

Abstract

Synthesis of a novel, stable reagent (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile from readily available ethyl α-(1,3-benzothiazol-2-ylsulfanyl)- α-fluoroacetate is reported. Aldehydes undergo condensations with (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile in the presence of DBU leading to α-fluoro acrylonitriles in high yields and with good Z-stereoselectivity. Lowering of reaction temperature increases the Z selectivity.
INTRODUCTION
Interest in fluorinated organic molecules stems from the altered biological and other properties excerted by fluorine atom introduction.1 As a consequence, a significant number of pharmaceuticals currently in the market contain fluorine atom.2 A convenient approach towards the synthesis of fluoroorganic compounds is through modular assembly via fluorinated building blocks.3 Functionalized fluoroolefins are attractive building blocks and in many instances they are biologically useful entities.4 For example, fluoroolefins act as peptide isosteres,1,5 whereas examples of enzyme inhibitors1,4 include fluorovinyl nucleoside,6a amino acid4 or steroid6b,cbased derivatives. Several methods have therefore been developed for the synthesis of variously functionalized vinyl fluorides, such as α-fluoro acrylates7 or fluorovinyl sulfones.6a,8 Synthetic routes to α-fluoro acrylonitriles, on the other hand, are scarce. Patrick and Nadji9 reported in situ preparation of diethyl α-cyano-α-fluoromethanephosphonate from fluoroacetonitrile that reacted as expected with aromatic aldehydes. On the other hand, aliphatic aldehydes were reported to give complex reaction mixtures resulting from aldol type reactions.9 Xu and DesMarteau10 prepared and isolated the Horner-Wadsworth-Emmons (HWE) reagent by fluorination of diethyl cyanomethanephosphonate using N-fluorobis(trifluoromethanesulfonyl)imide, a fluorinating agent that is not available commercially. Although the HWE reagent prepared in this manner was isolated, its use in condensation reactions with aldehydes and ketones was successful only when prepared in situ. Van der Gen et al.11 prepared (diphenylphosphinoyl)fluoroacetonitrile in situ from the fluoroacetonitrile anion, but attempts at its isolation were unsuccessful.11 Condensations proceeded with aldehydes as well as ketones in reasonable yields. Common to these methods is the necessity for in situ prepared reagent. Herein, we report preparation of a novel, stable reagent and its reactivity in α-fluoro acrylonitrile synthesis.
Modified Julia olefination12 has been reported as an alternative to Horner-Wadsworth-Emmons olefination for the preparation of fluoroalkylidenes.13,14 In this context, we have recently developed novel reagents for the synthesis of fluorinated stilbene and styrene derivatives,14 α-fluoro-α,β-unsaturated esters7j and phenyl sulfones8i via Julia olefination. Specifically, mild conditions, combined with high yields obtained in the synthesis of the latter two prompted our interest in exploring Julia fluoroolefination for the preparation of α-fluoro acrylonitriles.
RESULTS AND DISCUSSION
Our initial goal was synthesis of an appropriate heteroarylsulfonyl derivative as the reagent for a one-pot Julia-Kocienski olefination.15 We first focussed on the synthesis of (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile. Reaction of bromoacetonitrile (1) with the sodium salt of 2-mercapto-1,3-benzothiazole resulted in 2a16 in 88% yield, that was subjected to oxidation to 1,3-benzothiazol-2-ylsulfonyl (BT-sulfonyl) derivative 3 (Scheme 1). The use of m-CPBA gave a low yield of 3, whereas dimethyldioxirane or oxone led mainly to the sulfoxide derivative. Use of MoO24(NH4)6.4H2O/H2O217 gave 75% of 3, that increased to 90% upon use of H5IO6/CrO3.18 Fluorination of 3 with NaH and Selectfluor in THF resulted in a mixture of monofluoro derivative 4 and a difluoro amide byproduct 5 (2.2:1 ratio, respectively), along with starting material. Laborius chromatographic purification, that prevented synthesis on larger scales, gave (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile 4 in 32% isolated yield. In order to improve the efficiency of synthesis of 4, alternate synthetic routes were tested. Metalation and fluorination of benzylic nitriles using various strong bases (LDA, NaHMDS, KHMDS, LHMDS, n-BuLi) and NFSI gave fluorinated derivatives in 9-16%, and the reaction of the bases with the cyano functionality was suggested as the possible cause for the low yields.19 However, the use of t-BuLi and NFSI improved the yield of monofluoro derivative to 60%.19 Fluorination of 1,3-benzothiazol-2ylsulfanyl derivative 2a using tert-BuLi and NFSI was therefore performed, resulting in a complex reaction mixture. Since metalated 1-phenyl-1H-tetrazol-5-yl sulfone derivatives (PT-sulfones) were suggested to be more stabile and less prone to self condensation,15,20 PT-sulfide 2b was synthesized21 and subjected to tert-BuLi and NFSI fluorination to give 6b21 in 45% isolated yield (Scheme 1). Attempts at oxidizing 6b to corresponding sulfone using MoO24(NH4)6.4H2O/H2O2, or H5IO6/CrO3, were unsuccessful.
SCHEME 1.
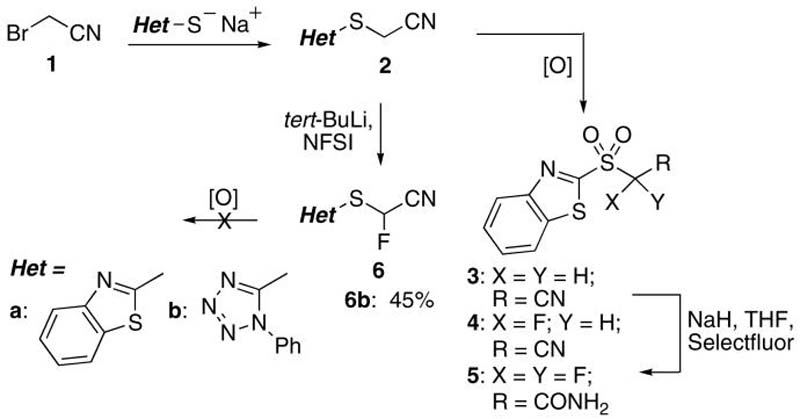
Attempts at an Efficient Synthesis of 4
Since little success was achieved with conversions of BT- or PT-derivatives containing a preinstalled cyano functionality, we decided on an alternate method, wherein fluorine introduction into the molecule would precede that of the cyano group. Olah and coworkers have reported high yielding conversions of amides to cyano derivatives using cyanuric chloride.22 Ethyl α(1,3-benzothiazol-2-ylsulfanyl)-α-fluoroacetate (8, 88% yield, Scheme 2) was therefore synthesized from commercially available ethyl bromofluoroacetate 7.7k Alternatively, unfluorinated precursor ethyl bromoacetate 9 (Scheme 2) was converted to ethyl (1,3-benzothiazol-2-ylsulfanyl)acetate 1023 (89% yield), followed by fluorination of 10 using LDA and NFSI in toluene to afford 8 in 57% yield.24 Ester conversion to amide 11 with NH3 in EtOH proceeded in high 96% isolated yield. Reaction of 11 using cyanuric chloride afforded (1,3-benzothiazol-2-ylsulfanyl)fluoroacetonitrile 6a and the crude reaction mixture was subjected to oxidation with H5IO6/CrO3without prior purification, to give the desired 4 in isolated yields ranging from 38%-42% (over two steps). Reversal of synthetic steps involving initial oxidation of the amide 11 to corresponding sulfone was tested as well, but in this case subsequent conversion to cyano derivative was unsuccessful.
SCHEME 2.

Successful Synthesis of 4
With the desired (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile 4 in hand, condensations with carbonyl compounds were undertaken. Initially, DBU mediated condensation of 4 with 2-naphthaldehyde under Barbier conditions15 was performed. Although αfluoro acrylonitrile was formed, the isolated yield of the product was only 32%. Even lower product formation was observed in the condensation of 4 and 2-naphthaldehyde in the presence of LHMDS, at -78 ºC and under Barbier conditions25 and no product was observed when NaHMDS was used as base.25 A much higher 98% yield of the product was obtained upon a slow, dropwise addition of 4 to solution of DBU and aldehyde (Table 1, entry 1). In a typical procedure, aldehyde (1 molar equiv) and DBU (4 molar equiv) were dissolved in CH2Cl2 (5 mL/mmol of aldehyde) and under stirring at room temperature, a solution of 4 (2 molar equiv) in CH2Cl2 (7.5 mL/mmol of 4) was slowly added dropwise to the reaction mixture. This resulted in the change of color from clear to black-brown. The conversions were checked by TLC after 1 hour and if the aldehyde was still present, 1 molar equiv of sulfone in CH2Cl2 was added and the reaction was allowed to proceed for another 0.5 h, checked by TLC and if starting aldehyde was not consumed, DBU (2 molar equiv) in CH2Cl2 was added (Table 1, entries 3, 4). Upon disappearance of the aldehyde (15- 150 min) the crude reaction mixtures were analyzed by 19F NMR for E/Z ratios of products, partially concentrated, then directly loaded on a dry silica gel column and the products were eluted (for purification of 21, see the Experimental Section). Although E/Z isomers were not separated, we wanted to assess the ease of separation by tlc. For this purpose, tlc analysis was conducted for several product mixtures. In all cases tested the separation of isomers was easily achieved (please see Supporting Information for details). The yields obtained in the condensation reactions with a series of aldehydes and the E/Z ratios are displayed in Table 1. Wherever applicable, comparison to literature data is included.
TABLE 1.
Efficient, Mild Route to α-Fluoro Acrylonitriles
| entry | aldehyde | temp | product: yield,a%E/Z ratiob | =C(CN)F: δ (ppm);cJ (Hz) | lit:d yield, E/Z ratio |
|---|---|---|---|---|---|
| 1 | 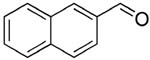 |
rt | 12: 98%, 15:85 | -122.1; 36.6 | -- |
| -78 °C | 12: 97%, 8:92 | -122.5; 15.3 | |||
| 2 | 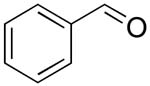 |
rt | 13: 93%, 19:81 | -121.9; 35.2 | 54%, 66:3310 |
| -122.6; 17.6 | 53%, 21:7911 | ||||
| 3 | 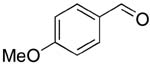 |
rt | 14: 95%, 16:84 | -126.2; 36.6 | 52%, 50:509 |
| -127.1; 18.3 | 50%, 32:6811 | ||||
| 4 | 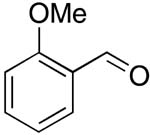 |
rt | 15: 91%, 37:63 | -122.3; 15.3 | -- |
| -78 °C | 15: 91%, 27:73 | -124.1; 36.6 | |||
| 5 | 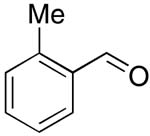 |
rt | 16: 94%, 17:83 | -120.4; 15.3 | -- |
| -123.0; 36.6 | |||||
| 6 | 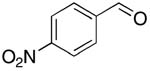 |
rt | 17: 72%, 17:83 | -115.3; 15.3 | 45%, 33:679 |
| -115.7; 33.6 | |||||
| 7 | 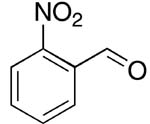 |
rt | 18: 60%, 15:85 | -117.0; 12.2 | 62%, 38:629 |
| -120.1; 30.5 | |||||
| 8 | 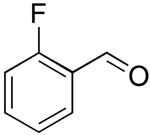 |
rt | 19: 91%, 16:84 | -118.8; 15.3 | -- |
| -119.8; 36.6 | |||||
| 9 | 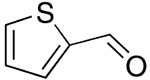 |
rt | 20: 96%, 17:83 | -122.8; 29.4 | -- |
| -129.0; 11.7 | |||||
| 10 | 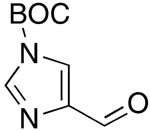 |
rt | 21: 59%, 18:82 | -117.9; 33.6 | -- |
| -125.3; 15.3 | |||||
| 11 | 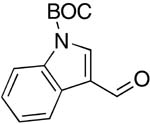 |
rt | 22: 86%, 20:80 | -119.2; 33.6 | -- |
| -125.9; 12.2 | |||||
| 12 | 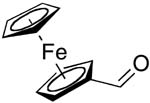 |
rt | 23: 92%, 17:83 | -128.5; 35.2 | -- |
| -129.6; 17.6 | |||||
| 13 | 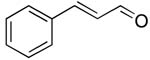 |
rt | 24: 81%, 17:83 | -126.6; 30.5 | 38%, 60:4010 |
| -127.7; 12.2 | 45%, 44:5611 | ||||
| 14 | n-heptyl-CHO | rt | 25: 97%, 23:77 | -123.9; 11.8 | Lit. data for n-heptanal:e 30%, 30:7010 50%, 26:7411 |
| -78 °C | 25: 90%, 16:84 | -125.8; 35.2 | |||
| 15 | 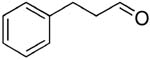 |
rt | 26: 80%, 23:77 | -122.6; 14.7 | 51%, 26:7411 |
| -78 °C | 26: 81%, 12:88 | -124.2; 32.3 | |||
| 16 | 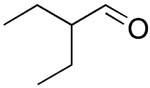 |
rt | 27: 76%, 15:85 | -122.8; 15.3 | -- |
| -78 °C | 27: 77%, 8:92 | -125.6; 30.5 |
Yields of isolated, purified products (reactions were performed under similar conditions but were not optimized for individual cases).
Relative ratio of diastereomers in the crude reaction mixture determined by 19F NMR prior to isolation. No change in ratio was observed after purification.
Referenced to CFCl3 as internal standard.
Where applicable, literature data and references are included for comparison.
Since no literature data for n-octanal are available, data for n-heptanal are included for comparison.
The reactions resulted in good to high yields of α-fluoroacrylonitriles. In the case of p- and onitrobenzaldehyde, Barbier conditions15 were found to give higher yields of products (72% and 60% respectively, entries 6 and 7, Table 1, see the Experimental Section for details).
Imidazole-4-carboxaldehyde and indole-3-carboxaldehyde gave high yields of products only upon N-Boc protection26 (Table 1, entries 10, 11).
In all the reactions studied, the Z-isomer predominated, and stereoselectivity was nearly independent of the structure of the aldehyde. For aromatic aldehydes E/Z ratio ranged from 15/85-20/80, and was independent of the electronic nature of the substituent (entries 1-3, 5-12). The only exception was o-methoxybenzaldehyde (entry 4), where the E/Z ratio dropped to 37/63. At this point, we are unable to offer any explanation for this. We do not believe that this is due to electronic or steric influence, since no changes in E/Z ratios were observed in the case of pmethoxy, o-methyl, o-nitro or o-fluoro substituents (E/Z ratios 16:84, 17:83, 15:85, and 16:84, respectively). In the case of α,β-unsaturated aldehyde a similar E/Z ratio of 17:83 (entry 13) was observed, whereas aliphatic aldehydes gave E/Z ratios of 23/77 for the unbranched (entries 14, 15) and 15/85 for the branched aldehyde (entry 16). The effect of temperature on E/Z ratio was tested in the condensation reactions of 4 with 2-naphthaldehyde, 2-methoxybenzaldehyde, octanal, 3-phenylpropanal and 2-ethylbutanal. In all cases tested, lowering of reaction temperature to -78 °C improved the stereoselectivity, whereas the yield remained unchanged (Table 1, entries 1, 4, 14, 15, 16).
Scheme 3 shows the mechanism that was proposed by Julia (for X = H).15,27,28 Reactions can proceed via competing pathways, leading to different stereochemical outcomes. The initial anti and syn addition product (I1 and I2) can either undergo retroaddition, if the BT-sulfone anion is stabilized, or collapse to products via the spirocyclic intermediates SI1 and SI2. Intermediate SI2 has been suggested to form faster than SI1 due to steric reasons. Thus, if retroaddition occurs, predominant stereoselectivity leading to cis arrangement of R1 and R (Scheme 3) can be expected. An additional pathway involving stabilized zwitterionic intermediates Z1 and Z2 has been proposed for aryl and vinyl aldehydes.15,27,28 In these cases formation of alkenes with a trans arrangement of R and R1 through Z2 is favored. This is in fact the case with Julia reactions of aryl and vinyl aldehydes leading to the unfluorinated acrylates.23 On the other hand, in our prior work on fluorinated acrylates a predominant cis arrangement of R and R1 was observed.7jThis led us to suggest that in the case of fluorinated sulfones, reaction involving zwitterionic intermediates Z1 and Z2 is disfavored due to the destabilizing effect a β-fluorine substituent has on carbocation stability.
SCHEME 3.
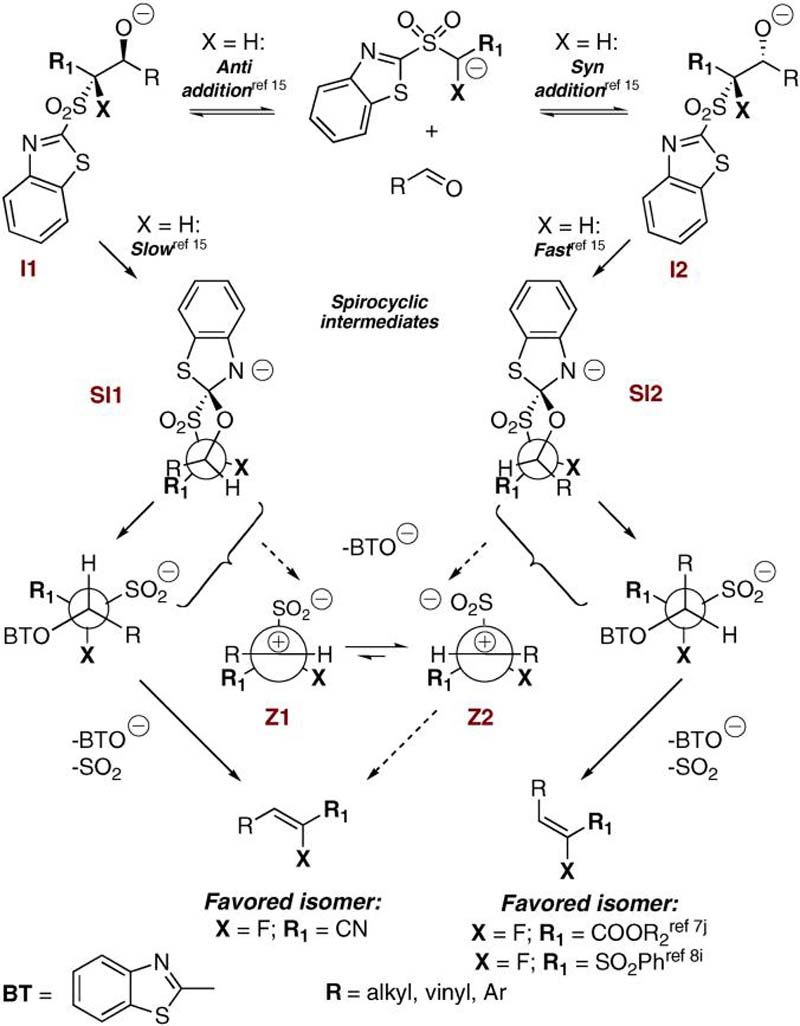
Fluoro-Julia Olefination Stereoselectivities
In connection to our previous work on the fluoro Julia olefination, we were interested in comparing the stereochemistry obtained in DBU mediated olefinations leading to α-fluoro acrylates7j and to phenyl α-fluorovinyl sulfones,8i to those observed in the present synthesis of α-fluoro acrylonitriles. Interestingly, alkenes with the cis disposition of R (from RCHO) and COOR2 or SO2Ph moiety were the major products in the synthesis of acrylates7j and phenyl sulfones.8i On the other hand, an opposite stereochemical outcome was observed in the present case. Assuming the lesser probability of Z1 and Z2 in fluoro Julia olefinations,7j due to lower stability of β-fluorocarbocations, the plausible reasons for the present outcome could be as follows. In the event that the initial addition products I1 and I2 do not equilibrate, that is addition/retroaddition of the initially formed β-alkoxy sulfone anion does not occur, then the E/Z product ratio reflects the initial ratio of I1/I2. Alternatively, if equilibration of I1 and I2 does occur, then collapse of adduct I1 through SI1 may not be unfavorable, due to a small steric effect of the cyano functionality. Supportive of the latter, the selectivity in the condensation of the branched aldehyde 2-ethylbutanal (Table 1, entry 16) increased from 15:85 (E/Z) at rt to 8:92 (E/Z) at -78 °C. This may imply a lower energy barrier to Smiles rearrangement for adduct I1 as compared to I2.
CONCLUSIONS
In conclusion, we have developed the synthesis of (1,3-benzothiazol-2-ylsulfonyl)fluoroacetonitrile, a stable, isolable reagent for the preparation of α-fluoro acrylonitriles via a modified Julia olefination. The reagent undergoes reactions with aldehydes under mild, DBU mediated conditions, to give α-fluoro acrylonitriles in good yields and with predominant Z-stereoselectivity. The structure of the aldehyde generally does not influence the stereochemical outcome of the olefination.
EXPERIMENTAL SECTION
(1,3-Benzothiazol-2-ylsulfonyl)acetonitrile (3)
A solution of periodic acid (H5IO6, 2.21 g, 9.71 mmol, 4.0 molar equiv) in CH3CN (39 mL) was allowed to vigorously stir for 30 min at rt. CrO3 (4.9 mg, 0.05 mmol, 0.02 molar equiv) was added, and the stirring was continued for another 5 min. A solution of 1,3-benzothiazol-2-ylsulfanyl)acetonitrile (2a, 500 mg, 2.43 mmol, 1 molar equiv) in CH3CN (5 mL) was slowly added dropwise and formation of precipitate was observed. After stirring at rt for 1 h, the reaction mixture was filtered, and the solid was washed with CH3CN. The filtrate was concentrated under reduced pressure, EtOAc was added to the residue and the organic layer was washed with water and brine. The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using CH2Cl2 as eluting solvent to yield 3 as a white solid (523 mg, 91% yield). Mp (recrystallized from 50% EtOAc in hexanes) 173-174 °C. 1H NMR (500 MHz): δ 8.27 (d, 1H, Ar-H, J = 7.9), 8.07 (d, 1H, Ar-H, J = 7.6), 7.69 (m, 2H, Ar-H, J = 7.6), 4.56 (s, 2H, CH2CN). HRMS (positive ion ESI): calcd. for C9H6N2O2S2Na+ (M+ + Na) 260.976290, found 260.976192.
Ethyl (1,3-Benzothiazol-2-ylsulfanyl)fluoroacetate (8) via Fluorination of Ethyl (1,3-Benzothiazol-2-ylsulfanyl)acetate (10)
A stirred solution of ethyl (1,3-benzothiazol-2-ylsulfanyl)acetate 10 (200 mg, 0.791 mmol, 1 molar equiv) in 1.3 mL of dry toluene, was cooled to -80 ºC (dry-ice/iso-PrOH) under nitrogen. LDA (0.455 mL, 0.909 mmol, 1.15 molar equiv of a 2 M solution in heptane/THF/EtPh) was added to the reaction mixture and the stirring was continued at -80 ºC. After 1.5 h solid NFSI (312 mg, 0.988 mmol, 1.25 molar equiv) was added, the reaction mixture was allowed to stir at -80 ºC for another 1 h and then warmed to room temperature over 1 h. Sat aq NH4Cl was added to the mixture and the layers were separated. The aqueous layer was extracted with EtOAc (3 x), and the combined organic layer was washed with sat aq NaHCO3 and brine. The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixtures was purified by column chromatography on silica gel using CH2Cl2 to yield 8 as a clear thick pale-yellow liquid (123 mg, 57%). 1H NMR (500 MHz): δ7.99 (d, 1H, Ar-H, J = 7.9), 7.83 (d, 1H, Ar-H, J = 7.9), 7.49 (t, 1H, Ar-H, J = 7.6), 7.39 (t, 1H, Ar-H, J = 7.6), 6.94 (d, 1H, CHF, 2JFH = 51.0), 4.34 (q, 2H, OCH2, J = 7.1), 1.33 (t, 3H, CH3, J = 7.1). 19F NMR (282 MHz): δ -161.7 (d, 2JFH = 47.0). HRMS (positive ion ESI): calcd. for C11H10FNO2S2Na+ (M+ + Na) 294.002919, found 294.002247.
For synthesis of 8 from 7, please see Supporting Information.
(1,3-Benzothiazol-2-ylsulfanyl)fluoroacetamide (11)
Into a solution of ethyl (1,3-benzothiazol-2-ylsulfanyl)fluoroacetate 8 prepared from 7 (3.88 g, 14.3 mmol) in ethanol (50 mL) at room temperature was bubbled a gentle stream of NH3 gas until disappearance of the starting material was observed by TLC (SiO2, 50% EtOAc in hexanes), and after 1 h the solvent was evaporated under reduced pressure. The product was dried overnight under vacuum to yield 3.33 g (96%) of 11 as a white solid that did not require any further purification. Mp (recrystallized from 50% EtOAc in hexanes) 134-135 °C. 1H NMR (500 MHz): δ 7.98 (d, 1H, Ar-H, J = 8.2), 7.82 (d, 1H, Ar-H, J = 7.9), 7.47 (t, 1H, Ar-H, J = 7.9), 7.39 (t, 1H, Ar-H, J = 7.9), 7.01 (d, 1H, CHF, 2JFH = 51.9), 6.53 (br s, 1H, NH2), 5.84 (br s, 1H, NH2). 19F NMR (282MHz):δ -158.4 (d, 2JFH = 52.8). HRMS (positive ion ESI): calcd. for C9H7FN2OS2Na+ (M+ + Na) 264.987604, found 264.987469.
(1,3-Benzothiazol-2-ylsulfonyl)fluoroacetonitrile (4)
Step 1
To a stirred solution of cyanuric chloride (3.89 g, 21.1 mmol, 1.7 molar equiv) in DMF (57 mL), was added (1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide (11, 3.00 g, 12.4 mmol, 1 molar equiv) at rt. The stirring was continued at rt for 1.5 h, until TLC (SiO2, 50% EtOAc in hexanes) showed disappearance of starting material. The reaction mixture was diluted with water and extracted with EtOAc (3x). The combined organic layer was thoroughly washed with water, brine, dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure to yield 2.49 g of the crude product 6a (see Supporting Information for analytical data on a purified sample), that was subjected to oxidation without further purification.
Step 2
A solution of periodic acid (H5IO6, 10.1 g, 44.5 mmol, 4.0 molar equiv) in CH3CN (100 mL) was allowed to vigorously stir for 30 min at rt, CrO3 (22.3 mg, 0.222 mmol, 0.02 molar equiv) was added, and the stirring was continued for another 5 min. A solution of 1,3-benzothiazol-2-ylsulfanyl)fluoroacetonitrile (2.49 g, 11.1 mmol, 1 molar equiv, crude reaction mixture from step 1) in CH3CN (20 mL) was slowly added dropwise and formation of precipitate was observed. After stirring at rt for 1 h, the reaction mixture was filtered and concentrated under reduced pressure. EtOAc was added to the residue and the mixture was washed with water and brine. The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using CH2Cl2 as eluting solvent to yield 4 as an off white solid (1.21 g, 38% yield over two steps). Mp (recrystallized from 50% EtOAc in hexanes) 149-150 °C. 1H NMR (500 MHz): δ 8.33 (d, 1H, Ar-H, J = 7.9), 8.09 (d, 1H, Ar-H, J = 7.6), 7.73 (m, 2H, Ar-H), 6.24 (d, 1H, CHF, 2JFH = 46.7). 19F NMR (282 MHz): δ-179.4 (d, 2JFH = 47.0). HRMS (positive ion ESI): calcd. for C9H5FN2O2S2Na+ (M+ + Na) 278.966868, found 278.966867.
Subsequent repetition of the two-step procedure using 5.00 g of 11 resulted in a 42% yield of 4.
(1,3-Benzothiazol-2-ylsulfonyl)fluoroacetonitrile (4) via Fluorination of (1,3-Benzothiazol-2-ylsulfonyl)acetonitrile (3)
A suspension of NaH (49.9 mg, 2.08 mmol, 1.1 molar equiv) in dry THF (6 mL) was cooled to 0 °C under nitrogen and a solution of (1,3-benzothiazol-2-ylsulfonyl)acetonitrile (3, 450 mg, 1.89 mmol, 1 molar equiv) in dry THF (6.5 mL) was added dropwise with stirring. After the addition, the reaction mixture was allowed to warm to rt and left to stir for 45 min. The mixture was then cooled to 0 °C, Selectfluor (804 mg, 2.27 mmol, 1.2 molar equiv) was added, the cooling bath was removed and the reaction mixture was allowed to stir at rt for 1.5 h, and then quenched with aqueous NH4Cl solution. The aqueous layer was extracted with EtOAc (3x), the combined organic layers were washed with aqueous NaHCO3, water and brine, dried over Na2SO4 and the solvent was evaporated. Separation of the crude reaction mixture by column chromatography on silica gel using CH2Cl2 as eluting solvent afforded 4 as an off white solid (155 mg, 32% yield).
General Procedure for Synthesis of 12-16, 19-20, 22-27 via Condensation of Aldehydes with Fluorinated Sulfone 4 at rt
A solution of 4 (2 molar equiv) in CH2Cl2 (7.5 mL/mmol of 4) was added slowly, dropwise to a stirring solution of aldehyde (1 molar equiv) and DBU (4 molar equiv) in freshly distilled CH2Cl2 (5 mL/mmol of aldehyde) at rt. Upon addition of 4, the reaction mixture turned black brown. The conversions were checked by TLC after 1 hour and if the aldehyde was still present, a solution of 1 molar equiv of sulfone in CH2Cl2 (7.5 mL/mmol of 4) was added dropwise and the reaction was allowed to proceed for another 0.5 h, checked by TLC and if starting aldehyde was still not consumed, DBU (2 molar equiv) in CH2Cl2 (1 mL/mmol of DBU) was added. The stirring was continued at rt until complete consumption of the aldehyde was observed by TLC (SiO2, CH2Cl2), usually 15-150 min. The E/Z ratio was determined by 19F NMR of an aliquot, the reaction mixture was concentrated to about 2 mL and directly loaded onto a dry silica gel column (200-300 mesh). The E/Z product mixture was obtained by elution with CH2Cl2 and the solvent was evaporated (except in the case of 21, please see detailed experimental procedure below).
Synthesis of 21 via Condensation of 4 with 1-Boc-imidazole-4-carboxaldehyde
A solution of 4 (522 mg, 2.04 mmol, 2 molar equiv) in CH2Cl2 (15 mL) was added slowly, dropwise (for about 10 min) to a stirring solution of 1-Boc-imidazole-4-carboxaldehyde (200 mg, 1.01 mmol, 1 molar equiv) and DBU (618 mg, 4.06 mmol, 4 molar equiv) in freshly distilled CH2Cl2 (5.1 mL) at rt. Upon addition of 4, the reaction mixture turned black brown. The stirring was continued at rt until complete consumption of the aldehyde was observed (1.5 h) by TLC (SiO2, 50% EtOAc in hexanes). The solvent was concentrated under reduced pressure to about 2-3 mL and the reaction mixture was loaded onto a dry silica gel column (200-300 mesh) and the E/Z product mixture was eluted with 50% EtOAc in hexanes. The solvent was evaporated and the E/Z ratio was determined by 19F NMR (18/82, Table 1, entry 15, the E/Z ratio does not change upon purification). Since the E/Z product mixture was contaminated with 1,3-benzothiazol-2-ol byproduct, the mixture was dissolved in CH2Cl2 (30 mL), aqueous NaOH was added (0.5 M NaOH, 6 mL) and the mixture was stirred at rt for 10 minutes, the organic layer was separated, washed with water and brine and dried over anhydrous Na2SO4. Upon removal of solvent under reduced pressure, 143 mg (59%) of 21 was isolated as an off-white solid. 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H, Ar-H, E-isomer), 8.09 (s, 1H, Ar-H, Z-isomer), 7.75 (s, 1H, Ar-H, Z-isomer), 7.71 (s, 1H, Ar-H, E-isomer), 6.94 (d, 1H, 2JFH = 14.6, E-isomer), 6.63 (d, 1H, 2JFH = 34.5, Z-isomer), 1.64 (s, 9H, t-Bu, Z-isomer), 1.635 (s, 9H, t-Bu, E-isomer). 19F NMR (282 MHz, CDCl3): δ -117.9 (d, 3JFH = 33.6, Z isomer), δ -125.3 (d, 3JFH = 15.3, E isomer). HRMS (positive ion ESI) calcd. for C11H12FN3O2Na+ (M+ + Na) 260.080576, found 260.080458.
General Procedure for Synthesis of 12, 15, 25, 26, 27 via Condensation of Aldehydes with Fluorinated Sulfone 4 at -78 °C
A solution of 4 (2 molar equiv) in CH2Cl2 (7.5 mL/mmol of 4) was added slowly, dropwise to a stirring solution of aldehyde (1 molar equiv) and DBU (4 molar equiv) in freshly distilled CH2Cl2 (5 mL/mmol of aldehyde) that was cooled to -78 °C under N2. Upon addition of 4, the reaction mixture turned black brown. The reaction mixture was stirred at -78 °C for 1 hour and then allowed to warm to rt. In all cases studied, TLC showed complete conversions. Analysis of reaction mixtures by 19F NMR and isolation of E/Z product mixtures was performed as described for the rt condensations.
General Procedure for the Synthesis of 17 and 18 via Condensation of Aldehydes with Fluorinated Sulfone 4
In the case of p-nitrobenzaldehyde and o-nitrobenzaldehyde, the condensations were performed under Barbier conditions as follows.
Aldehyde (1 molar equiv) and 4 (2 molar equiv) were dissolved in CH2Cl2 (19 mL/mmol of aldehyde), and under stirring at rt, DBU (4 molar equiv) in CH2Cl2 (1 mL/mmol of DBU) was slowly added dropwise to the reaction mixture. Upon addition of DBU, the reaction mixture turned black brown. The reaction was monitored by TLC and upon consumption of the aldehyde, the E/Z ratio was determined by 19F NMR. The reaction mixture was concentrated to about 2 mL in vacuo and directly loaded onto a dry silica gel column (200-300 mesh). The E/Z product mixture was eluted with CH2Cl2 and the solvent was evaporated.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NSF Grant CHE-0516557, infrastructural support and support for A.K.G. were provided by NIH RCMI Grant 5G12 RR03060. Support of M.d.S through the NIH-RISE program is gratefully acknowledged. Acquisition of a mass spectrometer was funded by NSF Grant CHE-0520963. We thank Dr. Andrew Poss (Honeywell) for a sample of NFSI and Dr. G. Sankar Lal (Air Products) for a sample of Selectfluor.
Footnotes
SUPPORTING INFORMATION Experimental details for synthesis of 2a, 8 from 7, 6b, analytical data of pure 6a, details of TLC separation of some E/Z isomer mixtures, HRMS and 19F NMR data and 1H NMR spectra of 2a, 3, 4, 6a, 6b, 8, 11-27 and 13C NMR spectrum of 4. This information is available free of charge via the Internet at http://pubs.acs.org.
This paper is dedicated to the memory of Dr. John W. Daly (1933-2008), an outstanding scientist and individual.
REFERENCES
- (1).(a) Welch JT, editor. Selective Fluorination in Organic and Bioorganic Chemistry. American Chemical Society; Washington, DC: 1991. [Google Scholar]; (b) Ojima I, McCarthy JR, Welch JT, editors. Biomedical Frontiers of Fluorine Chemistry. American Chemical Society; Washington, DC: 1996. [Google Scholar]
- (2).Bégué J-P, Bonnet-Delpon D. J. Fluorine Chem. 2006;127:992–1012. [Google Scholar]
- (3).J. Fluorine Chem. 2004;125:477–645. Special Issue on Fluorinated Synthons.. Soloshonok VA, editor. Fluorine-Containing Synthons. American Chemical Society; Washington, DC: 2005. . Soloshonok VA, Mikami K, Yamazaki T, Welch JT, Hoenk JF, editors. Current Fluoroorganic Chemistry: New Synthetic Directions, Technologies, Materials, and Biological Applications. American Chemical Society; Washington, DC: 2007. .
- (4).Welch JT. Tetrahedron. 1987;43:3123–3197. [Google Scholar]
- (5).See for example: Allmendinger T, Felder E, Hungarbühler E. Tetrahedron Lett. 1990;31:7301–7304.. Welch JT, Lin J. Tetrahedron. 1996;52:291–304.. Dutheuil G, Couve-Bonnaire S, Pannecoucke X. Angew. Chem. Int. Ed. 2007;46:1–4. doi: 10.1002/anie.200604246..
- (6).(a) McCarthy JR, Matthews DP, Stemerick DM, Huber EW, Bey P, Lippert BJ, Snyder RD, Sunkara PS. J. Am. Chem. Soc. 1991;113:7439–7440. [Google Scholar]; (b) Burkhart JP, Weintraub PM, Gates CA, Resvick RJ, Vaz RJ, Friedrich D, Angelastro MR, Bey P, Peet NP. Bioorg. Med. Chem. 2002;10:929–934. doi: 10.1016/s0968-0896(01)00354-6. [DOI] [PubMed] [Google Scholar]; (c) Weintraub PM, Holland AK, Gates CA, Moore WR, Resvick RJ, Bey P, Peet NP. Bioorg. Med. Chem. 2003;11:427–431. doi: 10.1016/s0968-0896(02)00434-0. [DOI] [PubMed] [Google Scholar]
- (7).For synthesis via Wittig and Horner-Wadsworth-Emmons see for example: Etemad-Moghadam G. Seyden-Penne. J. Bull. Soc. Chim. Fr. 1985:448–454.. Thenappan A, Burton DJ. J. Org. Chem. 1990;55:4639–4642.. Burton D,J, Yang Z, Qui W. Chem. Rev. 1996;96:1641–1715. doi: 10.1021/cr941140s.. Sano S, Ando T, Yokoyama K, Nagao Y. Synlett. 1998:777–779.. Bargiggia F, Dos Santos S, Piva O. Synthesis. 2002:427–437.. Sano S, Teranishi R, Nagao Y. Tetrahedron Lett. 2002;43:9183–9186.. Sano S, Saito K, Nagao Y. Tetrahedron Lett. 2003;44:3987–3990.. Suzuki Y, Sato M. Tetrahedron Lett. 2004;45:1679–1681.. Zoute L, Dutheuil G, Quirion J-C, Jubault P, Pannecoucke X. Synthesis. 2006:3409–3418.. For synthesis via Julia-Kocienski olefination see: Zajc B, Kake S. Org. Lett. 2006;8:4457–4460. doi: 10.1021/ol0616236.. Pfund E, Lebargy C, Rouden J, Lequeux T. J. Org. Chem. 2007;72:7871–7877. doi: 10.1021/jo070994c.. Alonso DA, Fuensanta M, Gómez-Bengoa E, Nájera C. Adv. Synth. Catal. 2008;350:1823–1829..
- (8).(a) Inbasekaran M, Peet NP, McCarthy JR, LeTourneau ME. J. Chem. Soc., Chem. Commun. 1985:678–679. [Google Scholar]; (b) Koizumi T, Hagi T, Horie Y, Takeuchi Y. Chem. Pharm. Bull. 1987;35:3959–3962. [Google Scholar]; (c) McCarthy JR, Matthews DP, Edwards ML, Stemerick DM, Jarvi ET. Tetrahedron Lett. 1990;31:5449–5452. [Google Scholar]; (d) Kunugi A, Yamane K, Yasuzawa M, Matsui H, Uno H, Sakamoto K. Electrochim. Acta. 1993;38:1037–1041. [Google Scholar]; (e) McCarthy JR, Huber EW, Le T-B, Laskovics FM, Matthews DP. Tetrahedron. 1996;52:45–58. [Google Scholar]; (f) Nakamura T, Guha SK, Ohta Y, Abe D, Ukaji Y, Inomata K. Bull. Chem. Soc. Jpn. 2002;75:2031–2041. [Google Scholar]; (g) Asakura N, Usuki Y, Iio H. J. Fluorine Chem. 2003;124:81–88. [Google Scholar]; (h) Wnuk SF, Garcia PI, Jr., Wang Z. Org. Lett. 2004;6:2047–2049. doi: 10.1021/ol049312n. [DOI] [PubMed] [Google Scholar]; (i) He M, Ghosh AK, Zajc B. Synlett. 2008:999–1004. doi: 10.1055/s-2008-1072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Patrick TB, Nadji S. J. Fluorine Chem. 1990;49:147–150. [Google Scholar]
- (10).Xu Z-Q, DesMarteau DD. J. Chem. Soc., Perkin Trans. 1992;I:313–315. [Google Scholar]
- (11).van Steenis JH, van den Nieuwendijk AMCH, van der Gen A. J. Fluorine Chem. 2004;125:107–117. [Google Scholar]
- (12).Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175–1178. [Google Scholar]
- (13).Chevrie D, Lequeux T, Demoute JP, Pazenok S. Tetrahedron Lett. 2003;44:8127–8130. [Google Scholar]
- (14).Ghosh AK, Zajc B. Org. Lett. 2006;8:1553–1556. doi: 10.1021/ol060002+. [DOI] [PubMed] [Google Scholar]
- (15).(a) Blakemore PR. J. Chem. Soc., Perkin Trans. 2002;I:2563–2585. [Google Scholar]; (b) Plesniak K, Zarecki A, Wicha J. Top. Curr. Chem. 2007;275:163–250. doi: 10.1007/128_049. [DOI] [PubMed] [Google Scholar]
- (16).Hou Y, Higashiya S, Fuchigami T. J. Org. Chem. 1997;62:9173–9176. [Google Scholar]
- (17).Ono K, Yoshida A, Saito N, Fujishima T, Honzawa S, Suhara Y, Kishimoto S, Sugiura T, Waku K, Takayama H, Kittaka A. J. Org. Chem. 2003;68:7407–7415. doi: 10.1021/jo034787y. [DOI] [PubMed] [Google Scholar]
- (18).Xu L, Cheng J, Trudell ML. J. Org. Chem. 2003;68:5388–5391. doi: 10.1021/jo030031n. [DOI] [PubMed] [Google Scholar]
- (19).Kotoris CC, Chen MJ, Taylor SD. J. Org. Chem. 1998;63:8052–8057. [Google Scholar]
- (20).(a) Blakemore PR, Cole WJ, Kociénski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]; (b) Albrecht BK, Williams RM. Proc. Natl. Acad. Sci. 2004;101:11949–11954. doi: 10.1073/pnas.0308432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zagipa B, Nagura H, Fuchigami T. J. Fluorine Chem. 2007;128:1168–1173. [Google Scholar]
- (22).Olah GA, Narang SC, Fung AP, Balaram Gupta BG. Synthesis. 1980:657–658. [Google Scholar]
- (23).Blakemore PR, Ho DKH, Nap WM. Org. Biomol. Chem. 2005;3:1365–1368. doi: 10.1039/b500713e. [DOI] [PubMed] [Google Scholar]
- (24).We have previously reported fluorination of ethyl (1,3-benzothiazol-2-ylsulfonyl)acetate using NaH/Selectfluor, that yielded the fluoro derivative in 71% yield.7j In the present case, due to the lower acidity of sulfanyl analog 10, stronger base LDA in combination with NFSI was chosen. Indeed, as anticipated fluorination of 10 using NaH/Selectfluor under conditions described for the fluorination of sulfonyl acetate derivative7j resulted in a complex reaction mixture. Proton NMR showed 39:61 % ratio of monofluoro derivative 8 and starting sulfide 10, whereas 19F NMR showed 74:26 % ratio of 8 and difluoro derivative, along with unidentified fluorinated byproduct. Chromatographic separation afforded 8 in 14% isolated yield.
- (25).LHMDS (2.4 molar equiv) mediated condensation of 4 (2.4 molar equiv) and 2-naphthaldehyde (1.0 molar equiv) was performed under Barbier conditions in THF at -78 ºC under N2. After stirring at -78 ºC for 3 h and at room temperature for 1 h followed by the usual workup, 1H NMR showed a 1:3 ratio of product 12 (24% isolated yield) and 2-naphthaldehyde, whereas %E/Z ratio of 12 was 30:70, as assessed by 19F NMR. Similar reaction using NaHMDS as base gave no product 12, although sulfone 4 was consumed.
- (26).(a) Kimura R, Nagano T, Kinoshita H. Bull. Chem. Soc. Jpn. 2002;75:2517–2525. [Google Scholar]; (b) Wasser J, Gaspar B, Nambu H, Carreira EM. J. Am. Chem. Soc. 2006;128:11693–11712. doi: 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- (27).Baudin JB, Hareau G, Julia SA, Lorne R, Ruel O. Bull. Soc. Chim. Fr. 1993;130:856–878. [Google Scholar]
- (28).Baudin JB, Hareau G, Julia SA, Ruel O. Bull. Soc. Chim. Fr. 1993;130:336–357. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.