

Abstract
Polymer membrane electrodes operated by pulsed chronopotentiometry have recently been introduced to replace traditional ion-selective electrodes for a number of applications. While ion-selective electrodes for the polycation protamine have been reported, for instance, a pulsed chronopotentiometric readout mode (called here pulstrode) provides improved stability and reproducibility while exhibiting sufficient selectivity for the direct detection of protamine in undiluted whole blood samples. Here, such protamine sensitive pulstrodes are applied for the real time detection of the activity of the protease trypsin and its soybean inhibitor. This is possible because small fragments produced by the trypsin digestion are not detectable by the protamine sensing membrane. The real time response to the proteolytic reaction is shown to exhibit good reproducibility and reversibility, and the initial reaction rate is dependent on the concentration of the protease and its inhibitor.
Introduction
Potentiometric polyion sensitive electrodes can be successfully used for the detection of enzyme activity if the enzyme used can cleave the polyion into shorter fragments that are no longer detectable by such sensors. Compared with traditional spectroscopic methods, electrochemical measurements may offer significant advantages if the sample possesses a high optical density or turbidity [1]. Yun et al. employed potentiometry with polymeric ion-selective electrode membranes that were doped with the ion-exchanger potassium tetrakis(chlorophenyl) borate (KTpClPB) to directly monitor the response to protamine and to analyze the enzymatic protamine digestion by trypsin [1]. The initial potential drop was found to be linearly dependent on the concentration of trypsin in a given concentration range. Researchers from the same group later applied the same methodology with dinonylnaphthalene sulfonate (DNNS) as the active component in the membrane to enhance its selectivity over common cations in the sample [2]. Consequently, the catalytic cleavage activity of chymotrypsin and renin on synthetic peptide substrates that are rich in diarginine or triarginine residues were studied in undiluted plasma and blood samples [3]. At the same time, the authors also found a very poor activity of such enzymes for substrates such as protamine, which lacks such active cleavage sites, corroborating their proposed approach [3].
Beyond the direct detection of enzyme activity, protamine-sensitive electrochemical sensors have also be used to monitor the activity of a corresponding enzyme inhibitor. Badr et al. demonstrated the feasibility of detecting trypsin-like protease inhibitors in real time, such as α1-antiproteinase inhibitor, α2-macroglobulin, aprotinin and soybean inhibitor [4]. The initial potential decrease upon addition of a mixture of enzyme and inhibitor was found to be dependent on the concentration of inhibitor. Recovery measurements of aprotinin in spiked treated plasma yielded recovery rates of 97–105% for blood samples containing 0.19 to 0.48 μg·mL−1 aprotinin [4, 5].
Potentiometric polyion sensitive electrodes of this type can also find applications in non-separation immunoassays, which employ labeled polyions or related enzymes as markers to detect analytes that can serve as a label through the competitive binding of free and labeled analytes with antibodies. The well-established avidin-biotin system was utilized as a model system to demonstrate the promise of such applications. [5–8]
Although potentiometry employing non-equilibrium ion extraction has been successful in polyion detection and associated applications [8–10], this technique has limitations. Since the non-equilibrium extraction process is generally not reversible, polyion sensitive electrodes based on this principle can typically only be used in a disposable design. Alternatively, a chemical regeneration of the membrane is possible [11], which seems most attractive via sample pH changes as demonstrated with chemically modified membrane compositions. [12]
Recently, a pulsed chrono-potentiometric control of similarly configured membrane electrodes, so-called pulstrodes, has afforded an instrumental control over the ion extraction process [13–16]. Because of a potentiostatic stripping pulse applied after a current-controlled ion extraction pulse, the sensing membrane is regenerated after each pulse cycle. This principle was used to develop operationally reversible polyion sensors that showed promise in the measurement of undiluted whole blood samples [13, 15]. In parallel work, other authors developed corresponding voltammetric techniques with the aim of improving sensing characteristics, and demonstrated a linear relationship between polyion concentration and electrochemical signal under certain conditions. [17, 18] Here, polyion pulstrodes are demonstrated to be useful in the reversible detection of the activity of a protease enzyme, and its inhibitor, that can cleave arginine rich polyions such as protamine into smaller fragments.
Experimental
Reagents
High molecular weight poly(vinyl chloride) (PVC), 2-nitrophenyl octyl ether (o-NPOE), tetradodecylammonium tetrakis(4-chlorophenyl) borate (ETH 500), tetrahydrofuran (THF), and all salts were purchased from Fluka Chemical Corp. (Milwaukee, WI). Protamine sulfate (from herring), trypsin (from bovine pancreas), and trypsin soybean inhibitor (type II-s, SI) were purchased from Sigma (St. Louis, MO). Aqueous solutions were prepared with Nanopure deionized water (18.2 MΩcm). The lipophilic salt DNNS-TDDA was prepared before in our group by metathesis of dinonylnaphthalene sulfonic acid (DNNS) and tetradodecylammonium chloride (TDDACl) according to reference [15].
Electrode Preparation
The ion-selective membranes (200 μm thick) contained PVC and o-NPOE, 1:2 by weight and 5 wt % lipophilic salt DNNS-TDDA. The membranes were prepared by solvent casting, using THF as solvent. The membranes were cut with a cork borer (6-mm diameter) from the parent membrane and incorporated into a Philips electrode body (IS-561, Glasbläserei Möller, Zürich, Switzerland). The inner filling solution consisted of 0.1 M NaCl in 10 mM Tris-HCl buffer (pH = 7.4) and was contacted with an internal Ag/AgCl electrode. The electrodes were conditioned overnight before experiments in the solution identical to inner filling solution. A double-junction Ag/AgCl electrode with a 1 M LiOAc bridge electrolyte was used as an external reference electrode.
Experimental Setup
Pulstrode measurements were conducted in a three-electrode cell system where the Philips body electrode that acted as the working electrode, an external reference electrode and a counter electrode (coiled platinum wire) were immersed into the sample. The pulsed galvanostatic/potentiostatic technique was utilized in the control of the ion-selective membrane, which was described in detail elsewhere [15, 16]. During the experiments, each applied constant current pulse with a current density of 0.5 μA/cm2 (of 0.5-s duration) was applied to induce the ion flux toward the membrane. This step was followed by a zero current pulse (of 0.5-s duration) to avoid any undesired iR drop if optinally measured there. After that, a constant potential pulse (of 15-s duration) was imposed to strip the ions out of the membrane so that the membrane could be electrochemically regenerated, which is the main reason for the good reproducibility of the technique. Sampled potentials, which represent the sensor response, were obtained as the average value during the last 50 ms of the first current pulse. A scheme showing this sequence is presented in Figure 1.
Fig. 1.
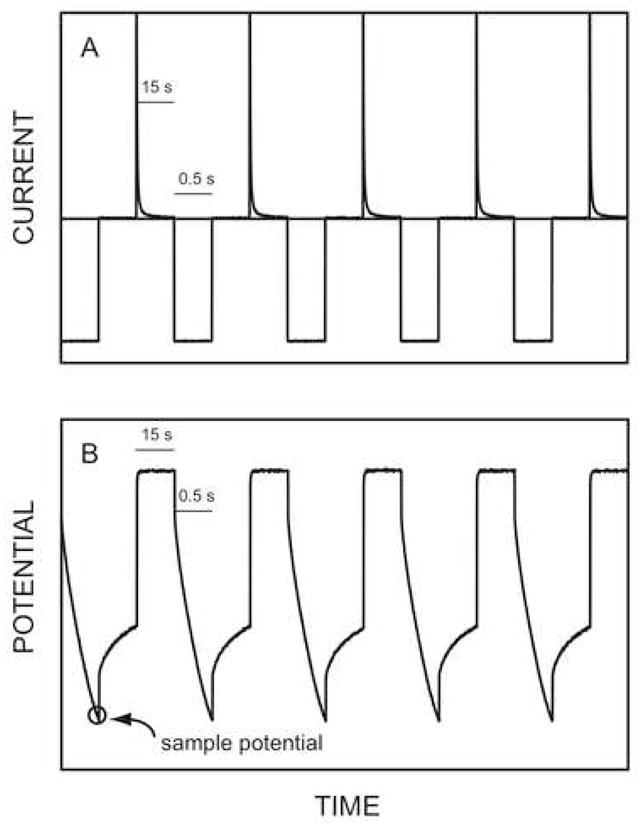
Observed typical time trace of potential and current during the pulstrode measurement. A) current response; B) potential response.
Results and discussion
It was shown earlier that pulsed chrono-potentiometric protamine sensors may possess reversible response characteristics that are maintained in undiluted whole blood samples with acceptable lifetimes and good stability [13, 15]. Figure 2 shows the calibration curve to protamine obtained here in a background of 0.1 M sodium chloride and 10 mM Tris-HCl buffer (pH 7.4). The sigmoidal shape is typical for such a polyion response and is indicative of a competitive extraction process with background cations that saturates at higher protamine concentrations. Unfortunately, an exact response theory that quantitatively agrees with experimental data is difficult to describe. For this reason, the modified standard sigmoidal equation shown here was used to fit experimental data in a semi-empirical manner to describe the pulsed chronopotentiometric sensor response:
| (1) |
Fig. 2.

Pulsed chrono-potentiometric protamine response of DNNS-TDDA protamine-sensitive membrane electrode in 0.1 M sodium chloride and 10 mM Tris-HCl buffer.
In equation 1, Eo represents the potential in a 0.1 M NaCl background (here, Eo = −0.267 V) and c is the concentration of protamine. The empirical constants a1,a2,a3 were chosen as 0.08, 1.65 and −5.95, respectively. Protamine is known to possess a high content of basic amino acids, of which about 50% are arginine residues. This makes it an attractive substrate to study trypsin digestion reactions. Indeed, the response to protamine decreased dramatically upon addition of a high concentration of the protease trypsin, indicative of the occurrence of a proteolytic reaction (see Figure 3). All experiments were performed using the same electrode, and the membrane was merely washed with the same buffer as the background solution between measurements. A total baseline shift for the entire series of experiments was found as about 7 mV, demonstrating the effective electrochemical renewal of the sensing membrane.
Fig. 3.
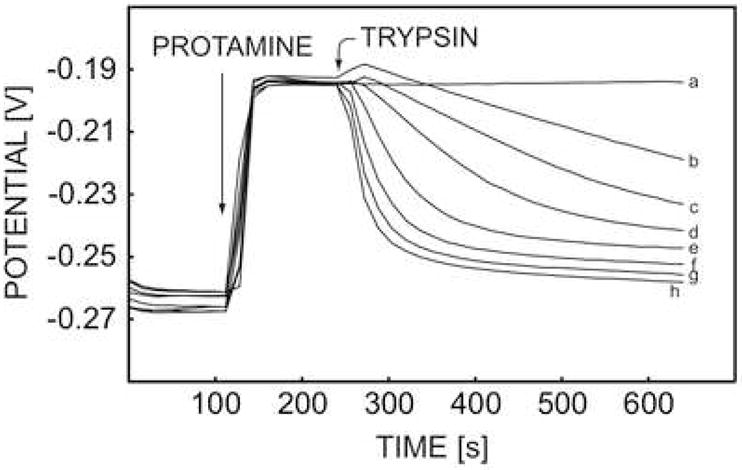
Pulstrode responses of DNNS-TDDA protamine-sensitive membrane electrode in 0.1 M sodium chloride and 10 mM Tris-HCl buffer toward 20 mg·L−1 protamine and the subsequent effect of adding different concentrations of trypsin (a) 0 U·mL−1, (b) 2.5 U·mL−1, (c) 5 U·mL−1, (d) 10 U·mL−1, (e) 30 U·mL−1, (f) 60 U·mL−1, (g) 100 U·mL−1, and (h) 200 U·mL−1.
The reproducibility of the enzyme activity measurement stated above was evaluated by repeating the same experiment three times, with 200 units/mL trypsin. As shown in Figure 4, the potential difference for the three consecutive measurements was less than 3 mV, again demonstrating excellent reversibility of the signal and renewal of the membrane.
Fig. 4.
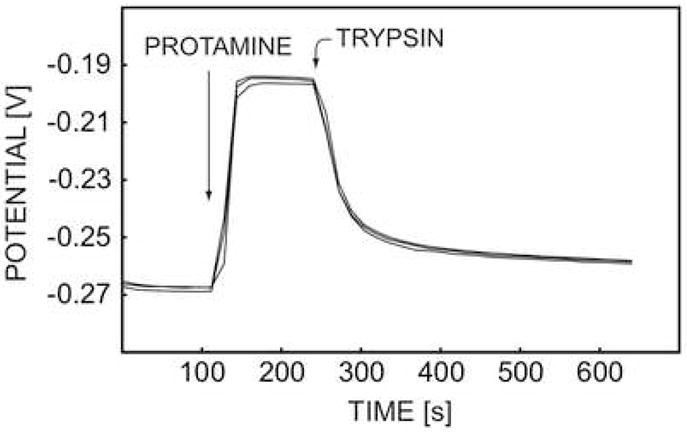
The reproducibility of pulstrode responses of DNNS-TDDA protamine-sensitive membrane electrode in 0.1 M sodium chloride and 10 mM Tris-HCl buffer toward 20 mg·L−1 protamine and 200 U·mL−1 trypsin.
The reaction rate for the digestion of protamine by trypsin may be estimated from the slope of the initial potential decrease upon trypsin addition. A gradually decreasing slope in Figure 3 implies an increasing reaction rate with increasing concentration of trypsin. In a separate experiment, the concentration of trypsin was fixed at 60 units per milliliter, and a change of protamine concentration from 8 mg L−1 to 20 mg L−1 did not induce a significant variation of trypsin activity (see Figure 5), indicating the rate limiting effect of trypsin under the experimental condition used here.
Fig. 5.
Pulstrode responses of DNNS-TDDA protamine-sensitive membrane electrode in 0.1 M sodium chloride and 10 mM Tris-HCl buffer toward 60 U·mL−1 trypsin and different concentrations of protamine (a) 8 mg·L−1, (b) 10 mg·L−1, (c) 15 mg·L−1, and (d) 20 mg·L−1.
To calculate the reaction rate, the average potential change rate and the rate of protamine concentration change during the first 96 seconds immediately after addition of trypsin were depicted against the concentration of trypsin in Figure 6a and Figure 6b, respectively. The protamine concentration change was calculated from the potential readings using the protamine calibration curve and equation 1. As shown in Figure 6, the proteolytic activity change of trypsin and its concentration exhibit an exponential relationship. The activity change levels off above ca. 100 units/mL trypsin, very similar to the rate change in Michaelis-Menten enzyme kinetics. The fitting of experimental data to Michaelis-Menten kinetics exhibited a good correlation with Vmax = 21.2 mg/L·s and Km = 8.28 mg/L.
Fig. 6.
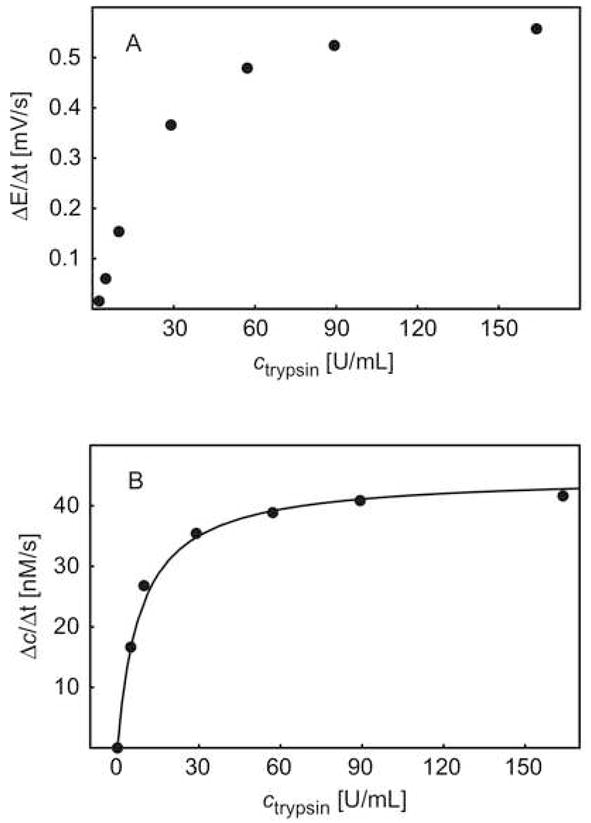
A) Observed initial rate of potential change (ΔE/Δt) as a function of trypsin concentration; B) initial rate of protamine concentration change (Δc/Δt) as a function of trypsin concentration, and the corresponding Michaelis-Menten kinetics (see text).
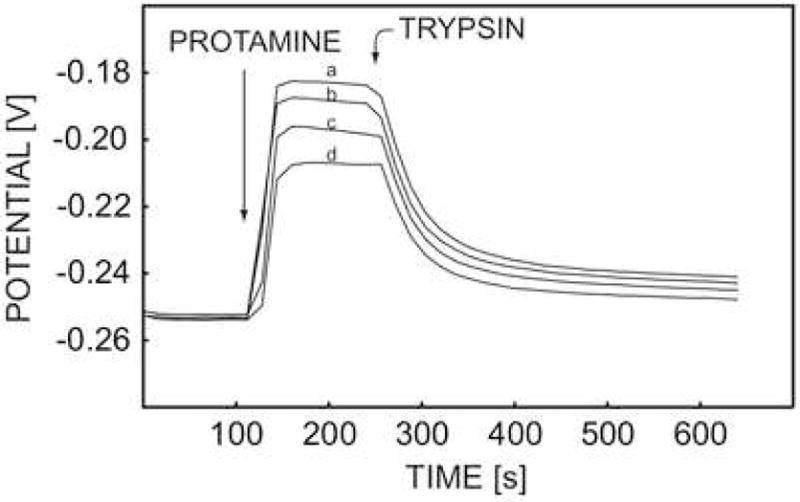
The activity of a protease inhibitor was detected earlier in pretreated plasma samples with the potentiometric protamine sensor and the trypsin-like inhibitor [4]. As stated above, every electrode was disposed after a single use since the protamine detection was not reproducible at the time. Here, the activity of trypsin soybean inhibitor (SI) was studied with the pulstrode protamine sensor. As depicted in Figure 7, the proteolytic reaction of trypsin was effectively inhibited by addition of soybean inhibitor. The membrane was regenerated effectively and repeated runs exhibited similar baseline potentials within 5 mV, allowing for the repeated use of the same membrane.
Fig. 7.

Pulstrode responses of DNNS-TDDA protamine-sensitive membrane electrode in 0.1 M sodium chloride and 10 mM Tris-HCl buffer toward 50 U·mL−1 trypsin and different concentrations of SI inhibitor at (a) 0 mg·L−1, (b) 0.5 mg·L−1, (c) 1 mg·L−1, and (d) 1.5 mg·L−1, (e) 2 mg·L−1, (f) 2.5 mg·L−1, (g) 3 mg·L−1.
The inhibited reaction rate was evaluated also by the observed potential change and calculated protamine concentration change in the course of the first 96 seconds after addition the mixture of trypsin and soybean inhibitor with a fixed trypsin concentration of 50 units/mL. The data are shown in Figure 8a and 8b versus the concentration of soybean inhibitor. The reaction rate decreased upon increased concentration of soybean inhibitor, as expected, and dropped to almost zero when the concentration of soybean inhibitor reached 3 mg·L−1. The concentration of inhibitor required to achieve 50% observable maximium inhibition (I50 value) was calculated as 76 nM by using a soybean inhibitor molar mass of 21,500 daltons. This corresponds well with previous work using zero potentiometric detection with membrane electrodes (85 nM) [4].
Fig. 8.
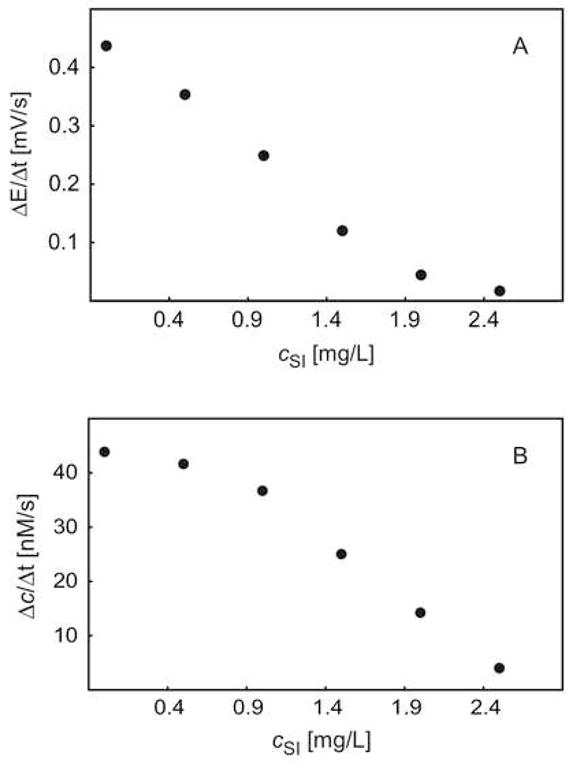
The observed initial rate of A) potential change (ΔE/Δt), B) protamine concentration change (Δc/Δt), as a function of SI inhibitor concentration, immediately after the addition of trypsin and SI inhibitor.
Conclusion
A pulsed galvanostatic protamine sensor has been employed to evaluate the activity of the protease trypsin that may cleave protamine into small fragments, as well as its soybean inhibitor. A good reproducibility and reversibility was demonstrated for the detection of enzyme activity and the calculated reaction rates correlate well with previous potentiometric trypsin soybean inhibitor activity measurement that used disposable electrodes. The use of pulsed chronopotentiometric polyion sensors exhibit favorable response characteristics that should make them attractive tools for the monitoring of suitable enzyme reactions in physiological samples.
Acknowledgments
The authors are grateful to the National Institutes of Health (GM071623) for financial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Yun JH, Meyerhoff ME, Yang VC. Anal Biochem. 1995;224:212. doi: 10.1006/abio.1995.1032. [DOI] [PubMed] [Google Scholar]
- 2.Ramamurthy N, Baliga N, Wahr J, Schaller U, Yang VC, Meyerhoff ME. Clin Chem. 1998;44:606. [PubMed] [Google Scholar]
- 3.Han IS, Ramamurthy N, Yun JH, Schaller U, Meyerhoff ME, Yang VC. FASEB Journal. 1996;10:1621. doi: 10.1096/fasebj.10.14.9002554. [DOI] [PubMed] [Google Scholar]
- 4.Badr IHA, Ramamurthy N, Yang VC, Meyerfhoff ME. Anal Biochem. 1997;250:74. doi: 10.1006/abio.1997.2188. [DOI] [PubMed] [Google Scholar]
- 5.Dai S, Esson JM, Lutze O, Ramamurthy N, Yang VC, Meyerhoff ME. J Pharm Biom Anal. 1999;19:1. doi: 10.1016/s0731-7085(98)00134-4. [DOI] [PubMed] [Google Scholar]
- 6.Dai S, Meyerhoff ME. Electroanalysis. 2001;13:276. [Google Scholar]
- 7.Ducey MW, Jr, Smith AM, Guo X, Meyerhoff ME. Anal Chim Acta. 1997;357:5. [Google Scholar]
- 8.Fu B, Yun JH, Wahr J, Meyerhoff ME, Yang VC. Adv Drug Deliv Rev. 1996;21:215. [Google Scholar]
- 9.Fu B, Bakker E, Yun JH, Yang VC, Meyerhoff ME. Anal Chem. 1994;66:2250. doi: 10.1021/ac00086a009. [DOI] [PubMed] [Google Scholar]
- 10.Fu B, Bakker E, Wang E, Yun JH, Yang V, Meyerhoff ME. Electroanalysis. 1995;7:823. [Google Scholar]
- 11.Mathison S, Bakker E. J Pharm Biom Anal. 1999;19:163. doi: 10.1016/s0731-7085(98)00185-x. [DOI] [PubMed] [Google Scholar]
- 12.Mathison S, Bakker E. Anal Chem. 1999;71:4614. doi: 10.1021/ac990387s. [DOI] [PubMed] [Google Scholar]
- 13.Shvarev A, Bakker E. J Am Chem Soc. 2003;125:11192. doi: 10.1021/ja037167n. [DOI] [PubMed] [Google Scholar]
- 14.Shvarev A, Bakker E. Anal Chem. 2003;75:4541. doi: 10.1021/ac034409t. [DOI] [PubMed] [Google Scholar]
- 15.Shvarev A, Bakker E. Anal Chem. 2005;77:5221. doi: 10.1021/ac050101l. [DOI] [PubMed] [Google Scholar]
- 16.Makarychev-Mikhailov S, Shvarev A, Bakker E. J Am Chem Soc. 2004;126:10548. doi: 10.1021/ja047728q. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Amemiya S. Anal Chem. 2004;76:6877. doi: 10.1021/ac048879e. [DOI] [PubMed] [Google Scholar]
- 18.Guo J, Amemiya S. Anal Chem. 2006;78:6893. doi: 10.1021/ac061003i. [DOI] [PubMed] [Google Scholar]