

Abstract
The gram-negative bacterium Escherichia coli offers a mean for rapid, high yield, and economical production of recombinant proteins. However, high-level production of functional eukaryotic proteins in E. coli may not be a routine matter, sometimes it is quite challenging. Techniques to optimize heterologous protein overproduction in E. coli have been explored for host strain selection, plasmid copy numbers, promoter selection, mRNA stability, and codon usage, significantly enhancing the yields of the foreign eukaryotic proteins. We have been working on optimizations of bacterial expression conditions and media with a focus on achieving very high cell density for high-level production of eukaryotic proteins. Two high-cell-density bacterial expression methods have been explored, including an autoinduction introduced by Studier (Protein Expr Purif 2005;41:207–234) recently and a high-cell-density IPTG-induction method described in this study, to achieve a cell-density OD600 of 10–20 in the normal laboratory setting using a regular incubator shaker. Several practical protocols have been implemented with these high-cell-density expression methods to ensure a very high yield of recombinant protein production. With our methods and protocols, we routinely obtain 14–25 mg of NMR triple-labeled proteins and 17–34 mg of unlabeled proteins from a 50-mL cell culture for all seven proteins we tested. Such a high protein yield used the same DNA constructs, bacterial strains, and a regular incubator shaker and no fermentor is necessary. More importantly, these methods allow us to consistently obtain such a high yield of recombinant proteins using E. coli expression.
Keywords: bacterial expression, recombinant protein, high cell density, Escherichia coli, expression condition and media
Introduction
Among many systems available for heterologous protein production, the gram-negative bacterium Escherichia coli remains one of the most attractive hosts.1,2 The advantages of fast growth at a high density in an inexpensive medium, the well-characterized genetics, and the availability of a large number of cloning vectors and mutant host strains enable E. coli to offer a mean for rapid, high yield, and economical production of recombinant proteins.3,4 However, in spite of the extensive knowledge on the genetics and molecular biology of E. coli, not every gene can be expressed efficiently and high-level production of functional eukaryotic proteins in E. coli may not be a routine matter, and sometimes it is quite challenging.3,5
Many factors contribute to this challenge, including: (1) the unique and subtle structural features of the gene sequence to be expressed, (2) the stability and translational efficiency of mRNA, (3) the ease of protein folding, (4) degradation of protein by host cell proteases, and (5) codon usage toxicity of the protein to the host.3,5 Techniques to optimize heterologous protein overproduction in E. coli have been explored that significantly enhanced the yield of the foreign eukaryotic proteins. Two recent excellent reviews summarized these optimizations (Table I in Refs.3 and4). Some empirical “rules,” for host strain selection, plasmid copy numbers, promoter selection, mRNA stability, and codon usage, have been derived from these optimizations that can be used to guide the design of expression system and to limit the unpredictability of protein expression in E. coli.3,4 However, an important optimization is cell growth conditions and media, which seems to be target protein dependent and there does not seem to be any empirical rules reported to date in this aspect.5
Table I.
The Optimized Recipe of the C-750501 Minimal Medium for Autoinduction and the Optimized High Cell Density Minimal Medium for Triple-Labeling Proteins
| Optimized autoinduction minimal medium | Optimized high-cell-density IPTG-induction minimal medium |
|---|---|
| 50 mM Na2HPO4 | 50 mM Na2HPO4·7H2O |
| 50 mM KH2PO4 (pH 8.0–8.2) | 25 mM KH2PO4 (pH 8.0–8.2) |
| 50 mM15NH4Cl | 10 mM NaCl |
| 5 mM Na2SO4 | 5 mM MgSO4 |
| 2 mM MgSO4 | 0.2 mM CaCl2 |
| 0.5–22× metals | 0.25× Metals |
| (Based on Studier's recipe) | 0.25× Vitamins |
| 0.4% 13C-glycerol | 0.1% NH4Cl or 15NH4Cl |
| 0.05% 13C-glucose | 1.0% Glucose or 13C-Glucose |
| 0.01% ß-lactose | |
| 12× Vitamins | |
| (100 × BME vitamins stock solution, SIGMA, MO) |
NMR structural studies of large proteins (molecular weight >30 kDa) require triple-labeled protein samples with 2H/13C/15N for recently developed TROSY techniques.6,7 To produce triple-labeled proteins, bacteria have to be grown in D2O, usually causing a significant reduction in protein yield. Our laboratory has been working on NMR structural studies of several large proteins and we frequently encountered these problems. To overcome these difficulties, we have performed optimizations of cell growth conditions and media. Our strategy mainly focuses on increasing cell density of bacterial expression, without manipulation of the bacterial expression vector, to enhance the protein production. However, a high cell density can frequently cause several major problems, including (1) plasmid loss from E. coli,8 (2) significant pH reduction because of cell metabolites, and (3) limited availability of dissolved oxygen.3 These problems often result in a low or even no protein production with a high cell density. Indeed, we frequently observed a low protein production from a high-cell-density bacterial expression, when we initially worked on enhancing cell density for the purpose of high-yield protein production. We developed several practical protocols that solved these problems. These protocols focused on colony selection for high-level protein production, optimization for bacterial expression condition, and better controls of the medium pH. In addition, these protocols also allow us to propose several empirical “rules” for the expression conditions that produce a very high yield of recombinant protein using E. coli.
In addition to these practical protocols, we also focused on optimization of cell growth conditions that allow for a very high cell density for production of high protein yield under a normal laboratory setting without fermentation. Recently, Studier9 introduced an autoinduction bacterial expression method, which provides several advantages over the traditional IPTG-induction method, including: (1) achieving a high cell density and (2) requiring minimal handling as there is no need to monitor cell growth for induction. This method has been used to prepare 13C/15N double-labeled proteins for NMR studies10 and selenomethionine-labeled proteins for X-ray crystallographic studies,11 both produced a moderate yield of target proteins (∼40 mg/L). However, no attempts have been reported to date using autoinduction in D2O for making triple-labeled proteins. We took advantage of the high cell density achieved using autoinduction and optimized cell growth condition in D2O using these protocols. Our optimization results in a very high protein production for triple-labeling proteins in D2O at a reasonable low cost.
Following the idea of autoinduction, we further developed a bacterial expression method that maintains the advantage of the tightly controlled induction by IPTG and utilizes both rich and minimal media to achieve a very high cell density for production of a very high yield of recombinant proteins. Unlike the autoinduction method, our high-cell-density method does not require longer time durations for achieving a high cell density, which is much more time efficient. This method starts cell culture with a rich medium that allows for a significantly enhanced initial cell-density at the OD600 values of 3–7 before IPTG-induction, depending on the rich medium used, while bacteria cells are still in the growing phase. After switching the cells into the minimal medium, the bacterial cells were cultured at a previously optimized temperature for 1.0–1.5 h and induced with IPTG for protein expression. With both autoinduction and the high-cell-density IPTG-induction methods, the final cell-density before cell harvest can reach to OD600 of 10–20, resulting in very high yields of protein production. We tested these bacterial expression methods and optimization protocols using seven different proteins. Our data indicated 9- to 85-fold enhancement in protein yields for all proteins. We routinely obtain 14–25 mg of triple-labeled proteins and 17–34 mg of unlabeled proteins from a 50-mL cell culture for all proteins. In addition, these methods allow us to consistently obtain such a high yield of recombinant protein using E. coli. Importantly, such a high protein yield used the same DNA constructs and the same bacterial strains that we previously used for the traditional IPTG method, and utilized regular incubator shakers under normal laboratory settings. Using our protocol, no fermentor is necessary to achieve a very high yield of pure recombinant protein. Thus, these methods and protocols can be applied by any laboratory for production of very high yields of recombinant proteins using bacteria.
Results and Discussion
E. coli offers a mean for the rapid and economical production of recombinant proteins. In recent years, the number of recombinant proteins used for therapeutic application increased dramatically. These demands drive the development of a variety of strategies for achieving high-level bacterial expression of proteins using E. coli. Optimizations in expression vector design, gene dosage, promoter strength (transcription regulation), mRNA stability, translation initiation and termination, E. coli host strain design, and codon usage have been performed, which result in significant enhancement of protein production and different commercial products,8 such as the pET expression vectors and pLysS plasmid by Novagen. The pLysS plasmid carries the gene for T7 lysozyme, which is a natural inhibitor of T7 RNA polymerase and serves to suppress basal expression of T7 RNA polymerase prior to induction, thus stabilizing recombinants encoding target proteins that may also affect cell growth and viability.8,12 In addition, empirical selection yields E. coli strains that are superior to the traditional BL21(DE3) host strain by overcoming the toxic effects associated with the overproduction of membrane and globular proteins under T7 transcriptional control.12,13
In contrast, optimization of bacterial expression conditions seems to be protein dependent.14 The general consideration is to increase cell-density of bacterial expression for the purpose of enhancing recombinant protein production. Much of the efforts have been centered on enhancement of cell-density in a fermentation setting, rather than in a general laboratory setting, because bacterial expression conditions, such as O2 level, pH, and nutrients, can be much better controlled using a fermentor to achieve a high cell density.15,16 In contrast, these expression conditions are difficult to control using a regular incubator shaker, and thus a much lower cell density can be achieved using this general laboratory setting. To achieve high cell density of bacterial expression in a general laboratory setting, we utilized autoinduction method developed by Studier.9 In addition, we also developed a bacterial expression method that utilizes rich medium to achieve a high cell density before IPTG-induction, while maintaining the advantage of the tightly controlled induction by IPTG in the minimal medium. With both the methods, the final cell density before cell harvest can reach an OD600 of 15–20, which is about 5- to 10-fold higher cell density comparing with that of the regular IPTG-induction method.
High-cell-density culture systems, especially under the nonfermentation, laboratory conditions, frequently suffer from several drawbacks, including plasmid loss,8 limited availability of dissolved oxygen, and increased carbon dioxide levels in the medium which causes significant reduction of medium pH.17 These problems often cause a low or even no protein production with a high-cell-density culture. Indeed, we frequently observed a low protein production from a high-cell-density bacterial expression before we implemented our protocols to solve these problems. The common practices in general laboratories to solve these problems are as follows: selecting high-expressing colonies and optimizing growth temperatures and time. We found that these common practices sometime produce inconsistent results that are not always repeatable. This is especially true for protein expression in D2O for production of the triple-labeled proteins.
We have designed several modifications to these common laboratory practices specifically for bacterial expression at high cell densities in a routine laboratory setting. The major modifications include (1) double colony selection and (2) proper preparation of a starting culture. Using these modifications, we have obtained repeatable very high yield of protein production for all the proteins tested, especially for triple-labeled proteins in D2O. Our data indicated a 9- to 85-fold enhancement of protein yields. Importantly, such a high protein yield used the same DNA constructs and the same bacterial strains that we previously used for regular IPTG-induction method. This provides a critical advantage of our method/protocols—a simple optimization in bacterial expression conditions can result in 9- to 85-fold enhancement of protein yields. Indeed, we routinely obtain 14–25 mg of triple-labeled proteins and 17–34 mg of unlabeled proteins from a 50-mL cell culture for all the proteins tested.
High-cell-density bacterial expression methods
Figure 1 shows a comparison of the procedures and final OD600 before cell harvest for three different bacterial expression methods used in this study. The traditional IPTG-induction method we used in the laboratory uses minimal medium for bacterial expression. This is because we frequently prepare isotopically labeled proteins for NMR studies, which requires minimal medium with 13C-glucose and 15NH4Cl in either H2O for double-labeled proteins or in D2O for triple-labeled proteins. As Figure 1 indicates the final OD600 of the traditional IPTG-induction expression before cell harvest is usually about 1.5–2.5.
Figure 1.
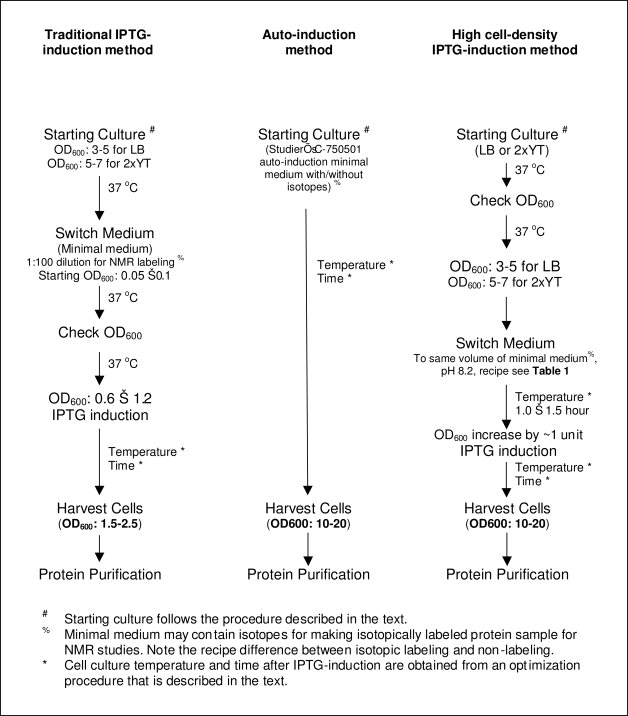
A schematic diagram of three expression methods used in this study.
We tested autoinduction expression for both unlabeled and triple-labeled proteins. Direct application of Studier's protocols using the C750501 recipe9 leads to inconsistent results. For some proteins, the yield was a two- to threefold increase compared with the traditional IPTG method, whereas for other proteins, bacteria either did not grow or only a poor yield was obtained. This is especially true when we grow bacteria in D2O for triple-labeling, which is not very surprising because different growth patterns for bacteria in D2O and H2O are expected. In addition, we frequently observed a phenomenon during autoinduction experiments: using minimal media, the OD600 reached quite high levels (usually 8–20), but no protein production was observed. We carried out experiments to optimize autoinduction conditions and developed several modifications, including: (1) double selection of high-level expression colonies, especially on D2O plates; (2) a proper starting culture for autoinduction; (3) a time course for the first attempt using autoinduction expression—monitoring the OD600, pH, and target protein yield; and (4) a modified autoinduction recipe (Table I), especially for expressing triple-labeled protein in D2O. These modifications, along with the other Studier's suggestion: multiple small volume expressions (5 × 50 mL) instead of a single large volume expression (250 mL), allow the final cell density before cell harvest to reach to OD600 at 10–20, with significantly enhanced protein yields. We are able to routinely obtain very high protein yields. More importantly, the autoinduction after these modifications produced consistent results that are always repeatable.
The third bacterial expression method uses rich medium, such as LB and 2× YT, to reach a high cell density before IPTG-induction. We then switch the culture medium by gently spinning down the cells and resuspending to an equal volume of minimal medium. A similar method was reported previously by Cai et al.18 and by Marley et al.19 for making double-labeled protein with 13C-glucose and 15NH4Cl. The procedure carried out by Cai et al.18 used a fermentor with carefully controlled O2 level and pH, whereas our method uses a regular incubator shaker that is commonly used in many laboratories for bacterial expression. Marley et al.19 generated a cell mass with unsaturated LB medium (OD600 = 0.7). They then concentrated the suspension (2×, 4×, and 8×) and transferred the bacteria into isotopically labeled minimal medium for expression. The cells were incubated for 1 h at 37°C to allow for the discharge of unlabeled metabolites and then induced with IPTG. They discovered that the 4× concentrated LB medium conferred maximal protein expression. We found that bacteria culture should not be saturated in the rich medium, because a saturated bacterial expression would not produce a high yield of protein production.9 Instead, the OD600 of bacterial cell culture in the rich medium should be an intermediate value, preferably in the middle of its growing phase, to ensure high-level protein expression. This will also avoid the problems associated with cells going into stationary phase, such as induction of proteases.5 For example, our experience suggested that a OD600 at 3–5 in LB medium and a OD600 at 5–7 in 2× YT medium were adequate before switching to minimal medium, because under these OD600 values, bacterial cells are in the middle of growing phase. After switching the medium, the bacterial cells were cultured at a previously optimized temperature for 1.0–1.5 h before IPTG-induction, to allow bacterial cells to adjust to minimal medium and to the new culture temperature. Using this high-cell-density method, we can easily achieve IPTG-induction within a “normal” working day, making this method a time-efficient method when comparing with the autoinduction method.
For isotopic labeling of proteins using the high-cell-density method, a slightly longer period of medium exchange time, such as 1.5–2.0 h, at a lower temperature might be preferred, because this not only allowed for the clearance of the unlabeled metabolites but also slowed down the bacterial growth during the exchange period, preserving the labeled nutrients for protein synthesis after IPTG-induction. At the end of this short period of medium exchange time, the OD600 of cell culture should increase, normally, by ∼1–2 OD600 units. For example, if we switch the LB medium at OD600 of 5.0 to an equal volume of minimal medium, the starting OD600 value in minimal medium should be close to 5.0. After 1.5–2.0 h cell culture period, an OD600 of 6.0–7.0 indicates that bacterial cells are in a healthy condition and will be induced for protein production with a previously optimized concentration of IPTG. After IPTG-induction, the bacterial cells are cultured at an optimized temperature for a previously optimized time period before harvest. With this method, the final cell density before harvest can reach OD600 of 10–20, which significantly enhances the protein yield. Figure 2 shows a comparison of the protein yields for human apoAI using three different expression methods described in Figure 1. It is clear that both high-cell-density expression methods produce greater than fivefold higher protein yields for apoAI as that obtained from the regular IPTG-induction method.
Figure 2.
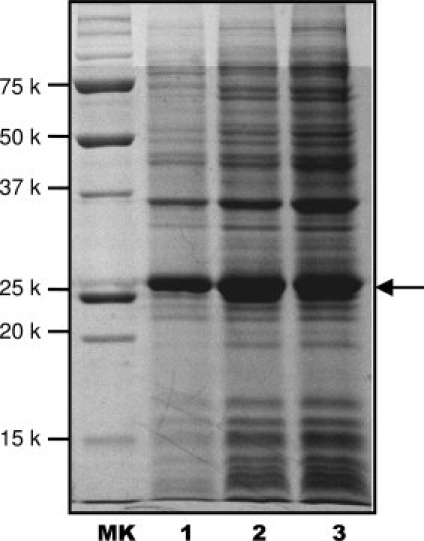
A 12% SDS-PAGE of protein expression levels of human apoAI in D2O. Lane 1: The traditional IPTG-induction method (0.4% 13C-glucose and 0.25 mM IPTG) at 20°C after IPTG induction. Lane 2: The high-cell-density IPTG-induction method (0.6% 13C-glucose and 0.25 mM IPTG). The cells were harvested at 12 h after IPTG-induction at 20°C. Lane 3: The autoinduction (0.4% 13C-glycerol) and the cells were harvested at 35 h at room temperature. MK—molecular weight marker. An arrow indicates the band position of human apoAI.
It is important to point out that there is no guarantee that a high cell density cell culture results in a high protein yield. As we described earlier, several drawbacks occur at high-cell-density bacterial expression, including plasmid loss, reduced medium pH, and limited dissolved molecular oxygen, causing either no protein production or a low protein yield. Indeed, when we initially worked on high-cell-density bacterial culture, we frequently encountered a situation that cell density became quite high, such as OD600 at 8–14, but protein yield was either very low or no protein production at all. In addition, the protein expression yield was not always repeatable. This is especially true when we expressed apoE(1-215)/pTYB1 vector in minimal medium at high cell density. We sometimes obtained an intermediate protein yield when we started with a freshly transformed colony. The other times we obtain a very low protein yield or no protein production at all even with a freshly transformed colony. When we started expression with freshly prepared glycerol stocks, most of the times we only obtained a very low protein yield. To solve these problems, we further developed the following practical protocols that ensure repeatable very high yield protein production using these high-cell-density bacterial expression methods.
Double selection of high-level expression colonies
We observed that colony selection was one of the most important factors for high-level protein production using high-density bacterial expression methods. This is especially true for bacterial expression in D2O for making triple-labeled proteins. As a common laboratory practice for high-level production of proteins, we routinely select high-level expressing colonies. However, we often found that a low yield of protein was obtained using the glycerol stock made with a selected colony, even though this glycerol stock previously produced high-yield protein. Such a situation happened quite often when we worked with human proteins that were toxic to the bacterial cells. This situation is also often observed when bacterial expression is carried out in D2O. To solve this problem, we have developed a double-colony selection protocol. In this protocol, the LB medium was inoculated with a single freshly transformed colony for a starting culture, which was grown to an OD600 of 0.7–0.9. The medium was then spread onto a plate, followed by selection of colonies from the plate. The selected colonies were checked for protein expression levels using the traditional IPTG-induced expression. After expression, 200–500 μL of cell suspension was spun down and the cell pellet was treated with SDS loading buffer for 20 min at 70°C. An SDS-PAGE was carried out to check the expression level. Only those colonies that displayed high-level expression will be used for the second selection. The second selection repeated the aforementioned procedure. If this double-colony selection is used for selecting high-level protein expression colonies in D2O, we will carry out all the aforementioned experiments in D2O, including D2O plates.
With this double-colony selection procedure, we were able to select several colonies for high-level expression of a protein, whereas our previous experiments showed very low protein production in D2O. Figure 3 shows SDS-PAGEs of expression levels of apoE(1-215), using a apoE(1-215)/pTYB1 expression vector, in D2O before (Panel A), during (Panel B), and after (Panel C) double selections. Panel A shows a poor protein production yield for all four colonies that were picked from a freshly transformed plate. Panel B shows a comparison of three different colonies from a single colony selection (Lanes 1–3) and another three different colonies from a double-colony selection (Lanes 4–6), suggesting that Colony 3 (Lane 3) gave the best protein expression level after the first-colony selection. Using Colony 3, we made a plate and picked three more colonies, Colonies 4–6. The second-colony selection indicated that Colony 6 gave the best protein expression level. With Colony 6, we made another plate and picked six colonies. It clearly demonstrates that all six colonies after double selection indeed solved the problem of low expression of apoE(1-215), resulting in a very high-level expression of target protein in D2O (Panel C). In contrast to single-colony selection, a glycerol stock prepared using a colony from double-colony selection can pass on for many generations and always give a consistent reproducible high-level protein production. Therefore, a double-colony selection procedure is recommended for colony selection of high-level expressing colonies.
Figure 3.
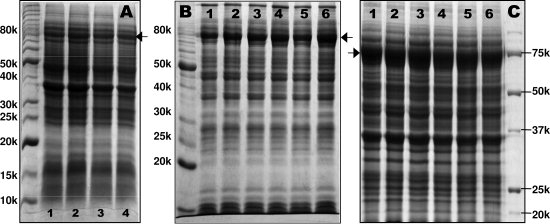
SDS-PAGEs of protein expression of apoE(1-215)/pTYB1 in D2O before (Panel A), during (Panel B), and after (Panel C) double-colony selections. Arrows indicate the expected protein band (∼80 kDa, apoE(1-215) + intein + CBD). Panel A shows four different colonies before colony selection. Panel B shows results of three different colonies selected from the single-colony selection (Lanes 1–3) and another three colonies selected from the double-colony selection (Lanes 4–6). The second-colony selection was based on Colony 3 (Lane 3) in the single-colony selection, because this colony gave a higher protein production. Panel C shows the results of six colonies from the double-colony selection, indicating a high protein expression level of all six colonies. Molecular weight markers are labeled with kDa.
A proper starting culture
The second important factor is the proper preparation of a starting culture in rich medium for scaling up in minimal medium. The general practice in the lab is to make a starting culture by growing an overnight culture using a rich medium, such as LB, at 37°C. We observed that an overnight starting culture in rich medium at 37°C usually reached saturation by the next morning. A saturated overnight culture might result in the plasmid instability because of the basal leakage of the T7 expression system.9,16 This usually resulted in a poor yield of target protein. To avoid this problem, we grew starting culture in a rich medium (H2O or D2O) for several hours at 37°C or overnight at 20–25°C until the OD600 was between 3 and 5 in LB and 5 and 7 in 2× YT (Fig. 1); this way, we were certain that the bacteria were still in the growing phase without a plasmid instability problem. We then gently spun down the cell and resuspended the cell pellet in minimal medium (H2O or D2O) for either regular IPTG-induction or high-cell-density IPTG-induction expression. For autoinduction bacterial expression, we usually directly started with either a single colony from a plate or with glycerol stock, both after double-colony selection. With such a starting culture, we can ensure that the majority of bacterial cells contain plasmid to begin the expression. We found that this was actually quite important, because our results showed a high yield target protein production using this starting culture.
Time courses and temperature optimizations
Another important step for high-level protein production using high-cell-density bacterial expression culture is to optimize expression conditions, such as culture temperature and the time after IPTG-induction or for autoinduction. This step is critical for the first time expression of a new protein using high-cell-density expression method. First, we carry out time courses at different temperatures, such as 15, 20, and 23 (room temperature), 28, 30, and 37°C. We closely monitor the following parameters: OD600, pH, and target protein production. We normally make a 10-mL culture either in D2O or in H2O for the time course. To check target protein yield, we take 200–500 μL of cell suspension after expression, spin down, treat the cell pellet with SDS loading buffer for 20 min at 70°C, and take 10 μL to run a SDS-PAGE. As an example, Figure 4, Left Panel shows an SDS-PAGE of an autoinduction time course of triple-labeled human apoAI expression in D2O at room temperature and Panel B shows a Western blot of the same time course. This figure clearly demonstrates the importance of the time course, indicating that either apoAI does not have enough time to be expressed under 30 h or the expressed apoAI starts to degrade after 40 h, both resulting in a low protein production. Table II lists the OD600, pH, and protein yield at each time point, suggesting that OD600 has indeed reached its maximum at 36 h (OD600:9.1), resulting in the highest protein yield. In contrast, the pH of the expression drops from the starting pH.7.2 to 6.01 after 36 h. Further reduction of pH may lead to significant instability of the plasmid, resulting in plasmid loss and significant reduction of the protein yield. In addition, for bacterial expressions of an ampicillin-resistant plasmid, further reduction in pH may also result in degradation of ampicillin in the medium. This could result in loss of ampicillin-resistant plasmids in the bacterial cells, thus the OD600 can further increase without protein production or a very low protein yield. To solve this problem, we constantly added ampicillin into the culture medium at regular intervals, because it can be degraded by β-lactamases and low pH.
Figure 4.
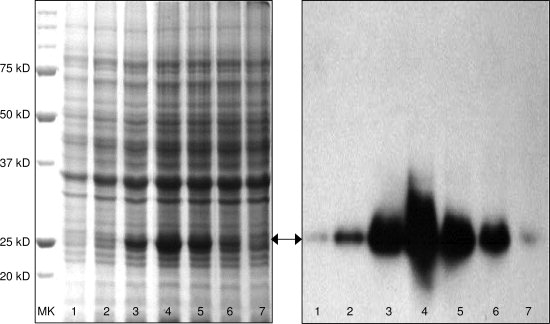
Left Panel: An SDS-PAGE showing autoinduction time course of triple-labeled human apoAI expression in D2O at room temperature. The expected apoAI band is indicated by an arrow. Lane 1:24 h, Lane 2:28 h, Lane 3:32 h, Lane 4:36 h, Lane 5:40 h, Lane 6:44 h, and Lane 7:54 h. Panel B: Western blot of the same time course using an anti-human apoAI monoclonal antibody, 5F6.
Table II.
Parameters for the Time Course of Autoinduction Expression of Human apoAI
| Time | 24 h | 28 h | 32 h | 36 h | 40 h | 44 h | 54 h |
|---|---|---|---|---|---|---|---|
| OD600 | 2.5 | 3.9 | 7.2 | 9.1 | 8.4 | 8.0 | 8.1 |
| pH | 6.6 | 6.5 | 6.3 | 6.0 | 6.0 | 6.0 | 6.1 |
| Protein yield | − | + | ++ | +++ | ++ | ++ | − |
For the seven proteins we tested, we found that different proteins require different temperatures for the optimized yield. For example, we did autoinduction at 37°C for two fragments of RAP, RAP(1-210) and RAP(91-323). For human apoE N-terminal domain, apoE(1-183), autoinduction was carried out at 28°C. For apoE(1-215)/pTYB1, high cell density IPTG-induction at 20°C after IPTG-induction and for the two apoAI proteins, experiments at room temperature provided the best yields. Nevertheless, time course experiments at different temperatures allow us to quickly optimize expression conditions for a high-level production of proteins.
Other important factors
Another modification is to change the pH of the culture medium to 8.2, so that the medium has a larger buffer capability. This is especially important for high-cell-density expression of an ampicillin-resistant plasmid, because a reduced pH results in degradation of ampicillin. In addition, a low pH medium may also cause stress to the bacterial cells, which results in plasmid loss from the high-density bacterial cells.16 Our result confirmed that an enhanced pH of the expression medium indeed helped to control medium pH at high cell density, thus at the end of bacterial expression, pH maintained at pH >6.2 for the cell culture of OD600 between 8 and 14. To solve the problem of limited availability of dissolved oxygen of high-cell-density culture, we used a smaller expression volume with a larger culture flask. For example, a 250-mL high-cell-density expression culture was divided into 5 × 50 mL cultures, and each culture used a 250-mL flask. This result in a better aeration in a small culture compared with a large culture, thus more molecular O2 will be dissolved in the medium.9
Optimization of 13C-glycerol usage for autoinduction expression
For triple-labeling protein using autoinduction, we have to modified Studier's C750501 recipe, because it requires 7.5 g of 13C-glycerol per liter of autoinduction medium, which is quite expensive. Our data indicated that autoinduction expression with 4 g of 13C-glycerol per liter of autoinduction medium produces a similar yield as that of the C750501 recipe, thus the cost for triple-labeling is nearly reduced by half. For optimization of 13C-glycerol usage, we carried out different time course experiments with 0.2, 0.3, 0.4, 0.5, and 0.75% glycerol concentrations. Figure 5 indicated that although the media containing 0.2 and 0.3% glycerol only produced a low yield of target protein, the media containing 0.4 and 0.5% glycerol produced a similar yield as those expressed in the original C-750501 medium (0.75% glycerol). However, for some proteins, we observed that a medium containing 0.4% 13C-glycerol only produced 70–80% of the triple-labeled protein produced in a medium containing 0.75% 13C-glycerol. In these cases, we used the medium with 0.75% 13C-glycerol, because the expression volume is only 50 mL.
Figure 5.
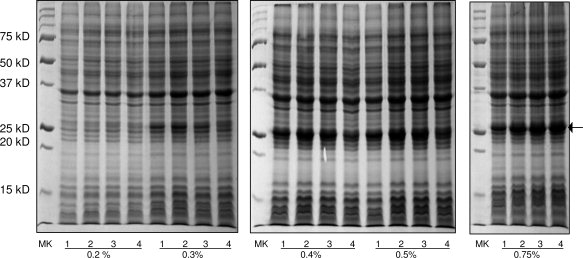
12% SDS-PAGEs of autoinduction expression of human apoAI using the modified recipe of C-750501 medium containing different concentrations of glycerol. The glycerol concentrations are indicated in the bottom of the figure. For 0.3, 0.4, 0.5, and 0.75% glycerol, Lane 1:36 h, Lane 2:38 h, Lane 3:40 h, and Lane 4:43 h. For 0.2% glycerol, Lane 1:27 h, Lane 2:30 h, Lane 3:33 h, and Lane 4:36 h. MK: molecular weight marker.
An optimized medium for high-cell-density IPTG-induction expression
High-density bacterial cells require more nutrition in the minimal medium, which usually uses NH4Cl as the nitrogen source and glucose as the carbon source. For making isotope-labeled protein, we use 15NH4Cl and 13C-glucose to replace normal NH4Cl and glucose for 13C and 15N-labeling the proteins. We intend to optimize both 15NH4Cl and 13C-glucose amounts for a low cost of production of isotope-labeled proteins. In most cases, our laboratory used 0.2–0.4% of 13C-glucose and 0.1% of 15NH4Cl for regular IPTG-induction bacterial expression. We found that this recipe did not work well with a high-cell-density IPTG-induction method, simply because of limited nutrition in the minimal medium, which limited bacterial cells to reach a high cell density and significantly reduced protein yield. We optimized different nutrition in the minimal medium for high-cell-density expression (Table I). As an example, Figure 6 shows glucose optimization of high-cell-density IPTG-induction expression of human apoE in D2O. These expressions started with a glycerol stock of apoE after double-colony selection and are carried out with an optimized time course and temperature. A cell culture containing 0.4% glucose could reach an OD600 of only 4.2 and the expression yield was also low. Increasing the glucose concentration increases the culture cell density and protein yield. With 1.0% glucose, the OD600 reached to 7.4 and protein yield seems enhanced by ∼10-folds (Table III). Marley et al. previously showed that increasing the amount of glucose to 0.8% only improved the protein yield modestly. They suggested that glucose concentration is not a critical factor in enhancing protein yield. Our results seemed to be different, indicating that the amount of glucose is critical for high-cell-density IPTG-induction expression. Our high-cell-density expression is based on several optimizations as described earlier, which may make a significant difference. We believe that at a high-cell-density bacterial culture, more nutrients, especially carbon source, are required for healthy cell growth, thus, the culture can reach to a high cell density, resulting in a higher protein production.
Figure 6.
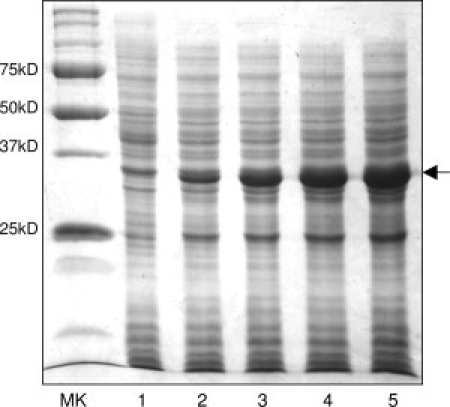
A 12% SDS-PAGE of glucose optimization of human apoE expression in D2O at 20°C using high-cell-density IPTG-induction bacterial expression: Uninduced (Lane 1), with 0.4% (Lane 2), 0.6% (Lane 3), 0.8% (Lane 4), and 1.0% glucose (Lane 5). Molecular weight marker is shown in left lane. Small-scale time course experiments with different glucose concentrations were also carried out to find the optimum protein expression time after induction of the culture.
Table III.
Glucose Optimization of High-Cell-Density IPTG-Induction Bacterial Expression of Human apoE in D2O
| Glucose (%) | 0.4 | 0.6 | 0.8 | 1.0 |
|---|---|---|---|---|
| OD600 at IPTG-induction | 2.0 | 2.5 | 3.0 | 3.3 |
| OD600 at harvest | 4.2 | 5.1 | 6.1 | 7.4 |
| pH at harvest | 6.7 | 6.6 | 6.4 | 6.3 |
| Time after IPTG-induction | 12 | 19 | 23 | 36 |
| Protein yield | + | ++ | +++ | ++++ |
A very high pure protein yield using high-cell-density bacterial expression methods
With these protocols, we routinely produced 14–25 mg of triple-labeled proteins and 17–34 mg of unlabeled proteins in a 50-mL cell culture for all the proteins we tested. Table IV lists the final yields of unlabeled and triple-labeled proteins using high-cell-density bacterial expressions and compared with the yields of the traditional IPTG-induction methods, which is also fully optimized, in a 50-mL cell culture, suggesting a 9- to 85-fold enhancement in protein yield. It is worth noting that we repeated the expressions of each protein more than three times and Table IV gives the average yields with standard deviations. This indicates that the protocols described earlier produce a consistent high-level triple-labeled protein production and is always reproducible. Table IV also gives the mass spectroscopic data of the triple-labeled protein, indicating that the efficiency of deuteration for triple-labeled protein using high-cell-density expressions. Overall, the deuteration efficiency is around 90% if we assume that the 13C and 15N-labeling are 100%. This is because we used 99.7% D2O and 13C-glycerol or 13C-glucose (not deuterated) in high-cell-density expressions. For the apoE(1-183) case, we only used 40% D2O and 13C-glycerol or 13C-glucose (not deuterated), the 89% deuteration level was based on 40% D2O (Table IV). This result is comparable with the deuteration efficiency of the traditional IPTG-induction expression with single-labeled 13C-glucose. To confirm the efficiency of triple-labeling by high-cell-density expressions, we carried out NMR experiments of these proteins. Figure 7 shows an example of the 1H-15N HSQC experiments of human apoE(1-183), for which the NMR samples were obtained using high-cell-density IPTG-induction expression (Panel C), autoinduction expression (Panel B), and traditional IPTG-induced expression (Panel A). This figure demonstrates that all three NMR samples produce an identical HSQC spectrum. In addition, with the proteins obtained using our high-cell-density IPTG method for apoE and apoAI, we have carried out NMR studies allowing us to completely assign NMR spectra of lipid-free apoE,20 lipid-free mouse apoAI(1-216),21 and human apoAI/preβHDL.22 In addition, we also determined NMR structures of lipid-free apoE(1-183)23 and mouse apoAI(1-216) (manuscript in preparation). Thus, we conclude that the high-cell-density expression produces a very high yield of triple-labeled, well-folded proteins for NMR studies (14–25 mg/50 mL for triple-labeled proteins; 17–34 mg/50 mL for unlabeled proteins). Table IV also indicates that the principles we described here for optimization of high-cell-density bacterial expression methods can be directly applied to other proteins, including membrane proteins in either H2O or D2O to obtain very high-level production of target proteins.
Table IV.
Final Yields (mg) of Triple-Labeled Proteins using High-Cell-Density Expression and Traditional IPTG-Induction Method in a 50-mL Culture
| Protein | High cell densityb (mg) | IPTGb (mg) | M.W. (Cal) (Dalton) | M.W. (MS) (Dalton) | %Dc |
|---|---|---|---|---|---|
| Triple-labeled | |||||
| RAP(1–210) | 20 ± 3 | 0.5 | 33,801 | 33,525 ± 195 | ∼92 |
| RAP(91–323) | 25 ± 3 | 0.8 | 36,633 | 36,376 ± 200 | ∼93 |
| ApoE(1–183)a | 18 ± 4 | 2 | 22,866 | 22,686 ± 116 | ∼89 |
| Mouse apoAI(1–216) | 15 ± 2 | 0.8 | 28,014 | 27,732 ± 125 | ∼90 |
| Human apoAI | 14 ± 1 | 0.6 | 32,814 | 32,401 ± 150 | ∼88 |
| Unlabeled | |||||
| Human apoAI | 34 ± 1 | 1.0 | |||
| Human apoE | 17 ± 2 | 0.2 | |||
M.W., molecular weight.
ApoE(1–183) was expressed in 40% D2O, the rest are expressed in 99.7% D2O.
High cell density: high-cell-density expression methods, including autoinduction and high-cell-density IPTG-induction; IPTG: the optimized traditional IPTG-induced expression; We repeated the expressions at least three times for all proteins, the yield shown is the average ± standard deviation.
%D: Estimated percentage of deuteration, assuming 100% 13C and 15N-labeling. For apoE(1–183), the %D is the estimated percentage of deuteration based on 40% D2O. For the other four proteins, the D% is the estimated percentage of deuteration based on 99.7% D2O.
Figure 7.
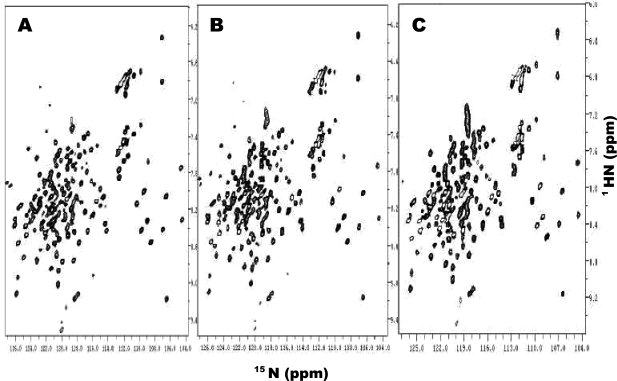
1H-15N HSQC spectra of triple-labeled human apoE(1-183) obtained using high-cell-density IPTG-induction expression (Panel C), autoinduction expression (Panel B), and traditional IPTG-induction expression (Panel A). All three samples contained 1.0 mM triple-labeled human apoE(1-183) in 100 mM phosphate buffer, 10 mM EDTA, 0.5 mM NaN3, 90 mM DTT, and 0.02 mM DSS, pH 6.80. The spectra were collected at 30°C on a 600 MHz NMR spectrometer with a cold probe.
Methods
Molecular cloning
Seven different proteins are tested, including two different constructs of receptor-associated protein, RAP(1-210), RAP(91-323), truncation mutants of the human apolipoprotein E, apoE(1-183) and apoE(1-214), full-length apoE, a truncation mutant of mouse apolipoprotein AI, apoAI(1-216), and full length human apoAI. The genes of these proteins were subcloned into different expression vectors as follows: RAP(1-210)/pET30a, RAP(91-323)/pET30a, human apoE(1-183)/pET22b, apoE/pET30a-sHT, human apoE(1-214)/pTYB1, mouse apoAI(1-216)/pET30a, and human apoAI/pET30a-sHT. The pET vectors were from Novagen, WI and the pTYB1 vector was from New England BioLabs, MA. We engineered the pET30a vector to introduce a Factor Xa site between the long his-tag and the target gene. The pET30a-sHT is also an engineered pET30a vector in which the long his-tag was replaced by a six histidine tag plus a two serine linker. The pET30a and pET30a-sHT are kanamycin-resistant vectors, whereas the pET22b and pTYB1 vectors are ampicillin-resistant vectors. The expression vectors were transformed into BL-21(DE3) bacterial strains.
Double-colony selection
First, LB agar plates were prepared either in H2O or in 70% D2O (for triple-labeled protein expression). For 70% D2O plates, the agar medium was not autoclaved, but microwaved until the agar dissolved. Three milliliters of agar was poured into a 35 mm ×10 mm petri plate (Corning, NY). Bacterial cells, either from a glycerol stock or 5 μL of a starting culture that has been diluted to an OD600 of ∼0.05–0.1, were streaked onto the LB agar plates. Several colonies were picked from the plates next morning, and the expression levels of these colonies were checked using the traditional IPTG-induction expression. Glycerol stocks were also prepared for each colony. We chose the colony with the highest protein expression and went through another round of selection, following the procedure described earlier. The colonies selected from the double selection were used for preparation of glycerol stocks and were stored in a −80°C freezer.
Traditional IPTG-induction expression
We use this expression method to either check protein expression levels of different colonies during double-colony selection or serve as an expression control. For double-colony selection, we used a small-scale expression with the following procedure: 2 mL of LB media was inoculated with a single colony from a freshly transformed plate as the starting culture and cultured at 37°C. When the OD600 of the starting culture reached between 2 and 3, the culture was gently spun down at 1500g for 5 min. The part of cell pellets was resuspended in 5 mL of the minimal M9 medium to obtain an initial O.D between 0.07 and 0.1. When the culture reached an OD600 of ∼1.0, it was induced with 0.5 mM IPTG and incubated at 20°C overnight [Human apoE(1-215), full-length apoE, mouse apoAI, human apoAI, RAP(1-210), RAP(91-323)] or at 28°C for 16–18 h [Human apoE(1-183)]. Two hundred fifty microliters of cell suspension was collected and spun down at 3300g for 5–10 min. The cell pellet was resuspended in 50 μL of 2× SDS gel loading buffer and heated at 70°C for 20 min. Cell debris and DNA molecules were pelleted by centrifuging at a maximum speed for 20 min. Finally, 10 μL of the supernatant was loaded into the SDS-PAGE to check the expression level. For the IPTG method as an expression control, we used a 50-mL expression with the following procedure: 10 mL of LB media was inoculated with glycerol stock (after double selection) as the starting culture and cultured at 37°C. When the OD600 of the starting culture reached between 2 and 3, the culture was gently spun down at 1500g for 5 min. Part of cell pellet was resuspended in 50 mL of the minimal M9 medium to obtain an initial O.D between 0.07 and 0.1. When the culture reached an OD600 of ∼1.0, it was induced with 0.5 mM IPTG and incubated at 20°C overnight [Human apoE(1-215), full-length apoE, mouse apoAI, human apoAI, RAP(1-210), RAP(91-323)] or at 28°C for 16–18 h [Human apoE(1-183)]. The cells were harvested by spin down and the cell pellet was used for protein purification.
Autoinduction expression
For autoinduction expressions, we followed a general procedure: Studier's C-750501 autoinduction minimal medium with 0.75% glycerol was used for the time course experiments and unlabeled protein production. A modified Studier's C-750501 autoinduction expression medium with 0.4% 13C-glycerol was used for triple-labeled protein production. For the time course experiments, the expression medium was made either with 99% D2O or with H2O, unlabeled glucose, glycerol, and ammonium chloride. For triple-labeled protein expressions, 13C6-glucose (0.05%), 13C-labeled glycerol, and 15NH4Cl in D2O were used. The medium was also supplemented with 0.2–2× trace metals as required and with vitamins if necessary. β-Lactose was used instead of α-lactose for induction, which contained 30% of α-lactose isomers and gave comparable expression levels. Starting cultures in LB media were made with the glycerol stocks from double selection and grown to an OD600 of 1–2 and then spun down at 1500g for 5–10 min. The cell pellets were resuspended in 25 or 50 mL autoinduction expression medium to obtain an initial OD600 of 0.3. Erlenmeyer flasks of 125 and 250 mL were used for 25 and 50 mL culture volumes, respectively, for better aeration of the cultures. Expressions were carried out at different temperatures for different proteins ranging from 20 to 37°C.
For the time course experiments, 250 μL culture samples were collected at different times. The OD600 and pH of the culture at these time points were monitored. The cell suspensions were spun down, resuspended in 50 μL of 2× SDS gel loading buffer, and ran on an SDS-PAGE to compare the expression levels at different times. Expression time usually varied between 24 and 64 h, depending on the culture temperature.
For unlabeled and triple-labeled protein expressions, we first do time courses at different temperatures to find the optimal expression time and temperature. For example, the time course of triple-labeled human apoAI indicated that an expression of 36 h at room temperature gave the best protein production. The triple-labeled apoAI expression was then carried out at room temperature with an initial OD600 of 0.3. At 36 h, the cells were harvested by centrifugation at 3500g for 15 min. The purifications of the triple-labeled proteins were carried out as described previously.24–26
High-cell-density IPTG-induction expression
This expression method uses rich medium for achieving high cell density. We started bacterial expression using a rich medium, such as LB or 2× YT, at 37°C. Once the cell density reaches a cell density that is in the middle of the growing phase before saturation, we switched the cell culture by gently spinning down cells and resuspending the pellet into the same volume of minimal medium. We found that bacterial cells reach saturation at cell densities of OD600 of ∼10–15 in LB and OD600 of ∼15–20 in 2× YT. Thus, bacterial cells are in the middle of the growing phase with a cell density of OD600 of 3–5 in LB and OD600 of 5–7 in 2× YT. After switching the medium, we cultured bacterial cells for another 1.0–1.5 h without adding IPTG, at the optimized temperature that is used for the cell culture after IPTG induction. During this period, the OD600 of the cell culture should increase by 1–2 U. IPTG was then added to induce protein production. The cell culture was incubated at the same temperature for a period that is optimized for different proteins before cell harvest. Usually, we found that the OD600 value at the end of cell culture increased by 2.0- to 2.5-fold compared with the OD600 value at IPTG-induction. Therefore, before cell harvest, the bacterial cells can reach to OD600 of 10–15 with a starting medium using LB and OD600 of 15–20 using 2× YT. This is about 5- to 10-fold higher OD600 than that of the regular IPTG-induction bacterial expression in minimal medium.
NMR spectroscopy
The NMR samples contained 100 mM phosphate buffer (pH 6.8), 0.01 mM NaN3, 10 mM EDTA, 50 mM DTT, 5% D2O, and 0.5–1 mM 40% 2H/15N/13C-labeled proteins. The 1H-15N TROSY-HSQC spectra were acquired at 30°C on 600 MHz Varian INOVA spectrometer with a cold probe. Proton chemical shifts were referenced to DSS. Data were processed and analyzed on a SGI workstation using nmrPipe27 and nmrview.28
Acknowledgments
The authors thank generous support from Isotec Inc. for providing 13C-labeled glycerol for this work.
Glossary
Abbreviations:
- ApoAI
apolipoprotein A-I
- ApoE
apolipoprotein E
- CBD
chitin-binding domain
- DSS
2,2-dimethyl-2-silapentane-5-sulfonic acid
- E. coli
Escherichia coli
- HSQC
heteronuclear single quantum coherence
- Intein
inducible self-cleavage activity of protein splicing elements
- IPTG
isopropyl-β-d-thiogalactopyranoside
- LB
Luria-Bertani
- mRNA
messenger RNA
- MS
mass spectroscopy
- NMR
nuclear magnetic resonance
- OD600
optical density at 600 nm
- RAP
receptor-associated protein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- 2× YT
two time of yeast extract tryptone.
References
- 1.Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol. 2001;12:195–201. doi: 10.1016/s0958-1669(00)00199-3. [DOI] [PubMed] [Google Scholar]
- 2.Hewitt L, McDonnell JM. Screening and optimizing protein production in E. coli methods. Mol Biol. 2004;278:1–16. doi: 10.1385/1-59259-809-9:001. [DOI] [PubMed] [Google Scholar]
- 3.Jana S, Deb JK. Strategies for efficient production of heterologous proteins in Escherichia coli. Appl Microbiol Biotechnol. 2005;67:289–298. doi: 10.1007/s00253-004-1814-0. [DOI] [PubMed] [Google Scholar]
- 4.Peti W, Page R. 2006 [Google Scholar]
- 5.Structural Genomics Consortium. Protein production and purification. Nat Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riek R, Fiaux J, Bertelsen EB, Horwich AL, Wuthrich K. Solution NMR techniques for large molecular and supramolecular structures. J Am Chem Soc. 2002;124:12144–12153. doi: 10.1021/ja026763z. [DOI] [PubMed] [Google Scholar]
- 7.Tugarinov V, Hwang PM, Kay LE. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev Biochem. 2004;73:107–146. doi: 10.1146/annurev.biochem.73.011303.074004. [DOI] [PubMed] [Google Scholar]
- 8.Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 9.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Tyler RC, Sreenath HK, Singh S, Aceti DJ, Bingman CA, Markley JL, Fox BG. Auto-induction medium for the production of [U-15N]- and [U-13C, U-15N]-labeled proteins for NMR screening and structure determination. Protein Expr Purif. 2005;40:268–278. doi: 10.1016/j.pep.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Sreenath HK, Bingman CA, Buchan BW, Seder KD, Burns BT, Geetha HV, Jeon WB, Vojtik FC, Aceti DJ, Frederick RO, Phillips GN, Jr, Fox BG. Protocols for production of selenomethionine-labeled proteins in 2-L polyethylene terephthalate bottles using auto-induction medium. Protein Expr Purif. 2005;40:256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 12.Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–222. doi: 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- 13.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 14.Sahdev S, Khattar SK, Saini KS. Production of active eukaryotic proteins through bacterial expression systems: a review of the existing biotechnology strategies. Mol Cell Biochem. 2008;307:249–264. doi: 10.1007/s11010-007-9603-6. [DOI] [PubMed] [Google Scholar]
- 15.Gschaedler A, Robas N, Boudrant J, Branlant C. Effects of pulse addition of carbon sources on continuous cultivation of Escherichia coli containing a recombinant E. coli gapA gene. Biotechnol Bioeng. 1999;63:712–720. doi: 10.1002/(sici)1097-0290(19990620)63:6<712::aid-bit9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 16.Chen HC, Hwang CF, Mou DG. High-density Escherichia coli cultivation process for hyperexpression of recombinant porcine growth hormone. Enzyme Microb Technol. 1992;14:321–326. doi: 10.1016/0141-0229(92)90159-l. [DOI] [PubMed] [Google Scholar]
- 17.Hannig G, Makrides SC. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998;16:54–60. doi: 10.1016/s0167-7799(97)01155-4. [DOI] [PubMed] [Google Scholar]
- 18.Cai M, Huang Y, Sakaguchi K, Clore GM, Gronenborn AM, Craigie R. An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J Biomol NMR. 1998;11:97–102. doi: 10.1023/a:1008222131470. [DOI] [PubMed] [Google Scholar]
- 19.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Chen J, Wang JA. Complete backbone spectral assignment of human apolipoprotein E. Biomol NMR Assign. 2008;2:207–210. doi: 10.1007/s12104-008-9122-8. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Wang J. A complete NMR spectral assignment of lipid-free mouse apolipoprotein A-I C-terminal truncation mutant, apoAI(1–216) Biomol NMR Assign. 2007;1:109–111. doi: 10.1007/s12104-007-9031-2. [DOI] [PubMed] [Google Scholar]
- 22.Ren X, Yang Y, Neville T, Sparks D, Wang J. A complete backbone spectral assignment of human apoAI/preβHDL particle. Biomol NMR Assign. 2007;1:69–71. doi: 10.1007/s12104-007-9020-5. [DOI] [PubMed] [Google Scholar]
- 23.Sivashanmugam A, Wang J. NMR structure of the complete receptor-binding domain of human apolipoprotein E. J Biol Chem. (in press) [Google Scholar]
- 24.Fisher CA, Wang J, Francis GA, Sykes BD, Kay CM, Ryan RO. Bacterial overexpression, isotope enrichment, and NMR analysis of the N-terminal domain of human apolipoprotein E. Biochem Cell Biol. 1997;75:45–53. [PubMed] [Google Scholar]
- 25.Ren X, Zhao L, Sivashanmugam A, Miao Y, Korando L, Yang Z, Reardon CA, Getz GS, Brouillette CG, Jerome WG, Wang J. Engineering mouse apolipoprotein A-I into a monomeric, active protein useful for structural determination. Biochemistry. 2005;44:14907–14919. doi: 10.1021/bi0508385. [DOI] [PubMed] [Google Scholar]
- 26.Cui C, Chen J, Zhao W, Wang J, Li Q. Elimination of in vivo cleavage between target protein and intein in the intein-mediated protein purification systems. Protein Expr Purif. 2006;50:74–81. doi: 10.1016/j.pep.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 27.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Johnson BA, Blevins RA. NMR view: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 29.Peti W, Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif. 2007;51:1–10. doi: 10.1016/j.pep.2006.06.024. [DOI] [PubMed] [Google Scholar]