

Abstract
Rationale
Hyperhomocysteinemia contributes to vascular dysfunction and risks of cardiovascular diseases. Stromal cell-derived factor-1 (SDF-1), a chemokine expressed by endothelial cells (ECs), is highly expressed in advanced atherosclerotic lesions. The interplays among homocysteine, chemokines and shear stress in regulating vascular endothelial function are not clearly understood.
Objective
To investigate the mechanisms for modulations of EC SDF-1 expression by homocysteine and shear stress.
Methods and Results
Homocysteine stimulation induced dose- and time-dependent SDF-1 expression and phosphorylation of mitogen-activated protein kinases (MAPKs) ERK, JNK, and p38. By using specific inhibitors, small interfering RNA (siRNA), and dominant negative mutants, we demonstrated that activation of JNK pathway is critical for the homocysteine-induced SDF-1 expression. Transcription factor ELISA and chromatin immunoprecipitation assays showed that homocysteine increased Sp1- and AP-1-DNA binding activities in ECs. Inhibition of Sp-1 and AP-1 activations by specific siRNA blocked the homocysteine-induced SDF-1 promoter activity and expression. Preshearing of ECs for 1-4 h at 20 dyn/cm2 inhibited the homocysteine-induced JNK phosphorylation, Sp1 and AP-1 activation, and SDF-1 expression. The homocysteine-induced SDF-1 expression was suppressed by nitric oxide (NO) donor. Inhibitor or siRNA for endothelial NO synthase (eNOS) abolished the shear-inhibition of SDF-1 expression.
Conclusions
Our findings serve to elucidate the molecular mechanisms underlying the homocysteine induction of SDF-1 expression in ECs and the shear stress protection against this induction.
Keywords: homocysteine, endothelial cells, stromal cell derived factor-1, shear stress, transcriptional regulation
Vascular endothelial cells (ECs) are constantly exposed to fluid shear stress, the nature and magnitude of which play a significant role in the homeostasis of blood vessels. The preferential development of atherosclerosis at arterial branches and curvatures, where local flow is disturbed, suggests a role of shear stress in atherogenesis. Adhesion of circulating monocytes and T-lymphocytes to ECs and their subsequent migration across the EC monolayer are early events in atherogenesis.1 Physiological levels of shear stress can modulate cellular signaling and EC function in ways that are protective against atherogenesis.2,3 A number of pathophysiologically relevant genes, such as adhesion molecules,4 growth factors,5 and chemokines,6 have been shown to be regulated by shear stress.
Chemokines play a significant role in atherosclerotic lesion development, primarily by inducing the transendothelial migration of leukocytes. Furthermore, chemokines may stimulate smooth muscle cell (SMC) proliferation and migration from media to intima, as well as angiogenesis, thus influencing plaque formation, progression and rupture.7 Chemokines are divided into CXC, CC, C, or CX3C based on their structural properties and primary amino acid sequence.8 Stromal cell-derived factor-1 (SDF-1), a member of the CXC family, is a strong chemoattractant in EPCs, lymphocytes, and monocytes9,10 and implicated in atherogenesis.11 SDF-1 has also been shown to induce platelet activation and aggregation via CXCR4 expressed on platelets, suggesting a link with atherothrombosis.12 SDF-1 expression can be regulated by extracellular factors derived from inflammatory or injured vascular tissues. The molecular mechanisms involved in the up-regulation of SDF-1 expression in pathophysiological conditions such as hyperhomocysteinemia is still unknown.
Homocysteine is a sulfur-containing amino acid formed during the metabolic demethylation of methionine. Elevated blood levels of homocysteine are related to higher risks of coronary heart disease, stroke and peripheral vascular disease.13 Homocysteine may promote vascular damage and atherothrombosis via several mechanisms, including release of proinflammatory mediators, induction of endothelial dysfunction, and activation of apoptotic pathways in vascular cells.14 The relationship between the plasma levels of homocysteine and SDF-1, the mechanisms underlying homocysteine-induced SDF-1 expression, and the role of shear stress in the modulation of homocysteine-induced gene expression still remain unclear.
We aimed to establish the relationship between plasma homocysteine and SDF-1 levels in human blood and the interplay between shear stress and homocysteine stimulation in modulating endothelial gene expression by analyzing the effect of shear stress on homocysteine-induced SDF-1 expression in human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs). We found that the induction of SDF-1 expression by homocysteine was mediated via phosphorylation of JNK and activation of the transcription factors activator protein 1 (AP-1) and Sp1. Furthermore, fluid shear stress attenuated the homocysteine-induced SDF-1 expressions at mRNA and protein levels, and ECs subjected to shear stress suppress the homocysteine-induced JNK phosphorylation and transcription factor activation. In addition, homocysteine-induced SDF-1 expression was modulated by nitric oxide (NO): An NO donor suppressed homocysteine-induced SDF-1 expression, and inhibition of endothelial NO synthase (eNOS) attenuated shear stress-inhibition of SDF-1 expression. These findings on the mechanisms of suppression of homocysteine-induced responses in ECs by shear stress provide new insights into the pathophysiology underlying the atheroresistancy of straight segments of vascular tree.
Materials and Methods
Materials and the procedures of subjects; measurement of plasma homocysteine; SDF-1 enzyme-linked immunosorbent assay (ELISA); endothelial cell culture; shear stress experiment; real-time quantitative PCR; western blot analysis; reporter gene construct, siRNA, transfection, and luciferase assays; adenoviral Infection; Sp1 and AP-1 transcription factor assays (TF ELISA assays); chromatin immunoprecipitation assay (ChIP); and statistical analysis are provided in Online Supplemental Document.
Results
The relationship between plasma levels of homocysteine and SDF-1
The mean plasma homocysteine concentration was significantly higher in high-risk patients (30.94 ± 4.13 μmol/L) than normal volunteers (14.08 ± 1.02 μmol/L). Linear regression shows a positive correlation between plasma homocysteine and SDF-1 levels in patients (Figure 1A, r = 0.81, p < 0.0001) and volunteers (Figure 1B, r = 0.51, p < 0.001). Linear regression of data combined the two groups was showed in Figure S1 (r = 0.79, p < 0.0001). We divided all subjects (patients + normal volunteers) according to their plasma homocysteine levels into normal (< 15 μmol/L, n=24, 10 from patients), mild (16-30 μmol/L, n=36, 11 from patients), and intermediate (31-100 μmol/L, n=12, 11 from patients) hyperhomocysteinemia.15 Plasma SDF-1 level was significantly higher in subjects with intermediate hyperhomocysteinemia than normal or mild hyperhomocysteinemia (Figure 1C), indicating that higher levels of plasma homocysteine were associated with higher SDF-1.
Figure 1.

Relationships between plasma levels of homocysteine and SDF-1 in (A) high risk patients, and (B) normal volunteers. (C) Subjects with intermediate hyperhomocysteinemia (int hhcy) had significantly higher levels of SDF-1 compared to subjects with normal or mild hyperhomocysteinemia. The results are shown as mean ± standard deviation (SD). *P < 0.01 versus normal subjects. #P < 0.01 versus mild hyperhomocysteinemia.
Homocysteine-induced SDF-1 expression is dose- and time-dependent
HUVECs and HAECs were stimulated with homocysteine at 100 μM for the times indicated, or different doses (0-500 μM) for 4 h (for mRNA expression by real-time PCR) and 12 h (for protein secretion by ELISA). In ECs, the SDF-1 mRNA level began to increase after 1 h of homocysteine stimulation and reached highest level at 4 h; thereafter it gradually reduced to a level that is not significantly different from the static control in HUVECs at 12 hr, but was still elevated at this time in HAECs (Figure 2A). SDF-1 protein in the conditioned medium also increased after stimulation (Figure 2C). The inductions of SDF-1 mRNA expression and protein secretion by homocysteine were dose-dependent (Figures 2B and 2D).
Figure 2.

Induction of SDF-1 expression in ECs by homocysteine stimulation. RNA samples were isolated at the indicated time periods or doses. All bar graphs represent folds of control ECs (CL) and normalized to 18S rRNA by real-time PCR analysis (A,B). The SDF-1 protein secretion was determined by ELISA analyses (C,D). ECs were stimulation with 100 μM homocysteine for the times indicated (A,C), or stimulated with homocysteine at various doses for 4 h (B) or 12 h (D). Data are shown as mean ± standard error of the mean (SEM). *P < 0.05 versus CL HUVECs. #P < 0.05 versus CL HAECs.
The viability of ECs was over 94% when cultured for 24 h with 20-200 μM homocysteine, and 87% with 500 μM homocysteine (data not shown).
Homocysteine-induced SDF-1 expression in EC was mediated by JNK pathway
Members of the MAPK superfamily are known to regulate EC gene expression and cellular functions.16 The phosphorylation of ERK, JNK, and p38 in HUVECs increased rapidly after homocysteine stimulation, reaching maximal levels at 2 min for ERK and JNK, and 5 min for p38 (Figure 3A). After transient increases, phosphorylation decreased to nearly basal levels (Figure S2 shows that JNK phosphorylation decreased to control at 60 min, but ERK and p38 phosphorylations were still elevated at 120 min).
Figure 3.

JNK pathway is required for homocysteine-induced SDF-1 expression. (A) HUVECs were kept as controls (CL) or stimulated with 100 μM homocysteine, and the phosphorylations of ERK, JNK, and p38 were determined by Western blotting. (B-D) HUVECs were kept as CL or stimulated with homocysteine for 4 h (B,D), or 12 h (C). Before being kept as CL or stimulated with homocysteine, ECs were (1) pretreated with PD98059 (PD), SP600125 (SP), or SB203580 (SB) individually for 1 h (B,C); (2) transfected with control siRNA (si-CL) or a specific siRNA of si-ERK, si-JNK, or si-p38 (D); or (3) infected with control adenovirus (Ad-GFP), DN-ERK, DN-JNK, or DN-p38 (D). All bar graphs represent folds of CL ECs and normalized to 18S rRNA (B,D). The SDF-1 secretion was determined by ELISA analyses (C). The results are shown as mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus vehicle control (DMSO in B,C), control siRNA (si-CL in D), or control adenovirus (Ad-GFP in D) with homocysteine stimulation.
HUVECs were incubated with specific inhibitors for ERK (PD98059; 30 μM), JNK (SP600125; 20 μM), or p38 (SB203580; 10 μM) for 1 h before and during stimulation with homocysteine. The homocysteine-induced SDF-1 mRNA expression (Figure 3B) and protein secretion (Figure 3C) were significantly inhibited by SP600125, but not by PD98059 and SB203580. The homocysteine-induced SDF-1 mRNA expression was also inhibited by transfection with JNK-specific siRNA or infection with Ad-DN-JNK, but not by ERK- or p38-specific siRNA (100 μmol/mL each), nor with Ad-DN-ERK or Ad-DN-p38 (Figure 3D). The effectiveness of these treatments was validated: ERK-, JNK-, and p38-specific siRNA (compared with control siRNA) caused a 75% reduction in ERK, JNK, and p38 protein expressions, respectively (data not shown). The DN-ERK, DN-JNK and DN-p38 caused at least 80% inhibitions in homocysteine-induced ERK, JNK and p38 phosphorylation (compared with control Ad-GFP) (data not shown).
AP-1 and Sp1 binding sites are essential for the homocysteine-induction of human SDF-1 promoter activity
The human SDF-1 gene promoter contains multiple transcription factor binding sites, which have been shown to be responsive to different stimuli.17 To elucidate the cis-acting elements in the SDF-1 gene promoter that mediate the homocysteine-induced SDF-1 transcription, luciferase assay was conducted with p-1010-Luc plasmid and several deletion promoters constructs (Figure 4A). Transient transfection of ECs showed that the 5′-flanking region of human SDF-1 (-1010/+122) could drive transcription of a luciferase reporter (Figure 4B). Construction and analyses of 5′-deletion mutants in the -1010/+122 region of SDF-1, which directed maximum luciferase activity, showed that the activity decreased to ∼45% following sequence deletion from -430 to -214, to ∼75% after further deletion to -111, and was nearly abolished following 5′-deletion to -20 (Figure 4B).
Figure 4.

The roles of Sp1 and AP-1 in homocysteine-induced SDF-1 mRNA expression and promoter activity. (A) Human SDF-1 promoter constructs. The arrows indicate the transcription factor binding sites. (B) HUVECs were cotransfected with 5′-deletion constructs and stimulated with homocysteine for 4 h. SDF-1 promoter activity was measured by luciferase assay normalized to β-galactosidase activity, and shown relative to that of HUVECs transfected with p1010-Luc (set to 100%). (C) SDF-1 mRNA and (D) SDF-1 p1010-Luc activity were determined in HUVECs pretreated with vehicle (DMSO) or Sp1 inhibitor mithramycin A (MMA), or transfected with si-CL, si-Sp1, or si-c-jun and then stimulated with homocysteine for 4 h. All bar graphs represent folds of CL ECs, mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus vehicle control (DMSO), or si-CL with homocysteine stimulation.
To test whether AP-1 and Sp1 activations are involved in the signal transduction pathway leading to the homocysteine-induced SDF-1 gene expression, we transfected HUVECs with siRNA of Sp1 or c-jun, or incubated them with the specific inhibitor for Sp1 (mithramycin A, 100 nM) for 1 h, prior to stimulation with 50 or 100 μM homocysteine for 4 h. The homocysteine-induced SDF-1 mRNA expression (Figure 4C) and SDF-1 promoter activity (Figure 4D) were significantly reduced by inhibition with mithramycin A, or siRNA of Sp1 or c-jun.
Homocysteine induced Sp1- and AP-1-DNA binding activities
To investigate whether Sp1 and AP-1 bind the SDF-1 promoter region in HUVECs, we performed quantitative analysis for Sp1 and AP-1 binding activities in vitro by using TF ELISA kits (Panomics). Treatment of HUVECs with 100 μM homocysteine caused both Sp1- and AP-1-DNA binding activities to increase at 30 min and remain elevated for at least 2 h (Figures 5A and 5B, respectively).
Figure 5.

Sp1 and AP-1 binding activities of the SDF-1 promoter region induced by 100 μM homocysteine stimulation in HUVECs. (A) Sp1 activation and (B) AP-1 activation were determined by TF ELISA assay. All bar graphs represent folds of control HUVECs (CL), mean ± SEM. *P < 0.05 versus CL. (C,D) ChIP assay was performed for Sp1 by using Sp1 antibody (C), and AP-1 by using c-jun antibody (D).
ChIP analysis was performed by subjecting immunoprecipitated chromosomal DNA with anti-Sp1 antibody to PCR using primers designed to amplify the SDF-1 promoter region harboring the Sp1 binding sites. Sp1 indeed bound to the SDF-1 promoter region containing the Sp1 sites (Figure 5C). Similarly, the DNA sequence including the AP-1 sites was specifically immunoprecipitated with anti-c-jun antibody (Figure 5D). These data suggested that the Sp1 and AP-1 binding sites play critical roles in the regulation of SDF-1 by homocysteine.
JNK signaling pathway was involved in homocysteine-induced SDF-1 promoter activity
To evaluate whether the inhibition of SDF-1 expression by JNK signaling pathway occurred at the transcriptional level, we studied the effect of SP600125, JNK siRNA, and DN-JNK on homocysteine-induced SDF-1 promoter activity with the use of p1010-luc reporter construct that contains -1010 bp of the proximal promoter sequences of human SDF-1 gene (p1010-Luc plasmid). Stimulation with homocysteine increases the luciferase activity significantly in HUVECs over the unstimulated control after normalization by transfection control (Figure 6A). Transfection of ECs with JNK siRNA or DN-JNK resulted in a marked inhibition of the homocysteine-induced SDF-1 promoter activity. However, transfection of cells with siRNA for ERK or p38, DN-ERK and DN-p38 had little inhibitory effect on this homocysteine inducibility (Figure 6A). In HUVECs, transfection with siRNA or promoter constructs under the concentrations used does not cause cytotoxicity based on the cell number and morphology (data not shown).
Figure 6.

JNK signaling pathway was involved in homocysteine-induced SDF-1 promoter activity. (A) SDF-1 p1010-Luc activity was determined in HUVECs transfected with si-CL, si-ERK, si-JNK, or si-p38, or infected with Ad-GFP, DN-ERK, DN-JNK, or DN-p38, and then stimulated with homocysteine for 4 h. (B) Sp1 and (C) AP-1 activations were determined by TF ELISA assays in HUVECs pretreated with vesicle (DMSO), transfected with si-CL, si-JNK, or infected with Ad-GFP, DN-JNK, and then stimulated with 100 μM homocysteine for 2 h. All bar graphs represent folds of CL ECs, mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus vehicle control (DMSO), si-CL, or Ad-GFP with homocysteine stimulation.
To explore whether JNK activates the SDF-1 promoter leading to SDF-1 transcription via activation of Sp1 and AP-1, HUVECs were pretreatment with MAPK inhibitors, transfection with JNK siRNA, or infection with Ad-DN-JNK, and followed by homocysteine stimulation, and then the Sp-1 and AP-1 activations were assessed by TF ELISA kits. Homocysteine-induced AP-1 and Sp1-DNA binding activities were significantly inhibited in cells pretreatment with SP600125, JNK siRNA, or Ad-DN-JNK, but not in pretreatment with PD98059, SB203580, control siRNA, or Ad-GFP (Figures 6B and 6C).
Pre-exposure of ECs to high shear stress (HSS) inhibited homocysteine-induced SDF-1 expression
Pre-exposure of ECs to HSS at 20 dyn/cm2 for 4 h significantly inhibited homocysteine-induced SDF-1 mRNA expression (Figure 7A), and this effect is shear time-dependent in HUVECs (Figure S3). Low shear stress (LSS) at 0.5 dyn/cm2 did not have such an effect. Both HSS and LSS alone had no significant effect on SDF-1 expression (Figure 7A). In addition, HUVECs cultured on fibronectin (FN) or collagen type I (COLI) and pre-exposed to HSS for 4 h had similar inhibitory effect on homocysteine-induced SDF-1 expression (Figure S4). Pre-exposure of HUVECs to HSS resulted in a marked inhibition of the homocysteine-induced JNK phosphorylation (Figure 7B). Pre-exposure of HUVECs to HSS also resulted in a significant inhibition of the homocysteine-induced SDF-1 promoter activity (Figure S5).
Figure 7.
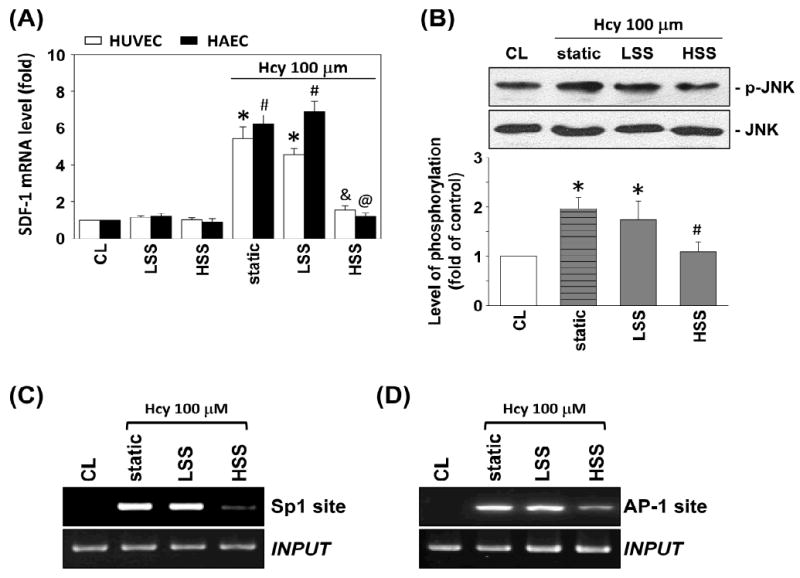
Pre-exposure of ECs to HSS inhibited homocysteine-induced SDF-1 expression. (A) ECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) or LSS (0.5 dyn/cm2) for 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). All bar graphs represent folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL HUVECs. #P < 0.05 versus CL HAECs. &P < 0.05 versus static HUVECs with homocysteine stimulation. @P < 0.05 versus static HAECs with homocysteine stimulation. (B) The phosphorylation of JNK after 30 min homocysteine stimulation in HUVECs was determined by Western blotting. Data are presented as percentage changes of CL HUVECs; normalized to JNK protein level. *P < 0.05 versus CL HUVECs. #P < 0.05 versus static HUVECs with homocysteine stimulation. (C) Sp1 and (D) AP-1 binding to SDF-1 promoter in HUVECs after 2 h homocysteine stimulation was analyzed by ChIP assay. ChIP assay was performed by using Sp1 and c-jun antibodies, respectively.
Preshearing of HUVECs at HSS for 4 h significantly inhibited the homocysteine-induced Sp-1- and AP-1-DNA binding activities in vivo by ChIP assays (Figures 7C and 7D) and in vitro by using TF ELISA kits (Figures S6A and S6B). These results provide additional evidence that shear stress plays an important role in the inhibition of the JNK-, Sp1- and AP-1-mediated SDF-1 expression in ECs induced by homocysteine stimulation.
Effect of NO on homocysteine-induced SDF-1 expression
A major effect of hyperhomocysteinemia-induced EC dysfunction appears to be related to decreased bioavailability of EC-derived NO.18 To investigate whether the homocysteine-induced SDF-1 expression is modulated by NO, both HUVECs and HAECs were incubated with different doses of NO donor S-nitroso-N-acetyl-penicillamine (SNAP) for 4 h before and during stimulation with homocysteine. The homocysteine-induced mRNA expression of EC SDF-1 was significantly inhibited by 20-100 ìM SNAP treatment (Figure 8A). Conversely, addition of 100 μM NG-nitro-L-arginine methyl ester (L-NAME) or transfection of eNOS siRNA before exposure to 4 h of HSS abolished the shear-mediated inhibition of SDF-1 expression (Figure 8B). These results indicated that NO plays an important role in the homocysteine-induction and shear-inhibition of SDF-1 expression in ECs.
Figure 8.
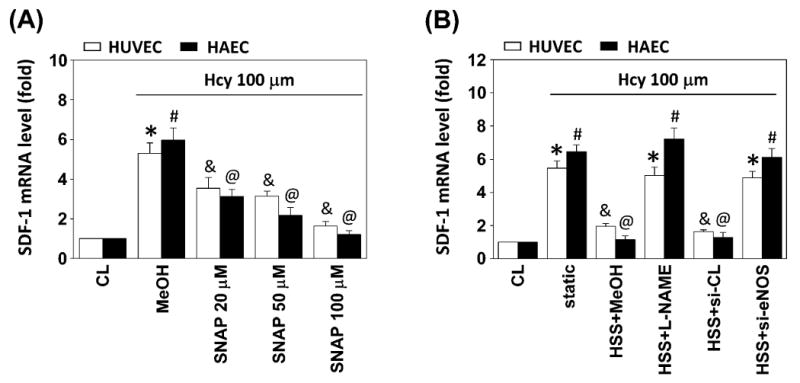
Effect of NO on homocysteine-induced SDF-1 expression. All bar graphs represent folds of control ECs (CL) and normalized to 18S rRNA. The results are shown as mean ± SEM. (A) ECs were kept as CL or stimulated with homocysteine for 4 h. Before being kept as CL or stimulated with homocysteine, ECs were pretreated with 20-100 μM SNAP for 4 h. *P < 0.05 versus CL HUVECs. #P < 0.05 versus CL HAECs. &P < 0.05 versus methanol (MeOH)-treated HUVECs (vehicle control) with homocysteine stimulation. @P < 0.05 versus MeOH-treated HAECs with homocysteine stimulation. (B) ECs were kept as CL or presheared at HSS (20 dyn/cm2) for 4 h before homocysteine stimulation. Before preshearing, ECs were pretreated with 100 μM L-NAME for 4 h or transfected with si-CL or si-eNOS. Static ECs were stimulated with homocysteine without preshearing (static). *P < 0.05 versus CL, vesicle control (HSS+MeOH), or control siRNA (HSS+si-CL) HUVECs with homocysteine stimulation. #P < 0.05 versus CL, vesicle control (HSS+MeOH), or control siRNA (HSS+si-CL) HAECs with homocysteine stimulation. &P < 0.05 versus L-NAME-treated (HSS+L-NAME) or eNOS siRNA-transfected (HSS+si-eNOS) HUVECs with homocysteine stimulation. @P < 0.05 versus L-NAME-treated (HSS+L-NAME) or eNOS siRNA-transfected (HSS+si-eNOS) HAECs with homocysteine stimulation.
Discussion
The production of proinflammatory factors in vascular cells play an important role in atherogenesis.19,20 SDF-1 is a potent chemokine that recruits mononuclear cells to the inflamed tissues,10,11 and has been shown to be highly expressed in human atherosclerotic plaques.12 Moreover, increased levels of blood SDF-1 have been associated with stable coronary disease,21 and SDF-1 is likely to be involved in neointimal hyperplasia or thrombus formation after injury.22 The ability of homocysteine to stimulate SDF-1 gene expression in ECs may lead to the elevation of SDF-1 in the circulation during hyperhomocysteinemia. The mechanism by which homocysteine regulates SDF-1 gene expression of ECs, however, remains unclear. The novel findings of the present study are (Figure S7): 1) plasma homocysteine is positively correlated with SDF-1 in both high-risk patients and normal volunteers, 2) homocysteine stimulates SDF-1 mRNA expression and protein secretion in ECs, 3) homocysteine-induced SDF-1 expression in ECs is mediated via JNK phosphorylation and Sp1, AP-1 activation, 4) shear stress attenuates the homocysteine-induced SDF-1 expression, and 5) the effect of homocysteine and shear stress on EC SDF-1 expression is mediated by NO.
The results of this study demonstrate that homocysteine not only promotes the secretion of SDF-1, but also induces their gene transcription and expression in human ECs. Analysis of the human SDF-1 promoter activity with different plasmid constructs revealed that AP-1 and Sp1 function as the cis-element for homocysteine responsiveness via JNK phosphorylation. SDF-1 promoter has different binding sites for various transcriptional factors.17 Previous studies have shown that Sp123 and AP-124 can be activated through the JNK pathway in ECs. Regulation of gene expression through the use of combinations of different transcription factors such as Sp1 and AP-1 has been reported,25,26 and JNK is involved in the phosphorylation and activation of c-Jun.27 In this study, we performed luciferase assays to show that Sp1 and AP-1 cooperate to activate the human SDF-1 promoter and we used TF ELISA and ChIP assays to demonstrate that the regulation of SDF-1 gene expression in ECs was mediated by increased Sp1- and AP-1-DNA binding activities following JNK phosphorylation. Previous study has shown that Sp1 induces a conformational change in the DNA that may contribute to the activation of AP-1 binding.28 The molecular details of Sp1 and AP-1 cooperation in activating the SDF-1 promoter need further investigations. Based on our results, we propose a possible signal transduction pathway in ECs in which homocysteine induces JNK phosphorylation, which activates Sp1 and AP-1, thus resulting in SDF-1 transcriptional activation.
Hyperhomocysteinemia has been defined as a plasma concentration of homocysteine exceeding 15 μmol/L, and is considered severe at levels beyond 100 μmol/L.15 Severe hyperhomocysteinemia produced by genetic disorders usually has plasma homocysteine elevated over than 100 μmol/L. When untreated, affected individuals have a 50% chance of developing a major vascular disease.18 Mild hyperhomocysteinemia is quite prevalent in the general population (plasma homocysteine 15-50 μmol/L), but also has been shown to be associated with increased risks for cardiovascular diseases.29,30 Previous studies have demonstrated that homocysteine might induce the expression of chemokines, e.g., monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8) in human monocytes and aortic ECs,31,32 and MCP-1 in human vascular SMCs.33 Homocysteine not only promotes the secretion of MCP-1 and IL-8 in human monocytes, but also enhances the responsiveness of monocytes to MCP-1 in hyperhomocysteinemia patients.31 The present findings of a positive correlation of plasma homocysteine with SDF-1 levels and the elevation of SDF-1 in patients group suggest that SDF-1 may play an important role in the pathogenesis of cardiovascular diseases. Furthermore, our results on human EC cultures showed that treatment with homocysteine concentrations characterizing mild to severe hyperhomocysteinemia (20-500 μmol/L) has stimulatory effects on SDF-1 expression. Therefore, hyperhomocysteinemia might provide an effective stimulus for SDF-1 accumulation in the arterial wall, thus promoting the recruitment of leukocytes, and contributing to proinflammatory responses.
The endothelial lining of the vasculature plays an important role in sensing blood flow perturbations leading to the modulation of gene expression in EC. At vessel bifurcations in an arterial tree, disturbed flow accompanied by LSS predisposes ECs to inflammation in which proinflammatory factors are involved; in contrast, laminar shear stress with a clear direction exerts atheroprotective effects.34,35 Our study showed that homocysteine-induced SDF-1 up-regulation was inhibited in ECs subjected to HSS with a clear direction. Pre-exposure of ECs to a high level of shear stress inhibited homocysteine-induced signal transduction and SDF-1 expression. Previous studies demonstrated that shear stress also induced JNK activation in ECs, and that this shear effect was dependent on the specific extracellular matrix.36,37 However, the shear-induced JNK activation seemed to be dependent on cell type and culture conditions. In our experiments, HUVECs cultured on FN or COLI had similar inhibitory effect on homocysteine-induced SDF-1 expression. It is likely that shear stress modulates inflammatory responses by other mechanisms. Berk et al.38 reported that shear stress induced greater extent of ERK activation, and that the ERK activation then repressed JNK activation in HUVECs, indicating that ERK pathway may mediate the shear stress suppression of JNK. Yoshizumi et al.39 suggested that the possible mechanisms for this ERK-induced suppression include the JNK phosphatases and JNK interacting proteins. The tyrosine phosphatase activity of SHP-2 appears to be required for tumor necrotic factor-α (TNF-α)-induced signaling pathways. Inhibition of the phosphatase activity by shear stress represents a presumable mechanism by which shear stress modulates inflammatory factor-induced signal transduction. Therefore, elucidation of the mechanisms in homocysteine- and shear-mediated signal transduction requires further studies on ECs by manipulating activities of other upstream or downstream signaling proteins.
NO has been recognized to be an anti-inflammatory molecule. It is produced by eNOS in response to physiological stimuli such as acetylcholine, thrombin, or shear stress.40 There is evidence in support of a role of NO in the regulating of gene expression in vascular cells. Endogenous NO production inhibited cytokine-induced expression of adhesion molecules as well as leukocyte adhesion.41 Exogenous addition of NO decreased MCP-1 expression in human ECs,42 and also inhibited the expression of MCP-1 in SMCs.43 Previously observations showed that homocysteine decreases NO bioavailability in cultured ECs.18,44 In addition, homocysteine-induced EC apoptosis was inhibited by NO.45 In this study, administration of a SNAP significantly suppressed homocysteine-induced SDF-1 expression. Furthermore, NO produced from eNOS in response to shear stress has been shown to play a crucial role in shear-mediated anti-atherogenic effects.46 Decreased bioavailability of NO may contribute to thrombosis and atherosclerosis, since the prominent effects of EC-derived NO produced in response to shear stress include vessel relaxation and inhibition of cytokine-triggered platelet and monocyte adhesion.46 Our findings on the effects of L-NAME and eNOS siRNA on the shear-induced inhibition in SDF-1 expression in ECs are in concert with the hypothesis that NO may play a role in the shear-mediated suppression of proatherogenic factor-regulated genes. Pre-exposure of ECs to 4 h HSS inhibited homocysteine-induced SDF-1 expression, and this shear effect was blocked by treatment of L-NAME and transfection of eNOS siRNA. Thus, our findings also indicate that EC-derived NO may mediate homocysteine-induced SDF-1 expression in human ECs.
In summary, the present study demonstrates for the first time an increased expression of SDF-1 in ECs and in the circulating blood during hyperhomocysteinemia. This study has identified a unique molecular mechanism of homocysteine-induced JNK phosphorylation, Sp1 and AP-1 activations, and SDF-1 expression in ECs. Our findings provide a molecular basis for the mechanisms underlying the protective function of laminar shear stress against this SDF-1 induction.
Supplementary Material
Figure S1. Relationships between plasma levels of homocysteine and SDF-1 in all subjects (n = 72). Black circles: patients recruited from outpatients at high risk for cardiovascular events. White circles: normal volunteers from routine physical examinations.
Figure S2. Stimulation with homocysteine induces HUVECs to increase their phosphoryalation of ERK (A), JNK (B), and p38 (C). HUVECs were kept as controls (CL) or stimulated with 100 μM homocysteine for the times indicated, and the phosphorylations of ERK, JNK, and p38 were determined by using Western blot analysis. Phosphorylated ERK, JNK, and p38 levels are presented as band densities (normalized to total protein levels) relative to CL. The results are mean ± SEM from 3 independent experiments. *P < 0.05 versus control EC (CL).
Figure S3. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) for 1 ∼ 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). Data are presented folds of control ECs (CL), mean ± SEM. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S4. HUVECs cultured on FN- or COLI-coated glass were presheared at HSS (20 dyn/cm2) for 4 h (HSS/FN or HSS/COLI) before homocysteine stimulation. Static ECs were ECs cultured on FN- or COLI-coated glass and stimulated with homocysteine without preshearing (static). Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static/FN or static/COLI) with homocysteine stimulation.
Figure S5. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) or LSS (0.5 dyn/cm2) for 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). The SDF-1 p1010-Luc activity after 4 h homocysteine stimulation was determined by luciferase assay normalized to β-galactosidase. Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S6. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) or LSS (0.5 dyn/cm2) for 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). Sp1 (A) and AP-1 (B) activation after 2 h homocysteine stimulation in HUVECs was performed by TF ELISA assay. Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S7. Schematic representation of the inhibitory effects of shear stress and the signaling pathway regulating the expressions of SDF-1 in ECs in response to homocysteine stimulation.
Acknowledgments
Sources of Funding: This work was supported by St. Martin De Porres Hospital (project no. P0703) (M.-L.S.); NIH (NHLBI) Research Grant HL-085159 (S.C); and National Science Council (Taiwan) (grant NSC97-2320-B-415-007-MY3) (C.-N.C.)
Non-standard Abbreviations and Acronyms
- SDF-1
stromal cell-derived factor-1
- EC
endothelial cell
- MAPK
mitogen-activated protein kinase
- siRNA
small interfering RNA
- NO
nitric oxide
- eNOS
endothelial nitric oxide synthase
- SMC
smooth muscle cell
- HUVEC
human umbilical vein endothelial cell
- HAEC
human aortic endothelial cell
- AP-1
activator protein 1
- HSS
high shear tress
- LSS
low shear stress
- SNAP
S-nitroso-N-acetyl-penicillamine
- L-NAME
NG-nitro-L-arginine methyl ester
- MCP-1
monocyte chemoattractant protein-1
- IL-8
interleukin-8
- TNF-α
tumor necrotic factor-α
Footnotes
Disclosures: None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 3.Chiu JJ, Chen LJ, Chang SF, Lee PL, Lee CI, Tsai MC, Lee DY, Hsieh HP, Usami S, Chien S. Shear stress inhibits smooth muscle cell-induced inflammatory gene expression in endothelial cells: role of NF-κB. Arterioscler Thromb Vasc Biol. 2005;25:963–969. doi: 10.1161/01.ATV.0000159703.43374.19. [DOI] [PubMed] [Google Scholar]
- 4.Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, Lin CW, Usami S, Chien S. Mechanisms of induction of endothelial cell E-selectin expression by smooth muscle cells and its inhibition by shear stress. Blood. 2007;110:519–528. doi: 10.1182/blood-2006-08-040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conklin BS, Zhong DS, Zhao W, Lin PH, Chen C. Shear stress regulates occludin and VEGF expression in porcine arterial endothelial cells. J Surg Res. 2002;102:13–21. doi: 10.1006/jsre.2001.6295. [DOI] [PubMed] [Google Scholar]
- 6.Tsai YC, Hsieh HJ, Liao F, Ni CW, Chao YJ, Hsieh CY, Wang DL. Laminar flow attenuates interferon-induced inflammatory responses in endothelial cells. Cardiovasc Res. 2007;74:497–505. doi: 10.1016/j.cardiores.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 7.Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–1959. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- 8.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 9.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 10.Chen CN, Chang SF, Lee PL, Chang K, Chen LJ, Usami S, Chien S, Chiu JJ. Neutrophils, lymphocytes, and monocytes exhibit diverse behaviors in transendothelial and subendothelial migrations under coculture with smooth muscle cells in disturbed flow. Blood. 2006;107:1933–1942. doi: 10.1182/blood-2005-08-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braunersreuther V, Mach F, Steffens S. The specific role of chemokines in atherosclerosis. Thromb Haemost. 2007;97:714–721. [PubMed] [Google Scholar]
- 12.Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res. 2000;86:131–138. doi: 10.1161/01.res.86.2.131. [DOI] [PubMed] [Google Scholar]
- 13.Refsum H, Ueland PM, Nygård O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 14.Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004;11:S56–S64. doi: 10.1038/sj.cdd.4401451. [DOI] [PubMed] [Google Scholar]
- 15.Schini-Kerth VB. Homocysteine, a proinflammatory and proatherosclerotic factor: role of intracellular reactive oxygen species. Circ Res. 2003;93:271–273. doi: 10.1161/01.RES.0000089561.87997.CF. [DOI] [PubMed] [Google Scholar]
- 16.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 17.García-Moruja C, Alonso-Lobo JM, Rueda P, Torres C, González N, Bermejo M, Luque F, Arenzana-Seisdedos F, Alcamí J, Caruz A. Functional characterization of SDF-1 proximal promoter. J Mol Biol. 2005;348:43–62. doi: 10.1016/j.jmb.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 18.Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost. 2005;3:1646–1654. doi: 10.1111/j.1538-7836.2005.01364.x. [DOI] [PubMed] [Google Scholar]
- 19.Trepels T, Zeiher AM, Fichtlscherer S. The endothelium and inflammation. Endothelium. 2006;13:423–429. doi: 10.1080/10623320601061862. [DOI] [PubMed] [Google Scholar]
- 20.Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. doi: 10.1161/01.ATV.0000133606.69732.4c. [DOI] [PubMed] [Google Scholar]
- 21.Damas JK, Waehre T, Yndestad A, Ueland T, Muller F, Eiken HG, Holm AM, Halvorsen B, Froland SS, Gullestad L, Aukrust P. Stromal cell-derived factor-1α in unstable angina: potential antiinflammatoy and matrix-stabilizing effects. Circulation. 2002;106:36–42. doi: 10.1161/01.cir.0000020001.09990.90. [DOI] [PubMed] [Google Scholar]
- 22.Schober A, Knarren S, Lietz M, Liehn E, Weber C. Crucial role of stromal cell-derived factor-1á in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation. 2003;108:2491–2497. doi: 10.1161/01.CIR.0000099508.76665.9A. [DOI] [PubMed] [Google Scholar]
- 23.Bein K, Odell-Fiddler ET, Drinane M. Role of TGF-beta1 and JNK signaling in capillary tube patterning. Am J Physiol Cell Physiol. 2004;287:C1012–1022. doi: 10.1152/ajpcell.00101.2004. [DOI] [PubMed] [Google Scholar]
- 24.Miyagi M, Miwa Y, Takahashi-Yanaga F, Morimoto S, Sasaguri T. Activator protein-1 mediates shear stress-induced prostaglandin d synthase gene expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:970–975. doi: 10.1161/01.ATV.0000159702.68591.0d. [DOI] [PubMed] [Google Scholar]
- 25.Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors alpha and beta. J Biol Chem. 2005;280:347–354. doi: 10.1074/jbc.M407879200. [DOI] [PubMed] [Google Scholar]
- 26.Vayalil PK, Iles KE, Choi J, Yi AK, Postlethwait EM, Liu RM. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1281–1292. doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banks EB, Crish JF, Welter JF, Eckert RL. Characterization of human involucrin promoter distal regulatory region transcriptional activator elements-a role for Sp1 and AP1 binding sites. Biochem J. 1998;331:61–68. doi: 10.1042/bj3310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet, and cardiovascular diseases: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999;99:178–182. doi: 10.1161/01.cir.99.1.178. [DOI] [PubMed] [Google Scholar]
- 30.Den Heijer M, Lewington S, Clarke R. Homocysteine, MTHFR and risk of venous thrombosis: a meta-analysis of published epidemiological studies. J Thromb Haemost. 2005;3:292–299. doi: 10.1111/j.1538-7836.2005.01141.x. [DOI] [PubMed] [Google Scholar]
- 31.Zeng X, Dai J, Remick DG, Wang X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ Res. 2003;93:311–320. doi: 10.1161/01.RES.0000087642.01082.E4. [DOI] [PubMed] [Google Scholar]
- 32.Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 33.Wang G, Siow YL, O K. Homocysteine stimulates nuclear factor kappaB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: a possible role for protein kinase C. Biochem J. 2000;352:817–826. [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol Genomics. 2002;9:27–41. doi: 10.1152/physiolgenomics.00075.2001. [DOI] [PubMed] [Google Scholar]
- 35.Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of vascular endothelial cells exposed to fluid mechanical forces: relevance for focal susceptibility to atherosclerosis. Endothelium. 2004;11:45–57. doi: 10.1080/10623320490432470. [DOI] [PubMed] [Google Scholar]
- 36.Li YS, Shyy JY, Li S, Lee J, Su B, Karin M, Chien S. The Ras-JNK pathway is involved in shear-induced gene expression. Mol Cell Biol. 1996;16:5947–5954. doi: 10.1128/mcb.16.11.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res. 2009;104:995–1003. doi: 10.1161/CIRCRESAHA.108.186486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann N Y Acad Sci. 2001;947:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 39.Yoshizumi M, Abe J, Tsuchiya K, Berk BC, Tamaki T. Stress and vascular responses: atheroprotective effect of laminar fluid shear stress in endothelial cells: possible role of mitogen-activated protein kinases. J Pharmacol Sci. 2003;91:172–176. doi: 10.1254/jphs.91.172. [DOI] [PubMed] [Google Scholar]
- 40.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 41.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai A, Zhao Y, Lankford HA, Warren JS. Nitric oxide suppresses EPO-induced monocyte chemoattractant protein-1 in endothelial cells: implications for atherogenesis in chronic renal disease. Lab Invest. 2006;86:369–379. doi: 10.1038/labinvest.3700396. [DOI] [PubMed] [Google Scholar]
- 43.Tsao PS, Wang B, Buitrago R, Shyy JY, Cooke JP. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96:934–340. doi: 10.1161/01.cir.96.3.934. [DOI] [PubMed] [Google Scholar]
- 44.Faraci FM. Hyperhomocysteinemia: a million ways to lose control. Arterioscler Thromb Vasc Biol. 2003;23:371–373. doi: 10.1161/01.ATV.0000063607.56590.7F. [DOI] [PubMed] [Google Scholar]
- 45.Lee SJ, Kim KM, Namkoong S, Kim CK, Kang YC, Lee H, Ha KS, Han JA, Chung HT, Kwon YG, Kim YM. Nitric oxide inhibition of homocysteine-induced human endothelial cell apoptosis by down-regulation of p53-dependent Noxa expression through the formation of S-nitrosohomocysteine. J Biol Chem. 2005;280:5781–5788. doi: 10.1074/jbc.M411224200. [DOI] [PubMed] [Google Scholar]
- 46.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Relationships between plasma levels of homocysteine and SDF-1 in all subjects (n = 72). Black circles: patients recruited from outpatients at high risk for cardiovascular events. White circles: normal volunteers from routine physical examinations.
Figure S2. Stimulation with homocysteine induces HUVECs to increase their phosphoryalation of ERK (A), JNK (B), and p38 (C). HUVECs were kept as controls (CL) or stimulated with 100 μM homocysteine for the times indicated, and the phosphorylations of ERK, JNK, and p38 were determined by using Western blot analysis. Phosphorylated ERK, JNK, and p38 levels are presented as band densities (normalized to total protein levels) relative to CL. The results are mean ± SEM from 3 independent experiments. *P < 0.05 versus control EC (CL).
Figure S3. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) for 1 ∼ 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). Data are presented folds of control ECs (CL), mean ± SEM. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S4. HUVECs cultured on FN- or COLI-coated glass were presheared at HSS (20 dyn/cm2) for 4 h (HSS/FN or HSS/COLI) before homocysteine stimulation. Static ECs were ECs cultured on FN- or COLI-coated glass and stimulated with homocysteine without preshearing (static). Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static/FN or static/COLI) with homocysteine stimulation.
Figure S5. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) or LSS (0.5 dyn/cm2) for 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). The SDF-1 p1010-Luc activity after 4 h homocysteine stimulation was determined by luciferase assay normalized to β-galactosidase. Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S6. HUVECs were kept as controls (CL) or presheared at HSS (20 dyn/cm2) or LSS (0.5 dyn/cm2) for 4 h before homocysteine stimulation. Static ECs were stimulated with homocysteine without preshearing (static). Sp1 (A) and AP-1 (B) activation after 2 h homocysteine stimulation in HUVECs was performed by TF ELISA assay. Data are presented folds of control ECs (CL), mean ± SEM. *P < 0.05 versus CL. #P < 0.05 versus static ECs (static) with homocysteine stimulation.
Figure S7. Schematic representation of the inhibitory effects of shear stress and the signaling pathway regulating the expressions of SDF-1 in ECs in response to homocysteine stimulation.