

Abstract.
The cell nucleus is a highly structured compartment where nuclear components are thought to localize in non-random positions. Correct positioning of large chromatin domains may have a direct impact on the localization of other nuclear components, and can therefore influence the global functionality of the nuclear compartment. DNA methylation of cytosine residues in CpG dinucleotides is a prominent epigenetic modification of the chromatin fiber. DNA methylation, in conjunction with the biochemical modification pattern of histone tails, is known to lock chromatin in a close and transcriptionally inactive conformation. The relationship between DNA methylation and large-scale organization of nuclear architecture, however, is poorly understood. Here we briefly summarize present concepts of nuclear architecture and current data supporting a link between DNA methylation and the maintenance of large-scale nuclear organization.
Keywords. Epigenetic, DNA methylation, nuclear architecture, chromosome territory, gene positioning
Introduction
The cell nucleus is the most prominent compartment in the eukaryotic cell. In this compartment many essential cellular activities take place, such as genome replication, control of gene expression and transcription, processing of transcripts and DNA repair. For a long time, however, the cell nucleus has been considered as a static and scarcely structured compartment. The nucleus was thought to be substantially altered only during cell division, following formation of metaphase chromosomes and partitioning of the chromosome complement into daughter cells. Another common image of the nuclear compartment has been that of a contingent cellular region where nuclear components are randomly located. This static view of the cell nucleus has drastically changed in recent years mainly due to technical advances in the field of microscopy. The cell nucleus is presently considered as a highly complex and organized compartment where nuclear components tend to occupy non-random positions, leading to a precise definition of the nuclear architecture concept (see [1–3] for recent reviews). The cell nucleus is an extremely dynamic structure where many components tend to rapidly and transiently interact with each other, giving rise to a highly ordered compartment. As these properties characterize open and self-replicating chemical systems, the idea of the cell nucleus as a self-organizing entity has been proposed [2].
The double strand of DNA, a major component of chromatin, and therefore of the nuclear compartment, can udergo chemical modification in the form of cytosine methylation of CpG dinucleotides. This biochemical modification does not alter the subjacent genetic information of the DNA molecule, and is considered to be an epigenetic mark in the genome. The impact of such DNA epigenetic modification within the scope of nuclear function and disease has been the subject of intense investigation (see [4–6] for recent reviews). Such work has clearly established that proper DNA methylation in definite regions of the genome, such as gene promoters and large repetitive sequences, is essential for the precise and orchestrated regulation of gene expression. However, the role of DNA methylation in maintenance of large-scale nuclear organization remains poorly understood. The aim of this review is to briefly summarize current knowledge about nuclear architecture and to discuss the potential role of epigenetic modification of DNA in the morphological and functional maintenance of global organization of the nuclear compartment.
The concept of nuclear architecture
Nuclear architecture is the result of the morphological and functional heterogeneity generated by the positioning of different subnuclear compartments inside the nucleus. A subnuclear compartment has been defined as a macroscopic region within the nucleus that is morphologically and/or functionally distinct from its surrounding [2]. Two types of subnuclear compartments are usually considered, nuclear bodies and chromosome territories, including associated chromatin domains.
Nuclear bodies are distinct subnuclear regions of different sizes lacking a lipidic membrane and usually characterized by a definite protein composition. The most prominent nuclear body is the nucleolus, factory for ribosome biogenesis and site of RNA Pol I-dependent rDNA gene transcription. There are many other nuclear bodies exclusively characterized by the presence of one or more specific proteins [7]. Of special relevance are the Cajal bodies, proposed site of snRNP assembly [8], and PML bodies, of unknown function and the main containers of the promyelocytic protein [9]. The existence of thousands of RNA Pol II ‘transcription factories’ dispersed in the nucleoplasm of mammalian cells has been also well documented [10, 11].
The chromatin bulk corresponding to each particular chromosome is not randomly distributed in the nuclear compartment but occupies a specific location known as chromosome territory or domain (Fig. 1), a feature which constrains the whole spatial organization inside the nuclear compartment. It has been proposed that chromosome territories are not compact structures. They are thought to be permeated by nucleoplasmic channels, creating a porous entity of enlarged surface area which is accessible to different nuclear factors [12]. Large-scale chromatin domains, belonging to one or more chromosome territories, are also morphologically defined as heterochromatic, highly condensed, genomic regions or as euchromatic, less-condensed regions.
Figure 1.
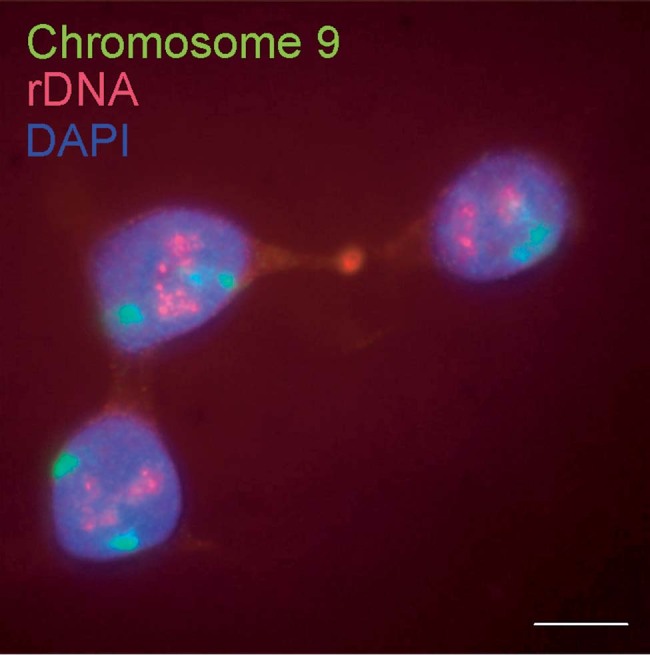
Localization by fluorescence in situ hybridization of two nuclear domains, the chromosome territories of the q-arm of chromosome 9 (green) and the nucleolar localization of rDNA gene repeats (red), in human HCT116 cells. Bar, 10 µm.
There are numerous examples showing that the nuclear compartment is non-randomly arranged in three-dimensional spaces. Polarization of chromosomes, where centromeres and telomeres occupy opposite locations in the nucleus (Rabl orientation), commonly takes place in Drosophila cells [13, 14]. Several reports also indicate a precise positioning of chromosome territories and chromatin domains relative to a radial orientation in mammalian cells [15–22]. In some cases the radial position has been correlated to the chromosome’s gene density [15–20], and in other cases to chromosome size [21, 22]. Positioning of chromosome territories relative to other chromosome territories has been also reported [23], and certain chromatin domains, such as heterochromatic regions, tend to associate to the nuclear membrane and to the nucleolus. Evidence also exists for a non-random positioning of some nuclear bodies. Formation of the nucleolar compartment is the result of a non-random association driven by RNA Pol I activity of several copies of tandemly repeated rDNA genes arising from different chromosomes. In the same way, Cajal bodies tend to associate to U2 snRNA (small nucleolar RNA)gene clusters [24–26], while PML bodies are preferentially found near active genes [27, 28]. In addition, PML and Cajal bodies are preferentially located outside chromosome territories [17].
Positioning of nuclear compartments is not exactly the same for all the cells in any particular model. In this sense, it is thought that chromosome positioning is not heritable, but rather established de novo at early G1 in human cells [29, 30], although global transmission of chromosome positions through mitosis has been reported in rat cells [31]. Positioning acquired in G1 is further maintained in subsequent interphase stages. In addition, chromosome positioning has been shown to be tissue and cell type specific [17, 19, 32]. Thus, non-random post-cell division positioning of nuclear compartments is viewed as a consequence of a stochastic and probabilistic process which results in fully functional organization of the nuclear compartment. It is assumed that this spatial organization should be broadly similar and equivalent for all growing cells in a cell or tissue type in order to maintain functional organization of the cell nucleus, which is the basis for a functional definition of nuclear architecture.
Nuclear architecture, gene positioning and gene function
The above observations question the importance of the nuclear architecture’s role in gene positioning and function. Many gene loci tend to localize inside their corresponding chromosome territories and have strong preferential positioning with respect to the nuclear centre [33]. This positioning is not directly related to gene activity, and probably reflects the nonrandom location of the corresponding chromosome territory. In some cases, such as CD4 locus activation during T-cell differentiation, gene activation implicates gene repositioning to the periphery of the correspondent chromosome domain [34].
Interestingly, large loops of chromatin protruding several microns from the resident chromosome territory have been described in mammalian cells (reviewed in [35]). These loop domains contain clusters of actively transcribed genes. For example, in differentiating ES cells, Hox1 and Hox9 genes loop from their chromosome territories upon activation [36], and two genes located 25 Mb away on chromosome 7 closely localize to pair and share a common transcriptional site [37]. These observations suggest that loops containing activated or transcriptionally competent genes are expelled from or moved to the external surface of chromosome territories to be near transcription factories. Assuming that chromosome territories are porous entities, acquisition of a transcriptionally competent state could be achieved by free diffusion of nuclear factors [12].
On the other hand, there are well-known examples of gene silencing after repositioning near or inside transcriptionally repressed heterochromatin domains. In the classical position effect variegation phenomenon (PEV), a gene locus becomes permanently silenced after placement near a heterochromatic domain [38]. A similar effect of transcriptional repression associated with gene repositioning close to heterochromatin blocks has been reported in many naturally occurring differentiation systems [39], although association with heterochromatin does not always result in gene inactivation [40]. It is clear that heterochromatin is characterized by a compacted chromatin structure in which transcription is inhibited and that loss of heterochromatinization may result in gene activation. However, repositioning of inactivated genes into heterochromatin domains might be a consequence rather than a cause of gene inactivation, and heterochromatin might be a sink for transcription factors rather than a repository of inactivated genes. In this sense, it has been recently shown that the correlation between chromatin structure and gene activity is not as strong as previously perceived [20]. In fact, a strong relationship was found between chromatin structure and gene density, whereas open chromatin regions were found to be enriched in gene loci, active or not, while condensed chromatin domains were associated with poor gene content [20].
Perhaps the most direct indication of the role of nuclear architecture in gene function is in disease states, which are very often characterized by altered gene expression patterns associated with aberrant nuclear morphologies, or vice versa. An immediate example is cancer. Many cancer cell types exhibit gross alterations of the nuclear architecture in the form of spatial organization changes, chromatin and chromosome domain textures, nuclear size and shape alterations, and changes in the number and size of nucleoli (summarized in [42]). In fact, morphological abnormalities of the nuclear compartment are used as key diagnostic features for many cancer types [42].
Other well-known examples of changes in gene expression associated with alterations in nuclear architecture are laminopathies [43]. These severe diseases are characterized by the loss of A-type lamin function, a major structural component of the nuclear envelope. As a consequence, the nuclear envelope is distorted and the whole nuclear organization is compromised. At the level of the organism level, patients suffering from laminopatic syndromes manifest muscular dystrophy, lipodystrophy, neurodystrophy and progeroid disorders.
DNA methylation machinery
Methylation of CpG dinucleotides is characterized by the transfer of methyl groups to the C-5 position of cytosine (5mC), and is catalyzed by members of the DNA methyltransferase (DNMT) protein family. To date, three families of DNMTs have been identified, DNMT1, DNMT2 and DNMT3 (DNMT3a DNMT3b and DNMT3L) [44]. DNMT1, DNMT3a, and DNMT3b are essential during the development of murine knockout models [44–46]. DNMT1, the most abundant DNA methyltransferase in somatic cells, has a strong preference for hemimethylated DNA, and is therefore believed to be the enzyme primarily responsible for copying and maintaining methylation patterns from the parental to the daughter strand following DNA replication [45]. DNMT3a and DNMT3b are highly expressed in embryonic and non-differentiated cells and have been proposed to be the enzymes responsible for de novo methylation [46]. Several lines of evidence, however, indicate that in addition to the cooperation between all three DNMTs, they may also possess both de novo and maintenance functions in vivo [47–49]. DNMT2 lacks the large N-terminal regulatory domain common to other eukaryotic methyltransferases and does not exhibit comparable DNA methyltransferase activity [50], although it does seem to have some residual activity in vitro [51]. DNMT3L lacks canonical DNA cytosine-methyltransferase motifs [44].
5mC in normal DNA constitutes 0.75–1% of all nucleotides, where 4–6% of all cytosines are methylated [52]. CpG dinucleotides are not randomly distributed throughout the genome but are enriched in regions known as CpG islands. CpG islands are usually hypomethylated and tend to embrace the 5′-end region (promoter, untranslated region and exon 1) of a wide number of genes [53]. In mammals, two waves of active demethylation of 5mC take place in early steps of embryo development, prior to the formation of a zygotic nucleus in germ cells and in preimplantation embryos [54]. Developmentally regulated re-methylation of specific CpG islands occurs at least in imprinted genes, X-chromosome-linked silenced genes in females, and in germline and tissue-specific genes [55]. Aberrant methylation of CpG islands leading to gene silencing is a common phenomenon during carcinogenesis [4]. Cytosine methylation is also observed outside CpG islands, where it is thought to play a key role in silencing parasitic DNA sequences, such as transposons and retroviruses [56]. It is well established that DNA methylation is associated with transcriptionally inactive states of chromatin, but the exact mechanism by which CpG methylation is translated into transcriptionally silent chromatin is still unclear. Three different hypotheses have been proposed to explain the way by which DNA methylation is interpreted by nuclear factors. The first possibility is that DNA methylation inhibits the binding of sequence-specific transcription factors to their binding sites [57]. CpG methylation would result in transcription factor release from the chromatin fibre. In this context, a protein with an affinity for unmethylated CpGs has been identified that is associated with actively transcribed regions of the genome [58]. In a second model, it is proposed that methylation may have direct consequences for nucleosome positioning, leading to the assembly of specialized nucleosomal structures on methylated DNA able to repress transcription [59]. The third possibility is that methylation results in the recruitment of nuclear factors that selectively recognize methylated DNA and either impede binding of other nuclear factors or have a direct effect on repressing transcription [60].
Although there are examples that support all three possibilities, the active recruitment of methyl-CpG binding activities appears to be the most widespread mechanism of methylation-dependent repression. MeCP1 and MeCP2 were the first two methyl-CpG binding proteins described [60]. It was shown years ago that MeCP2 represses the transcription of methylated DNA through the recruitment of a histone deacetylase-containing complex [61, 62], establishing for the first time a connection between DNA methylation and transcriptional repression. Characterization of MeCP2 led to the identification of a methyl-CpG-binding domain (MBD) [63], resulting in the further characterization of additional methyl-CpG-binding proteins containing this motif, namely MBD1, MBD2, MBD3 and MBD4 [64]. Moreover, it has been demonstrated that DNMTs and MBDs can also recruit histone deacetylases [65, 66] and histone methyltransferases that modify lysine 9 of histone H3 [67–69], a hallmark of heterochromatin. These observations have established a mechanistic link between DNA methylation and changes in the structural conformation of the chromatin fibre.
Epigenetic control of nuclear architecture
There is an emerging view of the eukaryotic nucleus as a three-dimensional region functionally divided into large heterochromatin compartments that repress transcription, and compartments in which transcription is permitted [70]. Accumulated evidence also suggests that a large-scale three-dimensional landscape is maintained in the nucleus by large genomic repeats, such as centromeres or telomeres, and heterochromatin blocks. In a nuclear volume in which small structures and particles move and diffuse following a random pattern [1, 71], large-scale structures should act as reference hallmarks for nuclear activity. In fact, heterochromatic compartments are presently considered as large repositories of repressor factors [1]. Positional or structural modifications of these large-scale hallmarks have key roles in cellular differentiation and transformation [39, 40, 42, 43, 70, 72].
The close relationship between DNA methylation and local chromatin structure is well known. Methylation of CpG nucleotides is associated with closed or compacted chromatin conformations and the formation of heterochromatin. DNA methylation of CpG islands contained in gene promoters results in chromatin compaction and transcriptional inactivation. Compacted chromatin states are also characterized by a well-defined pattern of biochemical modification of histone H3 and H4 tails [5]. Cross-talk between DNA methylation and modification of histone tails has been established in plants and animals [73–75], indicating that transition to the closed chromatin conformation is a coordinated phenomenon involving both DNA and histones. However, the precise role of DNA methylation in the maintenance of chromosome positioning and large-scale nuclear architecture is poorly understood. In any case, a few indicative examples are found in the literature. For example, in germinating wheat seeds, treatment with 5-azacytidine, which results in DNA hypomethylation, induces strong changes in the architecture of interphase chromosome arms [76]. In human chromosomes, 5-azacytidine treatment results in demethylation of heterochromatic regions [77]. It is also known that changes in nuclear architecture are closely associated with large-scale modification of the DNA methylation pattern during mammalian preim-plantation development [78] and in germ and Sertoli cells from developing mouse testis [79]. Similar changes in nuclear organization associated with changes in the DNA methylation pattern are found during normal development of the peach apical meristem [80]. Finally, chromosome instability and aberrant nuclear morphologies are tightly associated with DNA hypomethylation of discrete nuclear regions in cancer cells [81–83]. All these observations point out a causal relationship between DNA methylation, DNA methylation machinery and large-scale nuclear organization.
For the most part, dense DNA methylation regions in mammalian cultured cells can be spotted on discrete locations on metaphase chromosomes, such as secondary constrictions, juxtacentromeric regions and T-bands [84]. In the interphase nuclei, densely methylated DNA regions are found in discrete foci, frequently associated with the nuclear envelope and with heterochromatic regions (Fig. 2a and [75]). The distribution of 5mC in discrete heterochromatic foci associated with the nuclear envelope is best observed in tissues, where the spatial and functional organization of the nuclear architecture is constrained by the three-dimensional network of cell-cell and cell-substrate interactions that are required to maintain the homeostasis of the tissue (Fig. 2b). Many of these densely methylated regions correspond to large repetitive regions in the genome. In humans, such repetitive regions are typically found in classical satellites 2 and 3 at juxtacentromeric regions of chromosomes 1, 9 and 16. The ICF syndrome (for immunodeficiency, centromere instability and facial anomalies) is a recessive autosomal disorder involving abnormalities of genomic methylation patterns and mutations in both alleles of the DNMT3B gene [85]. ICF patients shown complete demethylation of specific repetitive sequences contained in satellites 2 and 3. This demethylation pattern is associated with decondensation of large blocks of juxtacentromeric heterochromatin, formation of multiradiate chromosome and gross alteration on the nuclear architecture in interphase nuclei [85]. Human cancer cells lacking both copies of the DNMT1 gene also show extensive and specific demethylation of satellite 2 repeats at chromosomes 1, 9 and 16 [75]. Concomitantly, these cells present distorted nuclear architecture and loss of heterochromatic organization [75]. Human cells lacking DNMT1 also show a specific demethylation pattern in a second type of genomic repeat, the rDNA genes [75]. Interestingly, these cells present profound disorganization of the nucleolar compartment [75]. These observations indicate that the DNA methylation machinery, which is required to maintain a specific pattern of methylation in large regions of the genome, is also required to maintain a particular organization of the nuclear architecture.
Figure 2.
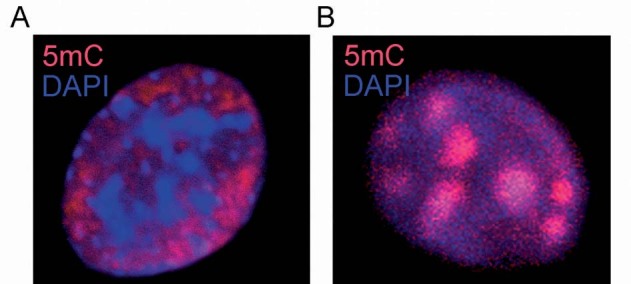
Confocal images showing the distribution of 5mC in the nucleus of (a) primary mouse fibroblasts and (b) keratinocytes of the interfollicular epithelium in a whole mount of mouse tail skin. Bars, 5 µm.
Interestingly, neither human cells lacking DNMT1 nor cells lacking DNMT3B show significant alterations in the DNA methylation pattern of promoter-contained CpG islands [47, 48]. In this scenario, an epigenetic modification of the chromatin fibre, specifically affecting large blocks of genomic repeats contained in heterochromatic regions, results in gross alterations of nuclear architecture. However, no significant changes are observed at the promoter level of regulation of gene expression. Since the output of these cells is a functionally altered state, it is tempting to speculate that large alterations of nuclear architecture have a direct effect on cell function. This observation constitutes, in turn, a change in common concepts of nuclear function, in which alterations of nuclear architecture are the result, rather than the cause, of dysfunction in local gene activities.
Albert Einstein famously said, ‘God does not play dice’. What Einstein was referring to was his own rejection of a chaotic universe. The increasing scientific amount of data obtained in recent years also shows that our DNA, chromosome and nuclear structure is not a random event occurring in the cell. There is a delicate superstructure of large chromatin domains, chromosomal territories and subnuclear compartments that require reliable, but, at the same time, dynamic caretakers. Epigenetic marks, such as DNA methylation and histone modification, are excellent candidates to assume this critical role.
Footnotes
Received 3 August 2006; received after revision 26 September 2006; accepted 22 November 2006
References
- 1.Taddei A., Hediger F., Neumann F. R., Gasser S. The function of nuclear architecture. A genetic approach. Annu. Rev. Genet. 2004;38:305–345. doi: 10.1146/annurev.genet.37.110801.142705. [DOI] [PubMed] [Google Scholar]
- 2.Misteli T. Concepts in nuclear architecture. Bio Essays. 2005;27:477–487. doi: 10.1002/bies.20226. [DOI] [PubMed] [Google Scholar]
- 3.Foster H. A., Bridger J. M. The genome and the nucleus: a marriage made by evolution. Genome organisation and nuclear architecture. Chromosoma. 2005;114:212–229. doi: 10.1007/s00412-005-0016-6. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Aberrant DNA methylation as a cancerinducing mechanism. Annu. Rev. Pharmacol. Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 5.Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr. Opin. Genet. Devel. 2005;15:490–495. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 7.Matera A.G. Nuclear bodies:multifaceted subdomains of the interchromatin space. Trends Cell Biol. 1999;9:302–309. doi: 10.1016/S0962-8924(99)01606-2. [DOI] [PubMed] [Google Scholar]
- 8.Stanek D., Neugebauer K. M. Detection of snRNP assembly intermediates in Cajal bodies by fluorescence resonance energy transfer. J. Cell Biol. 2004;166:1015–1025. doi: 10.1083/jcb.200405160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong S., Salomoni P., Pandolfi P. P. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2000;2:E85–E90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 10.Iborra F. J., Pombo A., Jackson D. A., Cook P. R. Active RNA polymerases are localized within discrete transcription ‘factories’ in human nuclei. J. Cell Sci. 1996;109:1427–1436. doi: 10.1242/jcs.109.6.1427. [DOI] [PubMed] [Google Scholar]
- 11.Jackson D. A., Iborra F. J., Manders E. M., Cook P. R. Numbers and organization of RNA polymerases, nascent transcripts, and transcription units in HeLa nuclei. Mol. Biol. Cell. 1998;9:1523–1536. doi: 10.1091/mbc.9.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cremer T., Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 13.Hochstrasser M., Mathog D., Gruenbaum Y., Saumweber H., Sedat J.W. Spatial organization of chromosomes in the salivary gland nuclei of Drosophila melanogaster . J. Cell Biol. 1986;102:112–123. doi: 10.1083/jcb.102.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiraoka Y., Agard D.A., Sedat J.W. Temporal and spatial coordination of chromosome movement, spindle formation, and nuclear envelope breakdown during prometaphase in Drosophhila melanogaster embryos. J. Cell Biol. 1990;111:2815–2828. doi: 10.1083/jcb.111.6.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croft J. A., Bridger J. M., Boyle S., Perry P., Teague P., Bickmore W. Differences in the localization and morphology of chromosomes in human cells. J. Cell Biol. 1999;145:1119–1131. doi: 10.1083/jcb.145.6.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridger J.M., Boyle S., Kill I. R., Bickmore W. Remodelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr. Biol. 2000;10:149–152. doi: 10.1016/S0960-9822(00)00312-2. [DOI] [PubMed] [Google Scholar]
- 17.Boyle S., Gilchrist S., Bridger J. M., Mahy N. L., Ellis J. A., Bickmore W. The spatial organization of human chromosomes within the nucleus of normal and emerin-mutant cells. Hum. Mol. Genet. 2001;10:211–219. doi: 10.1093/hmg/10.3.211. [DOI] [PubMed] [Google Scholar]
- 18.Tanabe,H, Muller S., Neusser M., von Hase J., Calcagno E., Cremer M., Solovei I., Cremer C., Cremer T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl Acad. Sci. USA. 2002;99:4424–4429. doi: 10.1073/pnas.072618599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cremer M., Kupper K., Wagler B., Wizelman L., von Hase J., Weiland Y., Kreja L., Diebold J., Speicher M. R., Cremer T. Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 2003;162:809–820. doi: 10.1083/jcb.200304096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilbert N., Boyle S., Fiegler H., Woodfine K., Carter N. P., Bickmore W. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2005;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Sun H. B., Shen J., Yokota H. Size-dependent positioning of human chromosomes in interphase nuclei. Biophys. J. 2000;79:184–190. doi: 10.1016/S0006-3495(00)76282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolzer A., Kreth G., Solovei I., Saracoglu K., Fauth C., Müller S., Eils R., Cremer C., Speicher,M.R, Cremer T. Complete 3D maps of chromosome positions in human male fibroblast nuclei and prometaphase rosettes demonstrate a chromosome size dependent, probabilistic arrangement. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parada L., McQueen P., Misteli T. Conservation of relative chromosome positioning in normal and cancer cells. Curr. Biol. 2002;12:1692. doi: 10.1016/S0960-9822(02)01166-1. [DOI] [PubMed] [Google Scholar]
- 24.Schul W., van Driel R., de Jong L. Coiled bodies and U2 snRNA genes adjacent to coiled bodies are enriched in factors required for snRNA transcription. Mol. Biol. Cell. 1998;9:1025–1036. doi: 10.1091/mbc.9.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shopland L. S., Byron M., Stein J. L., Lian J. B., Stein G. S., Lawrence J.B. Replication-dependent histone gene expression is related to Cajal body (CB) association but does not require substantial CB contact. Mol. Biol. Cell. 2001;12:565–576. doi: 10.1091/mbc.12.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frey M. R., Matera A. G. RNA-mediated interaction of Cajal bodies and U2 snRNA genes. J. Cell Biol. 2001;154:499–509. doi: 10.1083/jcb.200105084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shopland L. S., Johnson C.-V., Byron M., McNeil J., Lawrence J. B. Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighbourhoods. J. Cell Biol. 2003;162:981–990. doi: 10.1083/jcb.200303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., Shiels C., Sasieni P.,Wu,^P. J. Islam,^S.A., Freemont P. S., Sheer D. Promyelocytic leukaemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Biol. 2004;164:515–526. doi: 10.1083/jcb.200305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter J., Schermell L., Cremer M., Tashiro S., Cremer T. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 2003;160:685–697. doi: 10.1083/jcb.200211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson I., Gilchrist S., Bickmore W., Chubb J. R. The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 2004;14:166–172. doi: 10.1016/j.cub.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 31.Gerlich N., Beaudouin J., Kalbfuss B., Daigle N., Eils,R, Ellenberg J. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell. 2003;112:751–764. doi: 10.1016/S0092-8674(03)00189-2. [DOI] [PubMed] [Google Scholar]
- 32.Parada L., McQueen P., Misteli T. Tissue-specific spatial organization of genomes. Genome Biol. 2004;7:R44. doi: 10.1186/gb-2004-5-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozubek S., Lukasova E., Jirsova P., Koutna I., Kozubek M., Ganova A., Bartova E., Falk M., Pasekova R. 3D structure in human genome: order in randomness. Chromosoma. 2002;111:321–331. doi: 10.1007/s00412-002-0210-8. [DOI] [PubMed] [Google Scholar]
- 34.Kim S. H., McQueen P. G., Litchman M. K., Shevach E. M., Parada L., Misteli T. Spatial genome organization during T-cell differentiation. Cytogen. Genome Res. 2004;105:292–301. doi: 10.1159/000078201. [DOI] [PubMed] [Google Scholar]
- 35.Chambeyron S., Bickmore W. Does looping and clustering in the nucleus regulate gene expression? Curr. Opin. Cell Biol. 2004;16:256–262. doi: 10.1016/j.ceb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Chambeyron S., Bickmore W. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osborne C. S., Chakalova L., Brown K. E., Carter D., Horton A., Debrand E., Goyenechea B., Mitchell J. A., Lopes S., Reik W., et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Gen. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 38.Schotta G., Ebert A., Dorn R., Reuter G. Position-effect variegation and the genetic dissection of chromatin regulation in Drosophila. Semin. Cell Dev. Biol. 2003;14:67–75. doi: 10.1016/S1084-9521(02)00138-6. [DOI] [PubMed] [Google Scholar]
- 39.Kosak S. T., Groudine M. Form follows function: the genomic organization of cellular differentiation. Genes Dev. 2004;18:1371–1384. doi: 10.1101/gad.1209304. [DOI] [PubMed] [Google Scholar]
- 40.Dillon N., Festenstein R. Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 2002;18:252–258. doi: 10.1016/S0168-9525(02)02648-3. [DOI] [PubMed] [Google Scholar]
- 41.Gilbert N., Gilchrist S., Bickmore W. Chromatin organization in the mammalian nucleus. Int. Rev. Cytol. 2005;242:283–336. doi: 10.1016/S0074-7696(04)42007-5. [DOI] [PubMed] [Google Scholar]
- 42.Zink D., Fischer A. H., Nickerson J. A. Nuclear structure in cancer cells. Nature Rev. Cancer. 2004;4:677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- 43.Burke B., Stewart C. L. Life at the edge: the nuclear envelope and human disease. Nat. Rev. Mol. Cell. Biol. 2002;3:575–585. doi: 10.1038/nrm879. [DOI] [PubMed] [Google Scholar]
- 44.Bestor T.H. the DNA methyltransferases of mammals. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 45.Li E., Bestor T. H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-F. [DOI] [PubMed] [Google Scholar]
- 46.Okano M., Bell D. W., Haber D. A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 47.Rhee I., Jair K.W., Yen R.W., Lengauer C., Herman J. G., Kinzler K. W., Vogelstein B., Baylin S. B., Schuebel K. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 48.Rhee I., Bachman K. E., Park B. H., Jair K.W., Yen R.W., Schuebel K., Cui H., Feinberg A. P., Lengauer C., Kinzler K. W., Baylin S. B., et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2004;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 49.Kim G.D., Ni J., Kelesoglu N., Roberts R. J., Pradham S. Co-operation and communication between human maintenance and de novo DNA (cytosine-5) methylatransferases. EMBO J. 2002;21:4183–4195. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoder J. A., Bestor T. H. A candidate mammalian DNA methyltransferase related to pmt1p of fission yeast. Hum. Mol. Genet. 1998;7:279–284. doi: 10.1093/hmg/7.2.279. [DOI] [PubMed] [Google Scholar]
- 51.Hermann A., Schmitt S., Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J. Biol. Chem. 2003;278:31717–31721. doi: 10.1074/jbc.M305448200. [DOI] [PubMed] [Google Scholar]
- 52.Ehrlich M. DNA hypomethylation and cancer. In: Ehrlich M., editor. DNA alterations in Cancer: Genetic and Epigenetic Changes. Natick, MA.: Eaton; 2000. pp. 273–291. [Google Scholar]
- 53.Bird A. P. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 54.Reik W., Dean W., Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 55.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 56.Yoder J.A., Bestor T.H. Cytosine methylation and the ecology of intragrnomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/S0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- 57.Tate P. H., Bird A. P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993;3:226–231. doi: 10.1016/0959-437X(93)90027-M. [DOI] [PubMed] [Google Scholar]
- 58.Lee J. H., Skalnik D. G. CpG-binding protein is a nuclear matrix- and euchromatin-associated protein localized to nuclear speckles containing human trithorax. Identification of nuclear matrix targeting signals. J. Biol. Chem. 2002;277:42259–42267. doi: 10.1074/jbc.M205054200. [DOI] [PubMed] [Google Scholar]
- 59.Kass S. U., Pruss D., Wolfe A. P. How does DNA methylation repress transcription? Trends Genet. 1999;13:444–449. doi: 10.1016/S0168-9525(97)01268-7. [DOI] [PubMed] [Google Scholar]
- 60.Lewis J. D., Meehan R. R., Henzel W. J., Maurer-Fogy I., Jeppesen P., Klein F., Bird A. Purification, sequence and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 2002;69:905–915. doi: 10.1016/0092-8674(92)90610-O. [DOI] [PubMed] [Google Scholar]
- 61.Jones P. L., Veenstra G. J., Wade P. A., Vermaak D., Kass S. U., Landsberger N., Strouboulis J., Wolffe A. P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 62.Nan X., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 63.Meehan R. R., Lewis J. D., Bird A. P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acid Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendrich B., Bird A. P. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fuks F., Burgers W. A., Godin N., Kasai M., Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robertson K. D., Ait-Si-Ali S., Yokochi T., Wade P. A., Jones P. L., Wolfe A. P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 67.Fuks F., Hurd P. J., Deplus R., Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone mehtyltransferases. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuks F., Hurd P. J., Wolf D., Nan X., Bird A. P., Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 69.Fujita N., Watanabe S., Ichimura T., Tsuruzoe S., Shinkai Y., Tachibana M., Chiba T., Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39 h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 2003;278:24132–24138. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 70.Francastel C., Schübeler D., Martin D. K. I., Groudine M. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 2000;1:137–142. doi: 10.1038/35040083. [DOI] [PubMed] [Google Scholar]
- 71.Görisch S. M., Lichter P., Karsten R. Mobility of multi-subunit complexes in the nucleus: accessibility and dynamics of chromatin subcompartments. Histochem. Cell Biol. 2005;123:217–228. doi: 10.1007/s00418-005-0752-y. [DOI] [PubMed] [Google Scholar]
- 72.Foster H. A., Bridger J. M. The genome and the nucleus: a marriage made by evolution. Chromosoma. 2005;114:212–229. doi: 10.1007/s00412-005-0016-6. [DOI] [PubMed] [Google Scholar]
- 73.Lehnertz B., Ueda Y., Derijck A. A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A. H. Suv39 h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003;13:1192–1200. doi: 10.1016/S0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 74.Tariq M., Saze H., Probst A. V., Lichota J., Habu Y., Paszkowski J. Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc. Natl. Acad. Sci. USA. 2003;100:8823–8827. doi: 10.1073/pnas.1432939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Espada J., Ballestar E., Fraga M. F., Villar-Garea A., Juarranz A., Stockert J. C., Robertson K. D., Fuks F., Esteller M. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J. Biol. Chem. 2004;279:37175–37184. doi: 10.1074/jbc.M404842200. [DOI] [PubMed] [Google Scholar]
- 76.Santos A. P., Arranches R., Stoger E., Beven A., Viegas W., Shaw P. J. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J. Cell Sci. 2002;115:4597–4605. doi: 10.1242/jcs.00160. [DOI] [PubMed] [Google Scholar]
- 77.de Capoa A., Mendez F., Poggesi I., Giancotti P., Grappelli C., Marotta M.R., Di Leandro M., Reynaud C., Niveleau A. Cytological evidence for 5-azacytidine-induced demthylation of the heterochromatic regions of human chromosomes. Chromosome Res. 1996;4:271–276. doi: 10.1007/BF02263676. [DOI] [PubMed] [Google Scholar]
- 78.Rougier N., Bourc'his D., Gomes D. M., Niveleau A., Plachot M., Paldi A., Viegas-Pequignot E. Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 1998;12:2108–2113. doi: 10.1101/gad.12.14.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coffigny H., Bourgeois C., Ricoul M., Bernardino J., Vilain A., Niveleau A., Malfoy B., Dutrillaux B. Alterations of DNA methylation patterns in germ cells and Sertoli cells from developing mouse testis. Cytogenet. Cell Genet. 1999;87:175–181. doi: 10.1159/000015460. [DOI] [PubMed] [Google Scholar]
- 80.Bitonti M.B., Cozza R., Chiappetta A., Giannino D., Ruffini Castiglioni M., Dewitte W., Mariotti D., Van Onckelen Hand Innocenti A. M. Distinct nuclear organization, DNA methylation and cytokinin distribution mark juvenile, juvenilelike and adult vegetative apical meristems in peach (Prunus persica) J. Exp. Bot. 2002;53:1047–1054. doi: 10.1093/jexbot/53.371.1047. [DOI] [PubMed] [Google Scholar]
- 81.Habib M., Fares F., Bourgeois C.A., Bella C., Bernardino J., Hernandez-Blazquez F., de Capoa A., Niveleau A. DNA global hypomethylation in EBV-transformed interphase nuclei. Exp. Cell Res. 1999;249:46–53. doi: 10.1006/excr.1999.4434. [DOI] [PubMed] [Google Scholar]
- 82.Vilain A., Bernardino J., Gerbault-Seureau M., Vogt N., Niveleau A., Lefrancois D., Malfoy B., Dutrillaux B. and instability in lymphoblastoid cell lines. Cytogenet. Cell Genet. 2000;90:93–101. doi: 10.1159/000015641. [DOI] [PubMed] [Google Scholar]
- 83.Hernandez-Blazquez F. J., Habib M., Dumollard J. M., Barthelemey C., Benchaib M., de Capoa A., Niveleau A. Evaluation of global DNA hypomethylation in human colon cancer tisúes by immunohistochemistry and image análisis. Gut. 2000;47:689–693. doi: 10.1136/gut.47.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barbin A., Montpellier C., Kokalj-Vokac N., Gibaud A., Niveleau A., Malfoy B., Dutrillaux B., Bourgeois C. A. New sites of methylcytosine-rich DNA detected on metaphase chromosomes. Hum. Genet. 1994;4:684–692. doi: 10.1007/BF00206964. [DOI] [PubMed] [Google Scholar]
- 85.Xu, G. L., Bestor, T.H., Bourc'his, D., Hsieh, C. L., Tommerup, N., Bugge, M., Hulten, M., Qu, X., Russo, J. J. and Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191. [DOI] [PubMed]