Supplemental Fig. 4). As demonstrated in Fig. 4b, PDCs are sufficient and required to generate detectable levels of type I IFN in vitro in response to a positive control isRNA (siβgal). We have observed that type I IFN response of PBMCs to immunostimulatory siRNA appears to be limited to production of IFN-α (Supplemental Fig. 3) as no IFN-β was detected by ELISA (data not shown), which is consistent with previous reports of siRNA stimulation specifically of PDCs through TLR7 (Hornung et al. 2005; Sioud 2006). Similarly, TLR7 and TLR9 agonists induced large amounts of type I IFN, but PBMC responses to a TLR8 agonist were not detected (Fig. 4a) implicating the crucial role of PDC activation in generating IFN-α responses. Although other typical isRNA responses may not be detected by the 293T-ISRE-RFP system, such as secretion of TNF-α from monocytes following engagement of TLR8 (Judge et al. 2005), measurement of type I IFN secretion is an appropriate surrogate for investigating isRNA-mediated stimulation of the innate immune system.
Detection of IFN has traditionally relied upon ELISA and similar methods based upon the specific interaction of antibodies. While ELISA is highly sensitive for detection and quantification, this methodology is time consuming, requires multiple steps, relies on antibodies (and is hence expensive), and typically exhibits 2–3 logs of dynamic range. Additionally, each ELISA usually detects only 1 subtype of IFN due to the low cross-reactivity of antibodies. The cell-based assay described herein addresses a number of limitations associated with ELISA-based assays. Through optimization of parameters for mRFP signal detection by flow cytometry (Fig. 2) and subsequent data analysis (Fig. 3), we can readily quantify the presence of even low amounts of both IFN-α and IFN-β through engagement of the IFNAR on the 293T-ISRE-RFP cell line. A list of advantages and disadvantages of this cell-based assay compared to standard ELISA is presented in Table I.
Table I.
Comparison of ELISA assay with 293T-ISRE-RFP assay
| Consideration | ELISA | 293T-ISRE-RFP | ||
|---|---|---|---|---|
| Fixed Costs | $ | Plate reader | $$$$ | HTS FACS Machine * |
| Disposable Costs | $$$$ | Antibodies, plates, reagents | $ | culture media and plates |
| Workflow | + | 4–6 man-hours per plate | ++++ | <1 man-hour per plate |
| Specificity | ++++ | mAb mediated ** | ++ | Ubiquitous Type 1 IFN receptor |
| Detection Limit | ++++ | <12.5 pg/mL | ++++ | <2.5 U/mL *** |
| Dynamic Range | ++ | 12.5 – 5000 pg/mL | ++++ | 2.5 – 100,000 U/mL |
| HTS compatible | Cost- and time-prohibitive | Yes |
FACS costs can be distributed over institution
Multitype ELISA for human IFN-α available; multiplex assays also possible
1 U ~ 4.3 pg recombinant IFN-α for samples tested
By merely changing the method of analysis of flow cytometry data, the same data set can be used to produce either low-range or high-range detection. We have achieved femtomolar detection limits while preserving quantification of type I IFN activity over a 5-log range. Utilization of a yellow laser, which emits light with a wavelength much closer to the absorption peak of mRFP (Campbell et al. 2002), was unable to increase detection limits (data not shown). Additionally, attempts at using fluorescent plate readers for quantification of mRFP were only successful at very high levels of mRFP expression (data not shown). Thus, the low detection limit of the 293T-ISRE-RFP assay is a result of the ability to distinguish weak RFP signal from autofluorescence at low IFN concentrations.
An added benefit of quantification using a cell-based assay, which relies on ligand-receptor activation, is the ability to simultaneously detect and integrate signals from multiple interferons including IFN-α and IFN-β (Fig. 3c) and IFN-ω (Supplemental Fig. 1). While it may be desirable at times to differentiate the individual signals, in the case of HTS screening the reduction of general type I IFN response into a single output may be more efficient than the multiple parallel assays that would be required using standard ELISA.
One drawback to the use of the 293T-ISRE-RFP assay is an observed discrepancy of detected IFN levels with ELISA. Although the detection of IFN correlates perfectly from well to well, a larger quantity of IFN activity was detected in supernatants from isRNA-transfected PBMC by cell-based assay than by ELISA (Supplemental Fig. 3). One possible explanation for this discrepancy is that the 293T-ISRE-RFP assay detects multiple type I IFNs (IFN-α, IFN-β, IFN-ω), while ELISA only detects one type of IFN (IFN-α). We investigated whether our results with the cell-based assay were skewed by the presence of an additional IFNAR ligand (IFN-β) in the supernatants, but no IFN-β was detected by ELISA (data not shown). Thus integrating the concentrations detected by independent ELISA does not recapitulate the concentrations as detected by the 293T-ISRE-RFP assay. Another possible explanation is the presence of an additional type I IFN not recognized by the ELISA kits we have employed in our investigations, such as IFN-ω. In addition, there are other possible cytokines which may activate a receptor-mediated Jak/Stat pathway that can increase mRFP expression through the ISREs. For example, type III IFNs have been shown to play an important role in antiviral immunity (Ank et al. 2008). It is possible that the 293T-ISRE-RFP cell has receptors for type III IFNs or other cytokines that lead to mRFP production. We have not characterized 293T-ISRE-RFP activation beyond IFN-α, IFN-β, and IFN-ω. Still, responses to TLR agonists that do not result in type I IFN production in PBMCs were not detected (Fig. 4a). Finally, we hypothesized whether leftover lipidoid-siRNA nanoparticles in the supernatant from PBMCs could directly transfect 293T cells to activate mRFP through engaging the RIG-I receptor. Direct transfection with up to 400 ng/well ( siRNA in complex with lipidoids or lipofectamine 2000 did not result in mRFP production (data not shown). While detection using the 293T-ISRE-RFP cell-based assay cannot replace ELISA detection when knowledge of specific IFN type is necessary, it is very useful for integrated detection of total type I IFN activity.
The development of a cell-based assay allows for the simple and cost-effective high throughput screening of novel materials for isRNA-mediated immune stimulation in vitro in a 3-step process (Fig. 5a). The utility of this process is not limited to only isRNA delivery, and could be applied to investigate the incompletely understood structure-function relationships of modified siRNAs and TLR interactions (Eberle et al. 2008; Judge et al. 2006), sequence-dependent interactions with TLRs (Hornung et al. 2005; Judge et al. 2005), or other investigations where detection of type I IFNs can be used as a surrogate for these processes.
As a proof of concept, we investigated the isRNA delivery properties of 10 novel materials developed originally for non-viral delivery of nucleic acids. Screening revealed distinct isRNA delivery activity for some lipidoid materials that was RNA-specific for siβgal (ND98-(5)1-(5)1, NC98-(5)1, and NA116-3) as well as high activity for both siβgal and siGFP with the NC100-3 lipidoid (Figs. 4b and 4c). These observations are an example of the potential information that could be gained from high throughput screening for isRNA delivery, namely assigning structure-function relationships to a library of materials. The other lipidoid cores (110, 111, 112, and 114) are all closely-related chemical iterations (Supplemental Fig. 2) that might a priori be expected to result in similar levels of isRNA activity. However, without a suitable method for in vitro screening, in vivo investigations would be the only way to determine the immunostimulatory properties of these materials. Our results demonstrate that delivery of siRNA with some materials is in fact not immunostimulatory in vitro (Fig. 5b) and highlights the utility of screening for isRNA activity.
Lipidoids based on the 98 amine core (Supplemental Fig. 2b) have been found to be very useful for delivery of siRNA molecules for RNAi both in vitro and in vivo (Akinc et al. 2008). Here we find that these materials also exhibit strong isRNA delivery potential (Figs. 4b and 5). Immune stimulation by TLR7/8 RNA agonists requires endosomal localization (Sioud 2005) where TLR7 and 8 are functional, and the final location of RNA delivery may explain the lack of isRNA properties observed with some delivery agents. Consequently, it appears ND98-(5)1-delivered siRNA can reach at least two intracellular sites of action, the late endosome to achieve isRNA activity and cytosol where RNA interference occurs. The dual nature of ND98-(5)1-delivered siRNA provides insight into the uptake mechanism. Some ND98-(5)1-siRNA nanoparticles must remain in the endosome or isRNA activity would not be as pronounced as it is, but delivery to the cytosol must also be robust in order to sustain an RNAi effect as previously observed (Akinc et al. 2008). Of note, both ND98-(5)1and NC98-(5)1 particles exhibited isRNA delivery potential. These materials both have the same 98 core with 5 alkyl-acrylamide tails 12 carbons in length for ND98-(5)1 and 11 carbons in length for NC98 (Akinc et al. 2008).
The differences between induction of type I IFN exhibited with the siβgal and the siGFP sequences clearly indicates that most of the lipidoids tested do not directly activate IFN responses. Interestingly, the NC100-3 compound exhibited interferon responses with both siRNAs, giving an example of the importance of the control siGFP sequence. It is possible that highly efficient delivery of siGFP to the endosome in the PDC could increase TLR-mediated responses independent of sequence. We are currently undertaking further investigations to differentiate this possibility from direct lipidoid-mediated activation of the interferon response or another mechanism such as toxicity. Utilizing synthetic siRNA incorporating chemical modifications such as 2′-O-methyl or 5-fluoro-U has been shown to abrogate TLR mediated interferon responses (Eberle et al. 2008; Robbins et al. 2008) and may provide additional insight in this screening system as a further control.
Most methods for controlling innate immune responses to siRNAs have focused on siRNA design of sequence and structure (Pei and Tuschl 2006; Sioud 2007). Because the site of immunostimulatory action in response to siRNA is indeed the endosome and not the cytosol or nucleus, controlling the delivery of RNA molecules preferentially to the endosome or cytosol can be an additional method to modulate the immunostimulatory nature of siRNA molecules. Low isRNA activity may be due to low uptake by PDCs, lack of delivery to TLRs in the endosome, or low overall delivery potential. Materials exhibiting low isRNA activity may be preferentially selected for further development for therapeutic siRNA delivery; materials exhibiting high activity may be useful for therapeutic isRNA delivery. By applying in vitro screening for isRNA delivery to the complete lipidoid library (Akinc et al. 2008), we hope to discover novel materials and elucidate design criteria that will allow control of immunostimulatory RNA potential via drug delivery.
Conclusions
The delivery of RNA molecules holds great therapeutic promise to regulate both gene function and immune activation. Combinatorial chemistry has been utilized by our group and others to explore new materials for RNA delivery for siRNA therapeutics. The immunostimulatory potential of siRNAs as it relates to intracellular location is an important consideration in the rational design and iteration of these materials. However, efficient screening methods for detecting immune stimulation by siRNAs have not been available. Our new method based upon cell-mediated detection of the RNA-induced type I IFN response is sensitive, quantitative, correlates well with conventional ELISA-based approaches, and high-throughput compatible. This method is not limited to isRNA delivery and may be applicable in a wide range of investigations of innate immune responses. By utilizing a highly immunostimulatory sequence and a control sequence with low isRNA potential, we developed a standardized method for determining isRNA delivery potential. We have applied this method in a proof-of-concept screening of a combinatorial material class for isRNA delivery. We characterized differential activation of IFN secretion depending on material chemistry. Although the results of screening of just 10 materials is not sufficient to infer all the complex structure-function relationships surrounding isRNA delivery, we have shown that the method outlined herein can be an important tool for defining materials properties in RNA drug delivery. Further characterization of the lipidoid library and other materials used for RNA delivery using the 293T-ISRE-RFP system may provide valuable insights into structure-function relationships of novel materials with the innate immune system.
Supplementary Material
Acknowledgments
The authors wish to acknowledge the expert help of Glenn Paradis of the David H. Koch Institute for Integrative Cancer Research Flow Cytometry Core Facility, valuable discussions with Prof. Jianzhu Chen, and funding support from NIH Grants EB000244, U19AI62623, and U54AI57158.
References
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, Bacallado SA, Nguyen DN, Fuller J, Alvarez R, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26(5):561–9. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, et al. An important role for type III Interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180(4):2474–85. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006;13(4):644–70. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99(12):7877–82. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle F, Giessler K, Deck C, Heeg K, Peter M, Richert C, Dalpke AH. Modifications in Small Interfering RNA That Separate Immunostimulation from RNA Interference. J Immunol. 2008;180(5):3229–3237. doi: 10.4049/jimmunol.180.5.3229. [DOI] [PubMed] [Google Scholar]
- Furset G, Sioud M. Design of bifunctional siRNAs: combining immunostimulation and gene-silencing in one single siRNA molecule. Biochem Biophys Res Commun. 2007;352(3):642–9. doi: 10.1016/j.bbrc.2006.11.059. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312(5775):879–82. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN, Chen J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci U S A. 2003;100(5):2718–23. doi: 10.1073/pnas.0437841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Mahon K, Anderson D. Combinatorial and rational approaches to polymer synthesis for medicine. Adv Drug Deliv Rev. 2008;60(9):971–8. doi: 10.1016/j.addr.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11(3):263–70. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168(9):4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, Hartmann G. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. 2004;173(10):5935–43. doi: 10.4049/jimmunol.173.10.5935. [DOI] [PubMed] [Google Scholar]
- Hubbell JA. Biomaterials science and high-throughput screening. Nat Biotechnol. 2004;22(7):828–9. doi: 10.1038/nbt0704-828. [DOI] [PubMed] [Google Scholar]
- Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. 2006;13(3):494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23(4):457–62. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Kabanov AV. Polymer genomics: an insight into pharmacology and toxicology of nanomedicines. Adv Drug Deliv Rev. 2006;58(15):1597–621. doi: 10.1016/j.addr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591–7. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn J. New approaches to biomaterials design. Nat Mater. 2004;3(11):745–747. doi: 10.1038/nmat1249. [DOI] [PubMed] [Google Scholar]
- Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. 2005;23(11):1399–405. doi: 10.1038/nbt1161. [DOI] [PubMed] [Google Scholar]
- Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2006;80(18):9192–9. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EG, Taylor JM. Small-interfering RNAs in the radar of the interferon system. Nat Cell Biol. 2003;5(9):771–2. doi: 10.1038/ncb0903-771. [DOI] [PubMed] [Google Scholar]
- Pei Y, Tuschl T. On the art of identifying effective and specific siRNAs. Nat Methods. 2006;3(9):670–6. doi: 10.1038/nmeth911. [DOI] [PubMed] [Google Scholar]
- Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, et al. 5′-triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008 doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- Robbins M, Judge A, Ambegia E, Choi C, Yaworski E, Palmer L, McClintock K, Maclachlan I. Misinterpreting the therapeutic effects of siRNA caused by immune stimulation. Hum Gene Ther. 2008 doi: 10.1089/hum.2008.131. [DOI] [PubMed] [Google Scholar]
- Scheel B, Aulwurm S, Probst J, Stitz L, Hoerr I, Rammensee HG, Weller M, Pascolo S. Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol. 2006;36(10):2807–16. doi: 10.1002/eji.200635910. [DOI] [PubMed] [Google Scholar]
- Sioud M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J Mol Biol. 2005;348(5):1079–90. doi: 10.1016/j.jmb.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Sioud M. Innate sensing of self and non-self RNAs by Toll-like receptors. Trends Mol Med. 2006;12(4):167–76. doi: 10.1016/j.molmed.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Sioud M. RNA interference and innate immunity. Adv Drug Deliv Rev. 2007;59(2–3):153–63. doi: 10.1016/j.addr.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Stram Y, Kuzntzova L. Inhibition of viruses by RNA interference. Virus Genes. 2006;32(3):299–306. doi: 10.1007/s11262-005-6914-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood A, Elvin SJ, Healey GD, Williamson ED, Eyles JE. Immunological responses after immunisation of mice with microparticles containing antigen and single stranded RNA (polyuridylic acid) Vaccine. 2006;24(11):1736–43. doi: 10.1016/j.vaccine.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Xiao T. Innate immune recognition of nucleic acids. Immunol Res. 2008 doi: 10.1007/s12026-008-8053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Marques JT, Gantier MP, Behlke MA, John M, Rayman P, Finke J, Williams BR. Determinants of cytokine induction by small interfering RNA in human peripheral blood mononuclear cells. J Interferon Cytokine Res. 2008;28(4):221–33. doi: 10.1089/jir.2007.0090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.