

Abstract
A disulfide-linked nitroxide side chain (R1) used in site-directed spin labeling of proteins often exhibits an EPR spectrum characteristic of a weakly ordered z-axis anisotropic motion at topographically diverse surface sites, including those on helices, loops and edge strands of β-sheets. To elucidate the origin of this motion, the first crystal structures of R1 that display simple z-axis anisotropic motion at solvent-exposed helical sites (131 and 151) and a loop site (82) in T4 lysozyme have been determined. Structures of 131R1 and 151R1 determined at cryogenic or ambient temperature reveal an intraresidue Cα—H···Sδ interaction that immobilizes the disulfide group, consistent with a model in which the internal motions of R1 are dominated by rotations about the two terminal bonds (Columbus, Kálai, Jeko, Hideg, and Hubbell, Biochemistry 2001;40:3828–3846). Remarkably, the 131R1 side chain populates two rotamers equally, but the EPR spectrum reflects a single dominant dynamic population, showing that the two rotamers have similar internal motion determined by the common disulfide-backbone interaction. The anisotropic motion for loop residue 82R1 is also accounted for by a common disulfide-backbone interaction, showing that the interaction does not require a specific secondary structure. If the above observations prove to be general, then significant variations in order and rate for R1 at noninteracting solvent-exposed helical and loop sites can be assigned to backbone motion because the internal motion is essentially constant.
Keywords: site-directed Spin Labeling, nitroxide anisotropic motion, nitroxide crystal structures
Introduction
Site-directed spin labeling (SDSL) is a general method for characterizing protein topography, local and global structure, and dynamics,1–6 and has recently been extended to include nucleic acids.7 SDSL can be applied to both soluble and membrane proteins of arbitrary molecular weight, and studies performed under biologically relevant conditions.8–12 In SDSL a unique cysteine residue is introduced into a recombinant protein via site-directed mutagenesis, and subsequently reacted with a sulfhydryl-specific nitroxide reagent to generate a paramagnetic side chain. The paramagnetic side chain designated R1 [Fig. 1(a)] is widely used in SDSL studies as it has been shown to be well-tolerated in proteins.14,15
Figure 1.
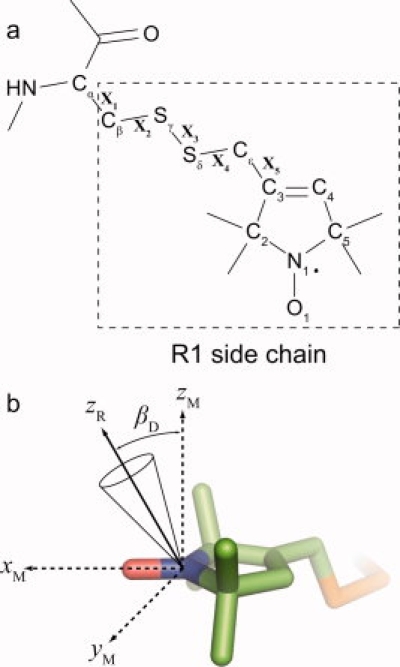
The R1 side chain and coordinate frames used in describing z-axis anisotropic motion. (a) Structure of the R1 side chain with designations of atoms and dihedral angles used in the text. (b) Molecular model of the nitroxide ring where xM, yM, and zM define the molecular frame, with xM along the NO bond of the nitroxide and zM along the 2p orbital of the nitrogen atom. zR is the z-axis of the diffusion frame, and βD the tilt angle with respect to the molecular frame (see Ref.13 for a compete description of the coordinate frames). Simple z-axis anisotropic motion arises from restricted diffusion of zR within a cone as indicated.
The EPR spectrum of a protein containing R1 reflects the overall motion of the nitroxide ring on the nanosecond time scale, with contributions from: (i) the internal motion of the side chain, modulated by interactions with the local environment; (ii) protein backbone dynamics; and (iii) rotational diffusion of the protein. The contribution of overall rotational diffusion to the nitroxide motion is negligible for membrane proteins or soluble proteins with a molecular weight greater than about 50 kDa; for smaller proteins, its effect can be minimized by increasing the viscosity of the solvent.14,15 One goal in the development of SDSL is to sort out the remaining contributions to nitroxide motion, which would enable the spectra to be interpreted in terms of local structure and conformational changes (via interactions with the environment) and backbone dynamics. To that end, crystallographic studies of spin-labeled T4 lysozyme (T4L) revealed interactions of the R1 nitroxide ring with the environment, providing an understanding of the complex EPR spectra in terms of the local structure.16–18
The present study is focused on R1 residues at the protein surface where there are apparently no or weak interactions of the nitroxide ring with the environment (noninteracting sites). Such sites are of interest because the motion of the nitroxide is determined solely by internal motion within the side chain and backbone dynamics. In fact, site-dependent contributions to R1 motion correlate with some expected characteristics of backbone dynamics.4,19 However, to put this strategy for measuring backbone dynamics on a more quantitative footing, the structural determinants of the internal motion must be known.
Information on the internal motion of R1 at noninteracting sites in α-helices, loops and edge strands of β-sheets has come from analysis of the EPR spectra. Generally, the spectra are well-fit by a model of simple anisotropic motion involving an order parameter for the z-axis of the nitroxide (0 ≤ S ≤ 1) and a correlation time (τ) for reorientation about the molecular axes [Fig. 1(b)].15,20 Typically, S varies from site-to-site, but is less than about 0.5; hence such motion will be referred to as “weakly ordered.” Mchaourab et al.21 showed that the weakly ordered motion for R1 at helix surface sites in T4 lysozyme (T4L) is essentially independent of the nearest neighbor identity within the helix (i.e. i ± 3 and i ± 4 residues), suggesting that the restricted motion arises from within the side chain itself. It was proposed that the anisotropic motion might result from an immobilization of the disulfide bond through an interaction with the protein backbone.14 Subsequently, this proposal was supported by crystallographic studies of the R1 side chain at several helix sites in T4L. In these structures, an intraresidue Cα—H···Sδ interaction was observed that effectively immobilized the entire Cβ—Sγ—Sδ group,16 allowing motion only about the disulfide (X3) and two terminal bonds [X4 and X5, Fig. 1(a)]. A first-order approximation assumed that motions of X4 and X5 are most important, and this “X4/X5 model” was found to account for the experimental spectra.20 Systematic variations of the nitroxide ring structure provided strong support for the X4/X5 model and identified motions about X5 as particularly important.20 Recent theoretical studies are consistent with the model.22
While support for the X4/X5 model of internal motion is strong, no crystal structure has yet been reported for an R1 residue having a weakly ordered z-axis anisotropic nitroxide motion, and the corresponding rotamers have not been identified. Here we report the structures of three R1 residues in T4L that have this motion, two in α-helices (131R1, 151R1), and one in a short loop connecting two helices (82R1). All three side chains extend into the solvent at sites where no contact is made with a symmetry related T4L molecule in the crystal lattice. In each case, including those determined at room temperature, the structures reveal immobilization of the R1 disulfide due to an intraresidue Cα—H···Sδ interaction, adding strong support for the X4/X5 model of internal motion for R1 in helices and loops. In addition, the structures presented here add to the growing rotamer library of R1 at solvent-exposed sites, and reveal that R1 adopts the same rotameric states (with respect to X1 and X2) at both non-contact and lattice-interacting sites. A complete rotamer library of R1 at solvent-exposed sites is important not only for understanding the internal motion of R1, but also for reliable modeling of the R1 conformational space for use in distance determinations.11,23
Results
To investigate the structural basis of weakly ordered, z-axis anisotropic nitroxide motion within the R1 side chain, T4L sites 131 and 151 were selected for crystallization as model α-helix surfaces sites, and site 82 as a model loop site (see Fig. 2). The EPR spectra of T4L mutants 131R1 and 82R1 have been reported,14 and are reproduced along with the spectrum of 151R1 in Figure 3. Each of these spectra can be fit reasonably well by a simple one-component z-axis anisotropic motion (dashed traces, Fig. 3), characterized by an order parameter of less than 0.5; thus the nitroxide motion is weakly ordered.
Figure 2.
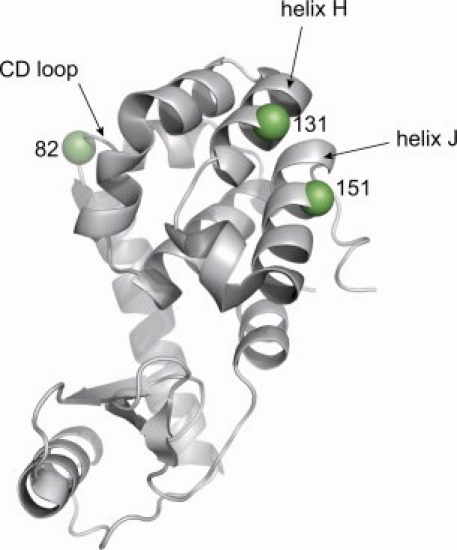
Ribbon model of the wild type T4L structure (PDB ID 1L63),24 highlighting sites 82, 131, and 151 with spheres at α-carbons. All three sites are solvent-exposed.
Figure 3.
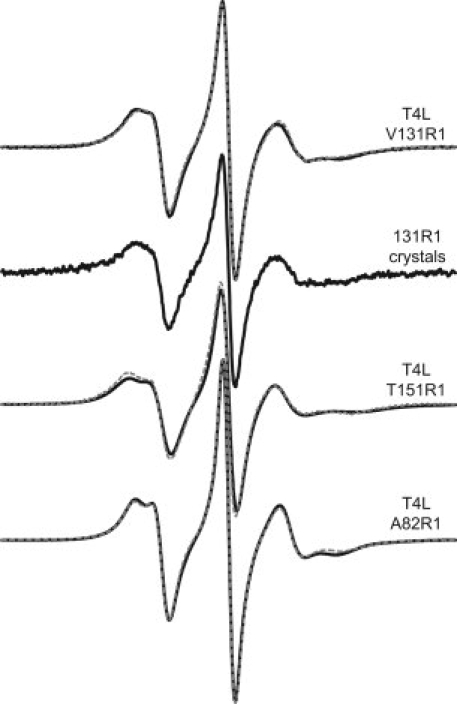
EPR spectra of T4L mutants A82R1, V131R1, and T151R1. All spectra were recorded with a 100 G scan width at 293 K, and are normalized to the area for ease of comparison. The spectrum designated “131R1 crystals” is that of a polycrystalline suspension of T4L 131R1 crystals in mother liquor. Other spectra were recorded in 30% sucrose to increase the solution viscosity. Overlaid on each solution spectrum (solid trace) is a nonlinear least squares fit of the EPR spectrum to the MOMD model (dashed trace; see Methods). Best fits of the 131R1 solution spectrum were obtained with S = 0.35 and 〈τ〉 of ∼2 ns, similar to previously published values.20 Fits of the 82R1 and 151R1 EPR spectra were obtained with S = 0.42, 〈τ〉 = 1.4 ns, and S = 0.39, 〈τ〉 = 2.4 ns, respectively.
Crystals of all three mutants were found to be isomorphous with wild type T4L crystals, with similar unit cell dimensions (Table I). Indeed, at the level of the backbone fold, each of the spin labeled mutants are similar to the pseudo wild type protein (WT*, PDB 1L63 from Ref.24, with an average backbone atom root mean square (RMS) deviation of 0.23, 0.29, and 0.21 Å for 131R1 (100 K), 151R1 (100 K), and 82R1 (100 K), respectively. For each of the structures reported here, no intermolecular interactions between the R1 side chain and a symmetry related lysozyme molecule were observed, indicating that the spin label conformations observed in the X-ray crystal structures are determined solely by intramolecular forces.
Table I.
X-ray Data Collection and Refinement Statistics for T4L mutants A82R1, V131R1, and T151R1a
| A82R1 | V131R1 LT | V131R1 RT | T151R1 LT | T151R1 RT | |
|---|---|---|---|---|---|
| Data Collection | |||||
| Reflections observed | 213,135 | 381,252 | 257,068 | 140,604 | 28,187 |
| Unique reflections | 22,843 | 21,086 | 12,597 | 18,230 | 9,516 |
| Temperature (K) | 100 | 100 | 291 | 100 | 291 |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Resolution (Å) | 80–1.70 | 100–1.75 | 80–2.10 | 80–1.80 | 80–2.30 |
| Highest Resolution Shell (Å) | 1.76–1.70 | 1.81–1.75 | 2.18–2.10 | 1.86–1.80 | 2.38–2.30 |
| Space group | P3221 | P3221 | P3221 | P3221 | P3221 |
| Rsym (%) | 5.7 (31.4) | 5.5 (36.8) | 12.3 (48.3) | 4.1 (16.2) | 15.9 (50.1) |
| I/σ | 41.2 (5.98) | 48.9 (8.1) | 31.2 (8.2) | 39.2 (6.1) | 9.6 (3.3) |
| Completeness (%) | 99.8 (99.9) | 99.8 (100) | 99.8 (100) | 97.8 (86.7) | 98.7 (98.3) |
| Unit cell dimensions | |||||
| a (Å) | 60.293 | 60.405 | 60.868 | 59.486 | 60.657 |
| b (Å) | 60.293 | 60.405 | 60.868 | 59.486 | 60.657 |
| c (Å) | 96.079 | 96.055 | 96.818 | 95.086 | 96.448 |
| Refinement | |||||
| Resolution (Å) | 52.20–1.70 | 51.99–1.75 | 35.70–2.10 | 31.7–1.80 | 26.27–2.30 |
| Reflections used | 21,562 | 19,921 | 12,558 | 18,197 | 9,410 |
| Rcrystb (%) | 18.7 | 19.2 | 15.2 | 18.6 | 17.3 |
| Rfreec (%) | 20.6 | 22.4 | 19.9 | 22.4 | 21.6 |
| Number of non-H atoms | |||||
| Protein | 1337 | 1351 | 1346 | 1329 | 1311 |
| Non-protein | 236 | 220 | 107 | 179 | 53 |
| RMS deviations | |||||
| Bond lengths (Å) | 0.009 | 0.010 | 0.007 | 0.004 | 0.004 |
| Bond angles (°) | 1.286 | 1.658 | 1.202 | 0.905 | 0.871 |
| Average B-factor (Å2) | |||||
| Protein atoms | 17.2 | 19.7 | 26.8 | 16.9 | 26.2 |
| Non-protein atoms | 27.8 | 29.6 | 37.4 | 24.4 | 31.9 |
| PDB accession code | 1ZYT | 2CUU | 3G3V | 3G3X | 3G3W |
Highest resolution shell data shown in parenthesis.
Rcryst = ∑||Fobs|−|Fcalc||/∑|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
Rfree is Rcryst calculated using 5% of the data, randomly chosen and omitted from refinement.
Structures of T4L 131R1 at 100 K and 291 K
The structure of 131R1 at 100 K was determined and refined to 1.8 Å resolution, with an R-factor of 19.2% (Table I). The 2Fo-Fc composite omit map of the electron density reveals two equally populated rotameric states of the 131R1 side chain [Fig. 4(a)]. In both states, only the —Cβ—Sγ—Sδ atoms of the side chain were resolved, allowing just the first two dihedral angles (X1 and X2) to be determined (Table II). The convention used here for designating the dihedral angles is based on the nomenclature proposed by Lovell et al.,25 in which m = −60°, p = +60°, and t = 180°; this notation is retained for X angles that are within about 20° of these nominal values. By this notation, one conformation of 131R1 is {m,m} (X1 = −69°, X2 = −60°) and the other {t,p} (X1 = 175°, X2 = 80°).
Figure 4.
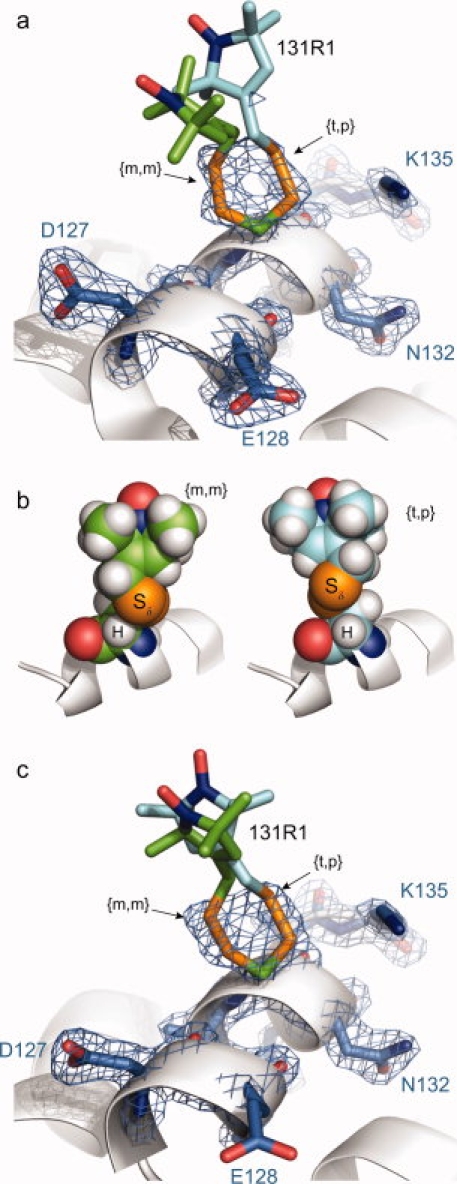
Structure of T4L 131R1 at 100 K and 291 K. (a) Electron density map for T4L 131R1 determined at 100 K calculated as an unweighted 2Fo-Fc composite omit map (marine mesh) and contoured at 1.3 σ. For clarity, only a stick model and electron density of 131R1 and selected nearby residues are shown (as indicated), with the protein backbone shown as a ribbon model. Two distinct conformations of the R1 side chain were observed, each with a unique orientation of the disulfide bond defined by the torsional angles X1 and X2 [Fig. 1(a)]. (b) Space-filling models of the {m,m} and {t,p} conformations of the 131R1 side chain with modeled H atoms, showing the proximity of the Sδ sulfur atom to the backbone Cα hydrogen atom in both states. (c) Electron density map for T4L 131R1 determined at 291 K calculated as an unweighted 2Fo-Fc composite omit map (marine mesh) and contoured at 1.5 σ. In each panel, the unresolved atoms of the nitroxide ring and the Cɛ carbon are not unique, and are modeled in a configuration free from steric interference.
Table II.
Summary of R1 Dihedral Angles
| T4L Mutant | Rotamer | Temp. (K) | X1 | X2 | X3 | X4 | X5 | Occupancy (%) |
|---|---|---|---|---|---|---|---|---|
| 82R1 | {m,m} | 100 | –68° | –56° | 101° | 100 | ||
| 131R1 | {m,m} | 100 | –69° | –60° | 50 | |||
| 131R1 | {t,p} | 100 | 175° | 80° | 50 | |||
| 131R1 | {m,m} | 291 | –75° | –57° | 50 | |||
| 131R1 | {t,p} | 291 | 175° | 83° | 50 | |||
| 151R1 | {m,m} | 100 | –83° | –72° | 50 | |||
| 151R1 | {m,m} | 291 | –82° | –72° | 70 |
When hydrogen atoms are modeled in the structure, the Sδ sulfur and Cα—H atoms are in proximity in both rotamers [Fig. 4(b)], a situation previously observed in X-ray crystal structures of R1 spin-labeled T4L mutants.16,17 The hydrogen–sulfur distance (Cα—H···Sδ) in {m,m} and {t,p} is 2.8 and 3.3 Å, respectively (Table III). For the {m,m} conformation, this distance is less than the sum of the hydrogen and sulfur van der Waals radii (3.1 Å), and is thus indicative of an attractive interaction, possibly a nonclassical C—H···S hydrogen bond (see Discussion). No other interatomic distances less than 4 Å were observed involving Sδ for either conformation. The absence of electron density for the remaining atoms of either side chain conformation, including the Cɛ carbon, precludes determination of the remaining dihedral angles (X3, X4, and X5). The nitroxide rings in Figure 4(b) are modeled based on previously observed values for X3–X5 in ordered R1 side chains (see Discussion section).
Table III.
Geometries Associated with {m,m} and {t,p} Conformations of R1a
| Conformation | Mutant | Cα···Sδ (Å) | Cα—H···Sδ (Å) | ∠Cα—H···Sδ (°) |
|---|---|---|---|---|
| 44R1CHAIN Ab | 3.4 | 2.7 | 123 | |
| 44R1CHAIN Bb | 3.3 | 2.7 | 124 | |
| 80R1c | 3.8 | 3.3 | 109 | |
| 82R1 | 3.4 | 2.8 | 117 | |
| 115R1 (RT)d | 3.6 | 3.0 | 121 | |
| {m,m} | 115R1 (LT)d | 3.7 | 3.2 | 112 |
| 119R1c | 3.3 | 3.0 | 111 | |
| 131R1 (LT) | 3.4 | 2.8 | 119 | |
| 131R1 (RT) | 3.5 | 2.9 | 116 | |
| 151R1 (LT) | 3.5 | 2.9 | 114 | |
| 151R1 (RT) | 3.5 | 2.9 | 116 | |
| 41R1b | 3.6 | 3.2 | 105 | |
| 65R1c | 4.0 | 4.0 | 87 | |
| {t,p} | 119R1c | 3.3 | 3.0 | 110 |
| 131R1 (RT) | 3.7 | 3.3 | 101 | |
| 131R1 (LT) | 3.7 | 3.3 | 105 |
In a previous study of 131R1 in solution, a relatively immobilized state was found to appear in the EPR spectrum at temperatures of about 288 K and below.20 The immobilized state identified in low temperature solution studies of 131R1 raises the possibility that conformations observed in the low temperature structure may result from interactions of the nitroxide ring stabilized at 100 K, a situation that was encountered in structural studies of T4L 115R1.17 To directly evaluate the role of temperature in possible rotamer selection of 131R1, the X-ray crystal structure of 131R1 was determined at 291 K (18°C).
Comparison of the unit cell dimensions (Table I) for the 291 K (RT) and 100 K (LT) crystal structures of 131R1 reveals that a 2% contraction of the unit cell volume occurs following flash-cooling of 131R1 crystals, a common result of flash-cooling protein crystals.26 The RT crystal structure of 131R1 was refined to 2.1 Å resolution and an R-factor of 15.2% (Table I), and is essentially identical to that at 100 K, with equal populations of {m,m} and {t,p} rotameric states (Table II) and lacking electron density for the nitroxide rings [Fig. 4(c)]. Although recent theoretical work predicts additional rotameric states of 131R1 at ambient temperature,27 no evidence for electron density corresponding to such states is present in the Fo-Fc composite omit map. The similarity between the low and room temperature structures of 131R1 demonstrates that the rotamers (with respect to X1 and X2) observed are not the result of low temperature selection, and conclusions drawn for the low temperature structure are applicable to the room temperature state.
Interactions of 131R1 in solution
Although the EPR spectrum of 131R1 in solution at room temperature is consistent with a single, weakly ordered motion (see Fig. 3), a relatively immobilized state was found to appear at reduced temperatures, as mentioned earlier. This is illustrated by the spectra in Figure 5(a), with the immobilized state identified by an arrow at 273 K. Presumably the new state results from favorable interactions of the nitroxide ring with nearby groups in the structure that are stabilized at lower temperatures, although the nature of such interactions was not revealed in the crystal structure determined at 100 K (i.e., the nitroxide ring was unresolved in both conformations observed).
Figure 5.
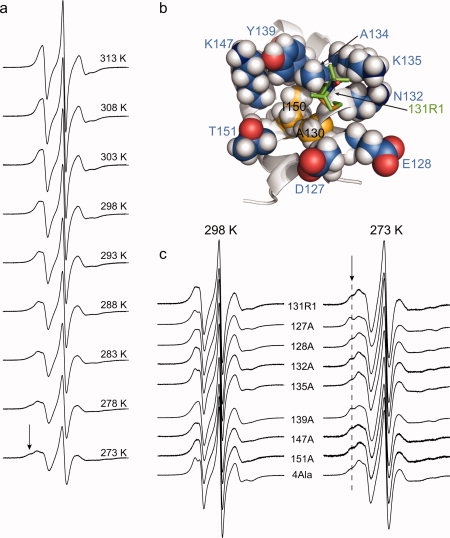
Interactions of 131R1 in solution. (a) EPR spectra of T4L 131R1 recorded at temperatures ranging from 273 K to 313 K in 5 degree increments, as indicated. (b) Model of T4L 131R1 showing the 131R1 side chain as a stick model, neighboring side chains in the structure as space-filling models, and the protein backbone as a ribbon model. Residues I150 and A130 form a small hydrophobic patch (highlighted in yellow). (c) Spectra of T4L 131R1 with WT* neighbors (131R1) and with the indicated Ala replacement (e.g., 127A) recorded at 298 K (left) and 273 K (right). The mutant designated 4Ala is a T4L 131R1 mutant with the following substitutions: D127A, E128A, N132A, and K135A. The arrows mark the position of a relatively immobilized component, and a dashed line has been added to guide the eye. All spectra were recorded for the protein in a 30% (w/v) sucrose solution.
To evaluate the role of nearby side chains in immobilizing 131R1 at low temperature, seven residues with potential for interaction with 131R1 were individually mutated to alanine (Ala) and their effects on the EPR spectrum studied in the range of 273–313 K. Specifically, solvent-exposed residues within helix H (D127, E128, and N132) and in close proximity to 131R1 in the tertiary structure (K135, Y139, K147, and T151) were selected for Ala substitution [Fig. 5(b)]. It was previously shown that T4L is thermodynamically and structurally tolerant to single and multiple alanine substitutions at solvent-exposed sites in the sequence 127–141, so it is likely that the native fold is retained for the Ala mutants in this region.28
Figure 5(c) shows the EPR spectra of 131R1 with WT* neighbors compared with those of the alanine mutants, recorded at 298 K (left panel) and 273 K (right panel). The EPR spectra for 131R1/E128A and 131R1/K135A at room temperature were previously reported,14,21 and spectra obtained in this study are similar. It is evident that the spectra for the mutants are all very similar to that for 131R1 with WT* neighbors at the same temperature, with subtle differences being seen only for E128A, K135A, and Y139A at both temperatures. The EPR spectra of weakly ordered states are exquisitely sensitive to subtle changes in motion,20,29 and the small spectral differences between the mutants and WT* reflect very small changes in nitroxide motion that could result, for example, from changes in local solvation due to the mutation. Collectively, these results indicate that the weakly ordered state of the nitroxide at 298 K and the immobilized state at 273 K are not the result of interaction with any single neighboring side chain, at least with chemical moieties beyond Cβ.
It is possible that the removal of one interacting side chain is compensated by a similar favorable interaction with another neighboring side chain, so the effect of replacing multiple residues near 131R1 with Ala on the spectrum of 131R1 was studied. In a polyalanine 131R1 mutant (designated 4Ala), residues 127, 128, 132, and 135, those physically closest to 131R1, were replaced by Ala. Remarkably, the EPR spectrum of this mutant [Fig. 5(c)] is again very similar to 131R1 with WT* neighbors at both 298 and 273 K, supporting the above conclusion that the side chains of these residues (exclusive of Cβ) contribute little-if at all-to the overall motion of 131R1. It is likely that the immobilized state populated at low temperature arises from interactions between the nitroxide of 131R1 and a hydrophobic patch formed in part by I150 and A130 [see Fig. 5(b) and Discussion].
Structures of T4L 151R1 at 100 K and 291 K
Like 131R1, the EPR spectrum of 151R1 in solution reflects weakly ordered motion (see Fig. 3). The X-ray crystal structure of 151R1 at 100 K was solved and refined to 1.8 Å resolution with an R-factor of 18.6% (Table I). In the 2Fo-Fc composite omit map of the electron density [Fig. 6(a)], the Cβ—Sγ—Sδ atoms of the 151R1 side chain were resolved in an {m,m} conformation (50% occupancy, X1 = −83°, X2 = −72°, Table II). As for the {m,m} conformation of 131R1, the atoms of the Sδ sulfur and Cα hydrogen of 151R1 reside in close contact [Fig. 6(b)]. Indeed, with hydrogen atoms modeled onto the 151R1 structure, the Cα—H···Sδ distance (2.9 Å, Table III) is less than the sum of the sulfur and hydrogen van der Waals radii, just as in the {m,m} conformation of 131R1.
Figure 6.
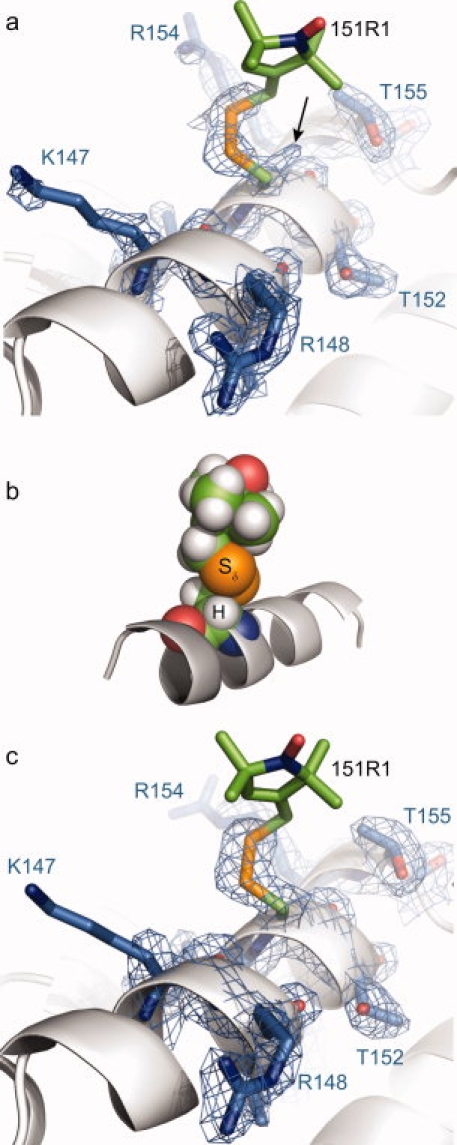
Structure of T4L 151R1 at 100 K and 291 K. (a) Electron density map for T4L 151R1 determined at 100 K calculated as an unweighted 2Fo-Fc composite omit map (marine mesh) and contoured at 1.3 σ. For clarity, only a stick model and electron density of 151R1 and selected nearby residues are shown (as indicated), with the protein backbone shown as a ribbon. (b) Space-filling model of the 151R1 side chain, showing the proximity of the Sδ sulfur atom to the backbone Cα hydrogen atom. (c) Electron density map for T4L 151R1 determined at 291 K calculated as an unweighted 2Fo-Fc map (marine mesh) and contoured at 1.5 σ. In each panel, the unresolved atoms of the nitroxide ring and the Cɛ carbon are modeled in a configuration free from steric interference.
In addition to the density for the {m,m} conformation, weak electron density was observed in the vicinity of 151R1 (unmodeled and denoted by an arrow in Figure 6(a). It is likely that this density corresponds to the Sγ of an unreacted cysteine, but in principle could be another minor X1 = t conformation of R1 with disorder beyond the Sγ sulfur atom. However, the nitroxide of such a rotamer would be expected to have a high mobility in solution, and there is no evidence for a second population in the solution EPR spectra. An interesting feature of the 151R1 structure is that the i + 3 arginine residue (R154) moves from its position in the WT* structure,24 possibly to avoid steric clash with the (unresolved) nitroxide ring.
As for 131R1, the question arises as to whether the {m,m} rotamer of 151R1 observed at 100 K is selected by attractive interactions favored at low temperature. In order to detect such interactions, the spectrum of 151R1 was studied as a function temperature (see Fig. 7). As the temperature is decreased, the rate and order of anisotropic motion decrease and increase, respectively. However, at the lowest temperature investigated (273 K) a new and relatively immobilized state is revealed as a shoulder in the spectrum (arrow, Fig. 7). Thus, low temperature does populate a new state, and to explore the role of temperature in possible rotameric selection of the 151R1 side chain the room temperature (291 K) crystal structure was determined.
Figure 7.
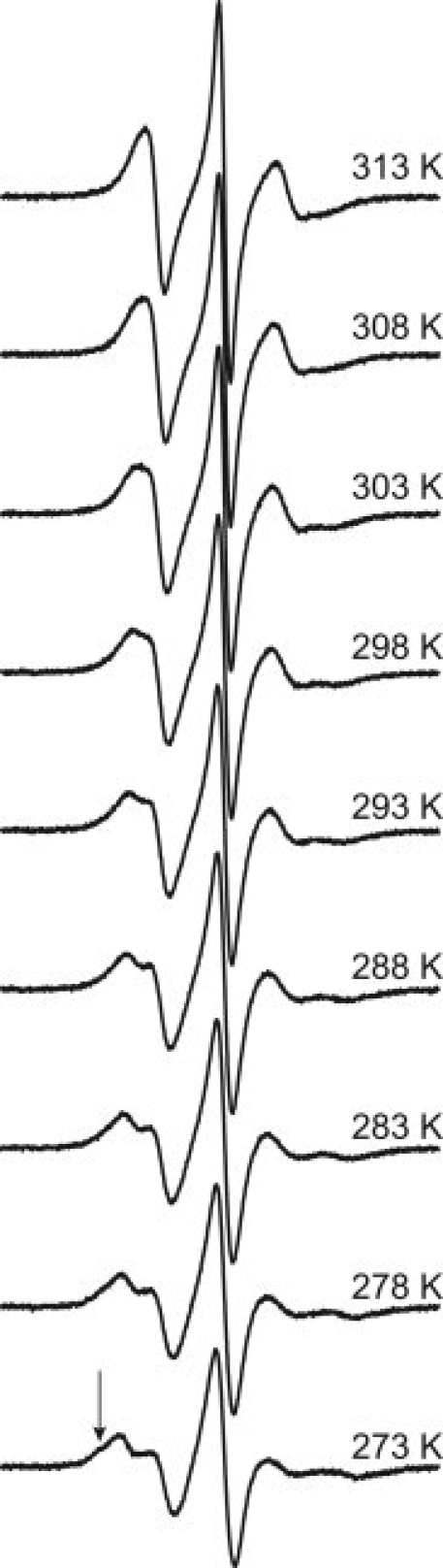
EPR spectra of T4L 151R1 recorded in 30% (w/v) sucrose solution at temperatures ranging from 273 to 313 K in 5 degree increments, as indicated. The arrow identifies a component arising from a relatively immobilized state.
The structure of 151R1 at 291 K was refined to 2.3 Å resolution and an R-factor of 17.3% (Table I). Like 131R1, comparison of the unit cell dimensions for 151R1 at 100 and 291 K reveals that a contraction of the unit cell occurs upon flash cooling of 151R1 crystals (Table I). The structures of 151R1 at 100 K [Fig. 6(a)] and 291 K [Fig. 6(c)] are remarkably similar, in that the —Cβ—Sγ—Sδ atoms of the side chain were resolved in a {m,m} conformation (70% occupancy, X1 = −82°, X2 = −72°, Table II) with the remaining side chain atoms unresolved. Thus conclusions drawn from the low temperature structure apply to that at room temperature.
Structure of T4L 82R1 at low temperature (100 K)
To investigate the structural origin of the weakly ordered nitroxide motion for R1 at a solvent-exposed loop site, the X-ray crystal structure of 82R1 was determined. The structure was solved and refined to 1.7 Å resolution with an R-factor of 18.7%. A previous study of R1 in helical peptides concluded that R1 has a high helical propensity similar to alanine, leucine, and methionine.30 However the R1 substitution at site 82 does not alter the native structure at this site, as the backbone torsional angles of 82R1 (φ = −68.6°, ψ = −19.6°) are similar to wild type A82 of T4L (φ = −67.3°, ψ = −23.7°).
In the crystal structure of 82R1, the Cβ—Sγ—Sδ—Cɛ atoms of the spin label side chain were resolved in a single conformation, allowing X1–X3 to be determined [Fig. 8(a)]. The dihedral angles X1 and X2 of 82R1 (−68° and −56°, respectively) are similar to those in other {m,m} rotameric states of R1 previously reported16,17 including 131R1 and 151R1. As noted above, in this conformation the Sδ sulfur and Cα hydrogen atoms lie in close contact [Fig. 8(b)], and, with hydrogen atoms modeled onto the structure, the Cα—H···Sδ distance is 2.8 Å (Table III), again closer than the sum of the hydrogen and sulfur van der Waals radii (3.1 Å). Although the Cα—H···Sδ interaction had previously only been observed for R1 at solvent-exposed helical sites, the structure of 82R1 demonstrates that this intraresidue interaction is also possible at solvent-exposed loop sites.
Figure 8.
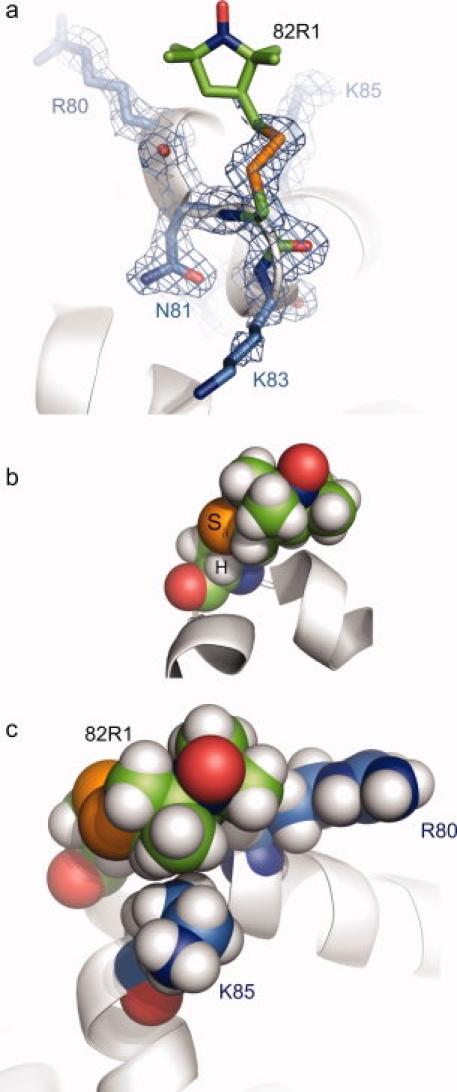
Structure of T4L 82R1 at 100 K. (a) Electron density of 82R1 (marine mesh), calculated as an unweighted 2Fo-Fc composite omit map contoured at 1.0 σ. For clarity, a stick model and the electron density for 82R1 (green carbons) and selected nearby residues (marine carbons) are shown, with the protein backbone displayed as a ribbon model. (b) Space-filling model of T4L 82R1, showing the proximity of the Sδ sulfur atom to the backbone Cα hydrogen atom. (c) Space filling model of 82R1 with nearby residues R80 and K85 shown. In each part, the unresolved atoms of the nitroxide ring and the Cɛ carbon are modeled in a configuration free from steric interference.
Although the nitroxide ring of 82R1 was not resolved, the dihedral angle X3 = 101° places the ring in van der Waals contact with R80 and K85 for any sterically allowed X4/X5 combination [a space-filling model of one possible conformation is shown in Fig. 8(c)]. Although interactions of the ring with either (or both) of these residues is expected, disorder of the nitroxide ring can be explained by the presence of nonspecific (i.e. hydrophobic) interactions that accommodate multiple X4/X5 states. If such interactions were significant for 82R1 in solution, a component of the EPR spectrum corresponding to an immobilized state would be expected, but none is observed at room temperature (see Fig. 3). However, decreasing the temperature below ca. 288 K reveals the appearance of an immobilized state in the EPR spectrum of 82R1 in solution (arrow, Fig. 9) that could correspond to weak interactions with R80 or K85.
Figure 9.
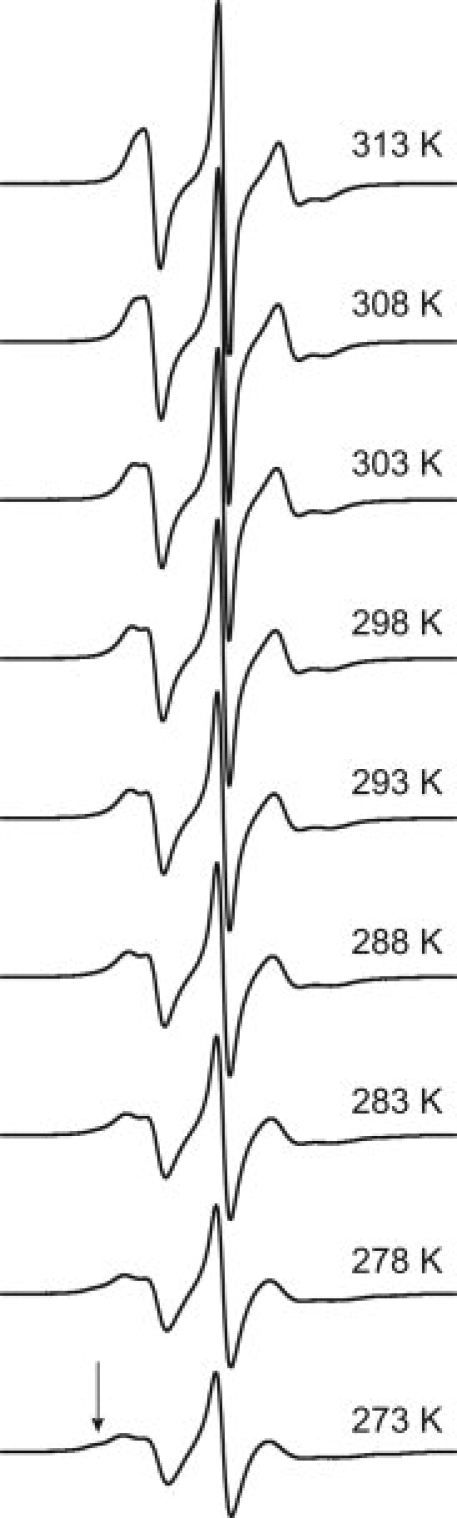
EPR spectra of T4L 82R1 recorded in 30% (w/v) sucrose at temperatures ranging from 273 to 313 K in five-degree increments, as indicated. At temperatures of 288 K and lower, a second, less mobile component of the EPR spectrum is observed (indicated by the arrow).
Discussion
Structural determinants of R1 internal motion at noninteracting sites and the X4/X5 model
The X4/X5 model for R1 internal motion was originally proposed to explain weakly ordered, z-axis anisotropic nitroxide motion observed for R1 at non-interacting helix surface sites.20 This first-order model posits that the motion arises primarily from torsional oscillations of X4 and X5, while torsions of X1 and X2 are restricted by immobilization of the disulfide group due to interaction with backbone atoms.
The structures reported here are the first for R1 side chains having a weakly ordered z-axis motion in solution (see Fig. 3) and serve to directly examine the structural basis of the X4/X5 model at solvent-exposed helical (131 and 151) and loop (82) sites in T4L. These sites were selected for crystallographic studies because each site is far from crystal contacts in the WT* crystal form, which minimizes the potential for the crystalline environment to influence the conformational state of the substituted R1 side chain. Indeed, no interactions between R1 and symmetry related lysozyme molecules were observed. Moreover, the EPR spectra of 131R1 in solution and a polycrystalline suspension are very similar (see Fig. 3), suggesting that the crystalline environment has little influence on the dynamic state of 131R1. Thus, it is highly likely that the conformations of R1 observed in the crystalline state (at 291 K) correspond to those in solution and give rise to the observed nitroxide dynamics reflected in the EPR spectrum.
Two features of the R1 side chains in both the low and room temperature structures of 131R1 and 151R1 are consistent with expectations of the X4/X5 model for R1 motion on helical surfaces. First, the well-resolved electron density for the Sγ—Sδ atoms reveals strong localization of the disulfide group, even at ambient temperature (Figs. 4 and 6); this requires a correspondingly strong constraint on torsions of X1 and X2. Second, the lack of electron density beyond Cɛ is due to disorder of the nitroxide ring that arises from torsional oscillations of X4 and/or X5 at ambient temperature. These are the basic features of the X4/X5 model. The similar EPR spectra and conformations for 82R1, 131R1, and 151R1 suggest generality of the model for R1 in ordered loops as well as helices.
Preferred rotamers of R1 at solvent-exposed sites and origins of disulfide bond immobilization
The preferred {X1,X2} rotamers of R1 at solvent-exposed helical sites, based on previously reported structures16–18 and those reported here, are shown in Figure 10. Consistent with a previous study,18 only {m,m}, {t,p}, and {t,m} rotamers are observed. It is interesting that although some of the data in the figure are derived from structures in which R1 interacts with neighboring T4L molecules in the crystal lattice (solid circles, Fig. 10), the rotamers are the same as those at non-contact sites (open circles, Fig. 10) suggesting that the {X1,X2} rotameric states of R1 are independent of such interactions.
Figure 10.
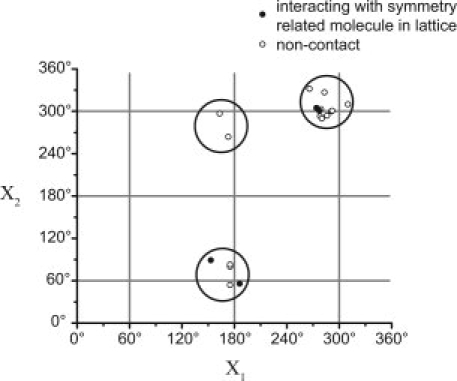
Preferred {X1,X2} rotameric states of the R1 side chain on solvent-exposed helical sites. Rotamers observed at lattice contact and noncontact sites are shown as filled and open circles, respectively. The figure includes data from twelve previously reported structures of the R1 side chain at helical surface sites in T4L,16–18 and the six structures of 131R1 and 151R1 observed in this study. This number includes structures of R1 obtained at a given site at two different temperatures (i.e., 115R1, 131R1, and 151R1), different rotamers at a given site (e.g., 131R1), and the structure of R1 at a given site differing by an alanine substitution at a nearby residue (i.e., 115R1/R119A).
The data of Figure 10 show that the {m,m} and {t,p} states are strongly preferred over the {t,m} state and the {m,t} and {t,t} states are absent despite recent theoretical work that predicted stability of these states for R1 at helical sites.22,31 These points are emphasized by the interesting structure of 131R1, in which the 131R1 side chain exists in equal populations of the {m,m} and {t,p} states. There is no electron density present in Fo-Fc omit maps that would correspond to other rotameric states of 131R1, even though the {m,t} and {t,t} rotamers are sterically allowed.
Why are the {m,m}, {t,p}, and to a lesser extent {t,m}, states favored while other sterically allowed rotamers have yet to be observed experimentally at solvent-exposed helical sites? Modeling reveals that of the nine possible {X1,X2} rotamers, the X1 = {p} and the {m,p} state are sterically forbidden on nearly all helical sites. Of the remaining five, only {m,m} and {t,p} allow the Cα—H···Sδ interaction (see Fig. 11), which is apparently responsible for stabilizing these states.16–18 The Cα—H···Sδ interaction is sufficiently strong to order the disulfide atoms in 151R1 and both rotamers of 131R1 at room temperature in the absence of lattice contacts or other intramolecular interactions [see Figs. 4(c) and 6(c)]. Further evidence for the existence of the Cα—H···Sδ interaction comes from variation of the side chain structure; elimination of one of the sulfur atoms in R1 would be expected to decrease nitroxide mobility, but, remarkably, a thioether linkage replacing the disulfide actually increases the mobility of the nitroxide although one fewer bond is present.21 The Sδ sulfur also has putative interactions with backbone atoms of the i + 1 residue in the {t,m} state of R1,17 but there is no such interaction to stabilize the remaining {m,t} or {t,t} states wherein Sδ is moved away from the backbone (see Fig. 11). The absence of the Cα—H···Sδ interaction would permit increased mobility about X2, and thus the {m,t} and {t,t} states would be expected to be disordered, but the EPR spectra of R1 at noninteracting helical surface sites reflect a strongly dominant weakly ordered motion at room temperature (see Fig. 3) consistent with disulfide immobilization and the X4/X5 model.20
Figure 11.
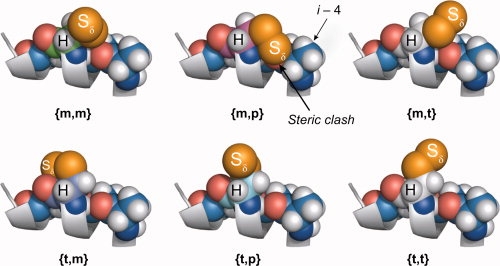
Models of the {m,m}, {m,t}, {m,p}, {t,m}, {t,t}, and {t,p} rotamers of the R1 side chain fragment (terminated at the Sδ sulfur) on a polyalanine helix. Rotamers with X1 = {p} (not shown) are sterically forbidden at most helical sites, as is the {m,p} state due to steric clashes involving the Sδ sulfur (as shown). The hydrogen atom of the Cα carbon and the Sδ sulfur are identified, and are only in contact in the {m,m} and {t,p} states.
The nature of the Cα—H···Sδ interaction in {m,m} and {t,p} is unknown, although it has been suggested that it might be a nonclassical hydrogen bond (H-bond).16 Current views of H-bonds in proteins have broadened the definition to include nonclassical H-bonds involving sulfur as an H-bond donor and acceptor, and C—H groups as H-bond donors.32 As shown in Table III, the geometry of some {m,m} but none of the {t,p} rotamers observed to date meet criteria set forth for a C—H···S hydrogen bond (C···S distance <3.5 Å and a C—H···S angle ≥120°).33 Nonetheless, the proximity of the Cα—H and Sδ atoms suggests an attractive interaction, especially as many of the Cα—H···Sδ distances are less than the sum of the van der Waals radii for sulfur and hydrogen (3.1 Å); simple hydrophobic interaction of the disulfide with the backbone also likely plays a role. Interestingly, the EPR spectrum of 131R1 in solution can be fit with a single dynamic component, so the internal motion that arises within the {m,m} and {t,p} conformations of 131R1 must be very similar. This suggests that the Cα—H···Sδ interaction must be similar in the two rotamers.
Given the equal populations of {m,m} and {t,p} rotamers of R1 at 131, it is of interest that only the {m,m} state is found at 82. The backbone dihedrals of 131R1, ϕ = −64° and ψ = −41°, are typical of an α-helical structure, while for loop residue 82R1 ϕ = −69° and ψ = −20°. For −40° < ψ < 0°, the carbonyl oxygen of R1 has a steric clash with the Sγ atom for X1 in a {t} configuration, thus favoring the {m,m} rotamer; variations in ϕ should have little influence on the relative stability of the rotamers. Indeed, only {m} conformers of cysteine are generally observed for −40° < ψ < 0°.34 This effect can also account for the single {m,m} rotamer of T4L 80R1 previously published,16 where ϕ = −85°, ψ = −8°.
The earlier discussion has emphasized the interaction of the disulfide with the R1 main chain atoms as the determinant of internal motion, and the similarity in this regard of the {m,m} and {t,p} rotamers. Were this the complete story, the EPR spectra of R1 at all noninteracting surfaces sites would be identical, dictated entirely by the internal motions that arise within the amino acid. In reality, the EPR spectra of R1 at noninteracting sites have been found to be diverse, reflecting site-to-site variation in the order and rate of nitroxide motion (see Fig. 3). It has been suggested that this variation is due to dynamic contributions from backbone fluctuations on the nanosecond time scale,4,20 which is apparently the case where large differences in backbone flexibility exist.19 Smaller site-to-site variations could have more subtle origins involving details of the local structure.
Probable conformations of R1 and the origin of immobilized states at low temperature in solution
The crystal structures presented earlier define X1 and X2 for each R1 side chain, with values for X3 being defined only for 82R1. Crystallographic studies of the R1 side chain at other sites where X3 could be determined suggest that the minimum energy conformations are near the expected values of ±90°.35 The disulfide rotamer adopted by 131R1 and 151R1 can be explored using modeling and constraints suggested by the motion of the nitroxide.
As discussed above, the room temperature EPR spectra of 82R1, 131R1, and 151R1 are all are consistent with the X4/X5 model, in which motions about X4 and X5 are unconstrained by interactions of the nitroxide ring with the protein. Models of R1 at a solvent-exposed helix site reveal that the {m,m,−90} and {t,p,+90} rotamers position the nitroxide ring away from the backbone where interactions of the ring are unlikely. Thus, these conformations can be invoked to account for the room temperature spectra of both 131R1 and 151R1. On the other hand, the {m,m,+90} and {t,p,−90} states position the ring close to the backbone where interactions are possible, and these rotamers could account for the immobilized states observed for 131R1 and 151R1 at low temperature (<288 K). In the case of 131R1, interaction of the nitroxide ring with a hydrophobic patch formed by Ile150 and Ala 130 is likely (see Fig. 5). Although a previous study has shown that an immobilized state can result from favorable interactions between R1 and neighboring side chains within the same helix,17 this does not seem to be the case for 131R1 because systematic mutations of the neighboring side chains to alanine have little effect on the nitroxide motion at any temperature [Fig. 5(c)].
For 82R1, dihedrals X1–X3 can be assigned from the 100 K structure (Table II), and the values place the nitroxide ring in close contact with the side chain of K85 and backbone atoms of R80 [Fig. 8(c)]. If this configuration exists at room temperature in solution, then the nitroxide would undoubtedly be immobilized rather than having the weakly ordered motion observed. An immobilized component is observed in the EPR spectrum of 82R1 only at temperatures below 288 K, so it is likely that this immobilized state corresponds to the configuration observed in the 100 K crystal structure. The environment around 82 is not highly restrictive, and modeling shows that isomerization of the 82R1 disulfide bond (from +90° to −90°) yields a conformational state fully compatible with the simple, non-interacting, anisotropic motion inferred from simulation of the room temperature EPR spectrum. As noted earlier, the nitroxide ring is positioned away from the protein surface in this {m,m,−90°} conformation.
Summary of the key points
Collectively, the data presented here suggest that the weakly ordered, z-axis anisotropic motion for the nitroxide in R1 at solvent-exposed sites in ordered helices and loops arises primarily from the internal motion of the side chain with minimal influence from interaction with the local environment at ambient temperature. The internal motion within the R1 side chain is dictated primarily by an intraresidue sulfur-backbone interaction (Cα—H···Sδ) that immobilizes the disulfide group and gives rise to the anisotropic nitroxide motion via torsions of X4 and X5. The disulfide immobilization is observed in crystal structures determined at both ambient and cryogenic temperatures, and in R1 at sites far from lattice contacts. The structures reveal that the Cα—H···Sδ geometry meets criteria established for a nonclassical C—H···S hydrogen bond in some cases, but regardless of its physical origin, it is sufficiently strong to completely immobilize the disulfide at room temperature without other interactions involving the side chain. Thus, it seems prudent for future modeling studies to recognize the importance of the Cα—H···Sδ interaction, and for molecular dynamic simulations of the R1 side chain to use force fields that reproduce it.
The common origin of disulfide immobilization in the {m,m} and {t,p} states suggests that the internal motion should be similar for these rotamers at noninteracting helical and loop sites, and the single dynamic mode for 131R1 supports this suggestion. If these conclusions prove to be general for R1 at noninteracting helical sites, then significant variation in motion from site-to-site can be assigned to backbone contributions on the nanosecond time scale.
The structures reported here add to the growing rotamer library for the R1 residue at solvent-exposed helical sites in which only three {X1,X2} rotamers are represented. Interestingly, these rotamers are the same whether or not the side chain is engaged in inter- or intramolecular interactions within the crystalline environment, and have been observed at both cryogenic (100 K) and ambient temperature. Thus, these three {X1,X2} rotamers permit rational modeling of the R1 side chain at solvent-exposed helical sites to interpret interspin distances and distributions determined from low temperature, pulse dipolar EPR spectroscopy36,37 in terms of protein structure.
Methods
Preparation of T4L cysteine mutants
The T4L mutants described here are in the “pseudo” wild type genetic background (WT*), which contains the mutations C54T and C97A.38 The genetic construct containing the pseudo wild type T4L gene was a gift of Dr. F.W. Dahlquist (University of California, Santa Barbara), and carries the bla gene for selection.39 Expression of the T4L gene is under the transcriptional control of a lacUV5 and multiple tac promoters, which is induced by isopropyl-thio-β-d-galactopyranoside (IPTG). The single cysteine mutants A82C, V131C, V131C/D127A, V131C/K135A, and T151C in the WT* background have been previously reported.14,40 The T4L mutants, V131C/E128A, V131C/N132A, V131C/Y139A, V131C/K147A, V131C/T151A, and V131C/R154A, and V131C/D127A/E128A/N132A/K135A were constructed in the WT* background using the QuikChange® site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to suggested protocol or by the overlap extension method.41
All mutants of T4L were expressed and purified as previously described.16 Briefly, single colony transformants of E. coli BL21 cells (Stratagene), transformed with a vector containing the mutant T4L gene, were grown overnight in ∼10 mL LB medium containing 100 μg/mL ampicillin at 37°C. The overnight culture was used to inoculate 1 L of LB medium (containing 100 μg/mL ampicillin), and the resulting culture grown at 37°C to an OD600 of 1.0–1.2. Cells were induced with 1 mM IPTG, shaken for 1.5 h at 37°C, and then harvested by centrifugation. A minimal volume of lysis buffer (25 mM Tris/MOPS, 0.1 mM EDTA, pH 7.6) was used to suspend cell pellets; mixtures were stored at −20°C.
Cell lysis was begun by allowing cells to thaw on ice. After thawing, the mixture was placed on ice and sonicated for 5 min (standard disruption horn, 50% output, duty cycle 4). The resulting cell lysate was centrifuged at 30,000g at 4°C for 30 min, and the soluble fraction loaded onto a cation exchange column (HiTrap™, SP Sepharose™ HP, GE Healthcare) equilibrated with lysis buffer containing 5 mM dithiothreitol. A 1M NaCl gradient was used to elute T4L, and fractions pooled that corresponded to its molecular weight (as judged by SDS-PAGE). Glycerol was added to the purified lysozyme preparation to a final concentration of 20% by volume, and the purified mixtures were stored indefinitely at −20°C.
Spin labeling of T4L mutants
T4L mutants purified as described above were passed over a desalting column (HiPrep™ 26/10, GE Healthcare) equilibrated with labeling buffer (50 mM MOPS, 25 mM NaCl, pH 6.8), diluted to a final concentration of approximately 10 μM, and immediately reacted with a 10 fold molar excess of 2,2,5,5-tetramethyl-pyrroline-1-oxyl methanethiosulfonate,42 a generous gift of Kalman Hideg (University of Pecs, Hungary). Labeling reactions were allowed to proceed overnight at 4°C. Excess spin reagent was removed using the above mentioned desalting column, eluting with labeling buffer. Spin-labeled T4L was concentrated to ∼1 mM in an Amicon® Ultra-15 concentrator fitted with Ultracel-10 membrane (Millipore). Samples for crystallization trials were further purified by passage over a gel filtration column (Superdex™ 75, GE Healthcare) equilibrated with labeling buffer. Final protein purity was greater than 95% as judged by SDS-PAGE.
X-band EPR spectroscopy and spectral simulations
For solution spectra, a 5 μL sample (typically 400 μM protein in labeling buffer containing 30% w/v sucrose) was contained in a quartz capillary (0.6 mm i.d. × 0.84 o.d., VitroCom, Mountain Lakes, NJ) sealed at one end. Polycrystalline suspensions in mother liquor were contained in the same capillary sealed at both ends with a non-paramagnetic sealant (X-seal, Bruker). All EPR spectra were collected at X-band over 100 G on a Bruker Elexys 580 fitted with a high sensitivity resonator using 20 mW incident microwave power and 1 G field modulation amplitude at 100 kHz.
Spectra were fit using the MOMD model of Freed and co-workers as implemented in the NLSL program.13 The experimental spectra to be fit are from R1 at solvent-exposed sites, and appropriate starting values for the elements of the A and g magnetic tensors in aqueous media were taken as: gxx = 2.0078, gyy = 2.0058, gzz = 2.0022 and Axx = 6.2, Ayy = 5.9, Azz = 37.0.43 In the MOMD model, anisotropic motion is simulated by a restoring potential that produces spatial ordering of the diffusion tensor. For noninteracting solvent-exposed R1 side chains, EPR spectra are adequately fit by an ordering potential dependent only on a single parameter, 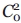 ; this corresponds to the familiar “diffusion in a cone” model for the z axis of the rotational diffusion tensor (zR), and defines the z-axis anisotropic motion. Reasonable fits are obtained with an axially symmetric motion where Rz ≡ Rpar and Rx = Ry ≡ Rperp, where Rpar and Rperp are (respectively) the parallel and perpendicular components of the rotational diffusion tensor.15 In a modified spherical representation, axially symmetric motion is specified by the isotropic diffusion rate
; this corresponds to the familiar “diffusion in a cone” model for the z axis of the rotational diffusion tensor (zR), and defines the z-axis anisotropic motion. Reasonable fits are obtained with an axially symmetric motion where Rz ≡ Rpar and Rx = Ry ≡ Rperp, where Rpar and Rperp are (respectively) the parallel and perpendicular components of the rotational diffusion tensor.15 In a modified spherical representation, axially symmetric motion is specified by the isotropic diffusion rate 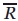 and the asymmetry parameter N, where
and the asymmetry parameter N, where 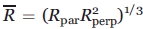 and N = Rpar/Rperp. For z-axis anisotropic motion [Fig. 1(b)], the diffusion tilt angles βD and γD were set to 36° and 0°, respectively.20
and N = Rpar/Rperp. For z-axis anisotropic motion [Fig. 1(b)], the diffusion tilt angles βD and γD were set to 36° and 0°, respectively.20
Least-square fits of the spectra were obtained with , , and N as adjustable parameters. Once these three parameters were optimized (i.e., yielded the lowest χ2 value), the principal values of the hyperfine and g tensors were allowed to vary slightly (±0.5 G or 0.0005, respectively) from their initial value if the fit could be improved. The effective correlation time (denoted 〈τ〉) is defined as 1/6 ; the order parameter (S ≡ S20) is computed directly from .13
Crystallization of R1 spin-labeled T4L
For crystallization trials, spin labeled T4L mutants were concentrated to 10–20 mg/mL with Amicon® Ultra-4 concentrators fitted with the Ultracel-10 membrane (Millipore). Crystals of 131R1 and 151R1 were grown by the hanging drop method by mixing equal volumes of spin-labeled T4L (in labeling buffer) and mother liquor to a total volume of 5 μL, and then suspended over 1 mL mother liquor. Diffraction quality crystals of 82R1 could not be obtained by the hanging drop method. Instead, crystals were grown by the sitting drop method, in which a drop was made by mixing equal volumes of protein and mother liquor (to a total volume of 2 μL), then overlaid by a 10 μL mixture of mineral and silicon oils (Hampton Research Corp., Aliso Viejo, CA). The mother liquor consisted of 1.8M to 2.2M mixtures of NaH2PO4 and K2HPO4 (ranging from pH 6.2 to 7.2), 0.25M NaCl, 0.04% NaN3, and saturated 2-hydroxy ethyl disulfide. The hanging drop plates were incubated at 4°C; crystals appeared in 2–4 weeks, and typically grew to a size of 200–300 microns (along the greatest dimension). Sitting drop plates were incubated at ambient temperature, and crystals appeared in 4–6 weeks.
X-ray diffraction studies and model refinement
For diffraction studies conducted at 100 K, single crystals were retrieved with a nylon loop (20 μ in diameter) fitted into a CrystalCap Copper Magnetic™ pin (Hampton Research Corp., Aliso Viejo, CA), cryoprotected in mineral oil, flash frozen under a stream of 100 K nitrogen gas, and then maintained in the cryostream during X-ray exposure. For diffraction studies at 291 K, single crystals were transferred into a glass capillary together with a small droplet of mother liquor. The structures of T4L 151R1 at 291 K and 100 K were obtained from crystals grown in the same hanging drop.
X-rays were generated from a rotating Cu/Kα anode (Rigaku, Corp.), and the diffraction collected on a RAXIS-IV++ image plate detector (Rigaku, Corp.). Diffraction data were processed with DENZO,44 and reduced with SCALEPACK44; data collection statistics are listed in Table I. All three spin-labeled mutants were found to crystallize in the same space group (P3221) and have similar unit cell dimensions as wild type T4L.
An initial set of phases was found by molecular replacement using rigid-body refinement or the program PHASER.45 For molecular replacement, a modified model for the pseudo wild type T4L structure was used-PDB accession code 1C6T46-wherein, at the site of R1 substitution, the native residue was replaced by glycine. Five percent of structure factors were set aside prior to the start of refinement; these structure factors were used to calculate Rfree in Table I. Structural models were iteratively refined with REFMAC47 or PHENIX48 and rebuilt with the graphics software O49 or Coot.50 After each model was refined to an R-factor of less than 22%, the spin label side chain was manually built-in and subject to further refinement iterations. Final refinement statistics for each model are reported in Table I.
Following refinement, each final structure was validated with the programs PROCHECK,51 ERRAT,52 WHAT_CHECK,53 and SFCHECK.54 The final models from refinement and the corresponding structure factors were deposited into the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank under the identification codes listed in Table I.
Acknowledgments
We thank Támás Kálai for synthesis of the 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate spin reagent, and Michael Bridges and Dr. Ned Van Eps for helpful comments on the manuscript. We also thank Evan Brooks for providing technical assistance in generating the 131R1/4Ala mutant. Figures 1b, 2, 4, 5b, 6, 8, and 11 were generated using the PyMOL Molecular Graphics System.55
References
- 1.Feix JB, Klug CS. Site-directed spin labeling of membrane proteins and peptide-membrane interactions. In: Berliner LJ, editor. Spin labeling: The next millennium. New York, NY: Plenum Press; 1998. pp. 251–281. [Google Scholar]
- 2.Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 3.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 4.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 5.Isas JM, Langen R, Haigler HT, Hubbell WL. Structure and dynamics of a helical hairpin and loop region in annexin 12: a site-directed spin labeling study. Biochemistry. 2002;41:1464–1473. doi: 10.1021/bi011856z. [DOI] [PubMed] [Google Scholar]
- 6.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Sowa GZ, Qin PZ. Site-directed spin labeling studies on nucleic acid structure and dynamics. Prog Nucleic Acid Res Mol Biol. 2008;82:147–197. doi: 10.1016/S0079-6603(08)00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanson SM, Francis DJ, Vishnivetsky SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differnetial interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oldham WM, Van Eps N, Preninger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13:772–777. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 10.Van Eps N, Oldham WM, Hamm HE, Hubbell WL. Structural and dynamical changes in an alpha-subunit of a heterotrimic G protein along the activation pathway. Proc Natl Acad Sci USA. 2006;103:16194–16199. doi: 10.1073/pnas.0607972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smirnova I, Kasho V, Choe JY, Altenbach C, Hubbell WL, Kaback HR. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci USA. 2007;104:16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson. 1996;120:155–189. [Google Scholar]
- 14.Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme: correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 15.Lietzow MA, Hubbell WL. Motion of spin label side chains in cellular retinol-binding protecorrelation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry. 2004;43:3137–3151. doi: 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- 16.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 17.Guo Z, Cascio D, Hideg K, Kálai T, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: Tertiary contact and solvent-inaccessible sites in helix G of T4 lysozyme. Protein Sci. 2007;16:1069–1086. doi: 10.1110/ps.062739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Z, Cascio D, Hideg K, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: Solvent-exposed sites in helix B of T4 lysozyme. Protein Sci. 2008;17:228–239. doi: 10.1110/ps.073174008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Columbus L, Hubbell WL. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4-58 bZip free and bound to DNA. Biochemistry. 2004;43:7273–7287. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- 20.Columbus L, Kálai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 21.Mchaourab HS, Kalai T, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme: Effect of side chain structure. Biochemistry. 1999;38:2947–2955. doi: 10.1021/bi9826310. [DOI] [PubMed] [Google Scholar]
- 22.Tombolato F, Ferrarini A, Freed JH. Dynamics of the nitroxide side chain in spin-labeled proteins. J Phys Chem B. 2006;110:26248–26259. doi: 10.1021/jp0629487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholsen H, Anderson DE, Dao-pin S, Matthews BW. Analysis of the interaction between charged side chains and the alpha-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry. 1991;30:9816–9828. doi: 10.1021/bi00105a002. [DOI] [PubMed] [Google Scholar]
- 25.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 26.Juers DH, Matthews BW. Reversible lattice repacking illustrates the temperature dependence of macromolecular interactions. J Mol Biol. 2001;311:851–862. doi: 10.1006/jmbi.2001.4891. [DOI] [PubMed] [Google Scholar]
- 27.Sezer D. Molecular dynamics and ESR spectra of a nitroxide spin label at solvent-exposed surfaces in proteins. 2008. PhD thesis, Cornell University.
- 28.Zhang XJ, Baase WA, Matthews BW. A helix initiation signal in T4 lysozyme identified by polyalanine mutagenesis. Biophys Chem. 2002;101/102:43–56. doi: 10.1016/s0301-4622(02)00193-x. [DOI] [PubMed] [Google Scholar]
- 29.Columbus L. Investigating backbone and side chain dynamics of α-helices in the nanosecond regime with site-directed spin labeling. 2001. pp. 51–58. PhD thesis, University of California, Los Angeles, pp.
- 30.Bolin KA, Hanson P, Wright SJ, Millhauser GL. An NMR investigation of the conformational effect of nitroxide spin labels on Ala-rich helical peptides. J Magn Reson. 1998;131:248–253. doi: 10.1006/jmre.1998.1365. [DOI] [PubMed] [Google Scholar]
- 31.Sezer D, Freed JH, Roux B. Parametrization, molecular dynamics simulation, and calculation of electron spin resonance spectra of a nitroxide spin label on a polyalanine alpha-helix. J Phys Chem B. 2008;112:5755–5767. doi: 10.1021/jp711375x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burley SK, Petsko GA. Weakly polar interactions in proteins. Adv Protein Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 33.Weiss MS, Brandl M, Suhnel J, Pal D, Hilgenfeld R. More hydrogen bonds for the (structural) biologist. Trends Biochem Sci. 2001;26:521–523. doi: 10.1016/s0968-0004(01)01935-1. [DOI] [PubMed] [Google Scholar]
- 34.Chakrabarti P, Pal D. The interrelationships of side-chain and main-chain conformations in proteins. Prog Biophys Mol Biol. 2001;76:1–102. doi: 10.1016/s0079-6107(01)00005-0. [DOI] [PubMed] [Google Scholar]
- 35.Fleissner MR. X-ray structures of nitroxide side chains in proteins: a basis for interpreting distance measurements and dynamic studies by electron paramagnetic resonance. 2007. PhD thesis, University of California, Los Angeles.
- 36.Berliner LJ, Eaton GR, Eaton SS. Distance measurements in biological systems by EPR. New York, NY: Kluwer Academic/Plenum Publishers; 2000. [Google Scholar]
- 37.Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 38.Matsumura M, Matthews BW. Control of enzyme activity by an engineered disulfide bond. Science. 1989;243:792–794. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]
- 39.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 40.Sathish HA, Stein RA, Yang G, McHaourab HS. Mechanism of chaperone function in small heat-shock proteins. Fluorescence studies of the conformations of T4 lysozyme bound to alpha B-crystallin. J Biol Chem. 2003;278:44214–44221. doi: 10.1074/jbc.M307578200. [DOI] [PubMed] [Google Scholar]
- 41.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 42.Berliner LJ, Grunwald J, Hankovszky HO, Hideg K. A novel reversible thiol-specific spin label: papain active site labeling and inhibition. Anal Biochem. 1982;119:450–455. doi: 10.1016/0003-2697(82)90612-1. [DOI] [PubMed] [Google Scholar]
- 43.Kusnetzow AK, Altenbach C, Hubbell WL. Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry. 2006;45:5538–5550. doi: 10.1021/bi060101v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode, in. Macromol Crystallogr A. 1997;267:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 46.Quillin ML, Breyer WA, Griswold IJ, Matthews BW. Size versus polarizability in protein-ligand interactions: binding of noble gases within engineered cavities in phage T4 lysozyme. J Mol Biol. 2000;30:955–977. doi: 10.1006/jmbi.2000.4063. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Grosse-Kunstleve RW, Hung WW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Jones TA, Bergdoll M, Kjeldgaard M. O: a macromolecular modelling environment. 1. An overview. In: Ealick S, Bugg CE, editors. Crystallographic and modeling methods in molecular design. Berlin: Springer-Verlag Press; 1990. pp. 189–195. [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model building tools for molecular graphics. Acta Cystallogr D Biol Crystallogr. 2004;60:2126–2133. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 52.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 54.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 55.DeLano WL. The PyMOL user's manual. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]