

Abstract
The type 1 repeat domain from thrombospondin has potent antiangiogenic activity and a structurally interesting fold, making it an attractive target for protein engineering. Chemical synthesis is an attractive approach for studying protein domains because it enables the use of unnatural amino acids for site-specific labeling and detailed structure-function analysis. Here, we demonstrate the first total chemical synthesis of the thrombospondin type 1 repeat domain by native chemical ligation. In addition to the natural domain, five sites for side chain modification were evaluated and two were found to be compatible with oxidative folding. Several challenges were encountered during peptide synthesis due to the functional complexity of the domain. These challenges were overcome by the use of new solid supports, scavengers, and the testing of multiple ligation sites. We also describe an unusual sequence-specific protecting group migration observed during cleavage resulting in +90 Da and +194 Da adducts. Synthetic access to this domain enables the synthesis of a number of variants that can be used to further our understanding of the biochemical interaction network of thrombospondin and provide insight into the structure and function of this important antitumorogenic protein domain.
Keywords: thrombospondin, protein synthesis, ligation, angiogenesis
Introduction
Thrombospondin-1 (TSP-1), a multi-domain secreted glycoprotein, influences cellular phenotype and regulates extracellular matrix structure through numerous interactions with effector proteins.1 TSP-1 influences angiogenesis through its regulation of endothelial cells, and its downregulation is associated with tumorogenesis. The primary antiangiogenic activity of thrombosopondin-1 has been localized to a region encompassing the type 1 repeat (TSR) domains,2 and the chemoprotective effect of full-length TSP-1 can be mimicked by a recombinant fragment consisting of three TSR domains (hTSP1 358–530).3 Further results suggest that the antitumorogenic activity of TSP-1 is directly related to the activity of the individual type 1 repeat domains. In isolation, the second and third TSRs of TSP-1 inhibit angiogenesis both in vitro and in vivo through the inhibition of endothelial cell migration, the induction of endothelial cell apoptosis, and the activation of transforming growth factor β.4 These properties make the TSR domains an attractive target for therapeutic development.
TSR domains, defined by a conserved sequence and unusual fold, are present in 41 known human proteins as well as a multitude of proteins in organisms ranging from Drosophila to C. elegans to apicomplexan parasites.5 TSR domains contain six conserved cysteines involved in three disulfide bonds, two to three conserved tryptophan residues, and two conserved arginine residues, all thought to be important for the three-dimensional structure of the domain. The crystal structure of TSR2 and TSR3 reveals an unusual fold consisting of two antiparallel β-strands and a third antiparallel strand that adopts a unique “rippled” structure. Cation-π stacking interactions between three tryptophans on the “rippled” strand and two arginine residues from the adjacent strand form the core of the folded domain, which is sandwiched by three disulfide bonds [Fig. 1(A)].6 In addition to these structural features, the second TSR domain of human TSP-1 can contain an O-linked disaccharide Glc-Fuc-O-Thr and a C-linked α-mannosyl residue on C2 of Trp420 and Trp423.7 However, these sugar residues are not essential for the antiangiogenic function of the domain, as recombinant proteins from bacteria lacking the necessary glycosyl transferases retain activity in vivo.8 The cation-π network forms a positive electrostatic groove on one face of the folded protein that is postulated to be an important binding site for protein–protein interactions.9 However, the unusual structure of this domain makes structure function studies challenging. It is likely that anything but very small modifications to the highly conserved core cation-π network will abolish folding of the domain.
Figure 1.

A: Structural representation of TSR2 based on the structural coordinates 1LSL.pdb. Structurally important residues are displayed in line form: disulfide bonds are indicated in yellow, with potential ligation site cysteines shown in red, and residues that were modified are indicated in blue. Image excludes N-terminal residues K412-Q416. B: Schematic diagram of the sequence of TSP-1(412–473), with structurally important residues shown. As in the structural representation, disulfide bonds are in yellow, potential ligation site cysteines are shown in red, and residues that were modified are circled in blue. The NleGly sequence replaces a native MetAsn sequence as described in the text.
The combination of therapeutic potential and unique structural features make thrombospondin type 1 repeat domains an attractive target for chemical synthesis. The use of chemical synthesis allows the introduction of unnatural amino acids to probe the structure and function of the domain. However, the TSRs are a challenging synthetic target due to the abundance of cysteine and tryptophan residues. To demonstrate the synthetic accessibility of the TSR domain, we have chemically synthesized the second TSR from TSP-1, TSR2. To demonstrate the utility of chemical access to this domain, we synthesized a biotinylated TSR2 designed as a tool to aid in efforts to identify the cellular receptor(s) for TSR2 and to elucidate the complex biochemical interaction network of the domain. In addition, a variant containing PEG-biotin was synthesized, as biotin-PEG-protein conjugates show improved binding to avidin when compared with biotin-protein conjugates.
Results and Discussion
Synthetic targets
The second TSR domain (TSR2) has been structurally defined as residues Q416 to I473 of TSP-1.6 However, we chose to focus on residues K412 through I473 [Fig. 1(B)] because the N-terminal basic KRFK(412–415) sequence is known to be responsible for TGF-β activation,10 and previous work has shown increased antitumorogenic effects of the domain when this sequence is included.4 We have also found that the inclusion of the KRFK residues decreases problems with aggregation during folding, making the extended domain a more robust target for future studies. Recombinant TSR domains have been expressed in several organisms and successfully refolded in good yield after reduction and purification.6 As a result, it was anticipated that the use of orthogonal protection on cysteines for directed disulfide bond formation would not be necessary.
Several minor modifications were made to the covalent structure of the TSR domain. Met451 was replaced with norleucine to prevent possible problems with oxidation, and Asn452 was replaced with glycine to eliminate the possibility of aspartimide side products.11 Glycine was chosen as a replacement for asparagine as it occupies the same position in the homologous and also biologically active third TSR of TSP-1. Finally, to mimic the larger protein context of the TSR domain and to reduce potential endopeptidase activity, the N-terminus was acetylated and the C-terminus was amidated.
Synthetic strategies
A two-piece ligation strategy was chosen for the synthesis of the 62 amino acid domain. This convergent approach allows for the introduction of variants in the C-terminal half of the domain without necessitating the resynthesis of the other half of the protein. All peptides were synthesized by manual solid-phase peptide synthesis (SPPS) using standard Boc protection and HCTU activation with in situ neutralization.12 Modifications that were made to standard protocols in order to improve yields are described later.
As the TSR2 domain contains six cysteines, there are many possible ligation sites (see Fig. 1). Initially, Leu443-Cys444 was chosen as the ligation site because of its location near the middle of the sequence. Although this route was successful, the synthesis of the N-terminal peptide, Ac-TSP-1(412–443)-COSR was problematic (see later), and low yields for this synthesis limited production of the ligated full-length product. Hence, two alternative ligation sites, Ser428-Cys429 and Thr432-Cys433, were also tested. Overall yields were highest with the Ser428-Cys429 ligation site, so this was used for later syntheses. Despite ligation rates being relatively slow with leucine or threonine as the C-terminal amino acid of the N-terminal thioester peptide,13 all three ligation reactions were complete after 18 h at 37°C, pH 7, and had comparable yields of 25–35% recovered following purification. The difficulties encountered in the synthesis of TSR2 occurred during SPPS and hydrogen fluoride (HF)-mediated cleavage of the peptide segments, whereas the ligation reactions to generate full-length product were straightforward.
Optimization of SPPS and cleavage conditions
C-terminal synthesis
Initial results indicated a number of increased-mass side products for the synthesis of 444Cys-TSP-1(445–473)-NH2. As it is likely that these adducts arise during HF cleavage and deprotection of the peptide, several HF scavengers were tested.14,15 Crude yields in the presence of 10% anisole were ∼40% by analytical HPLC. The addition of ethanedithiol (EDT) (0.6 mL/g resin) in the presence of 10% p-cresol improved crude yields to ∼57%. These modified conditions were used for all C-terminal fragments synthesized.
N-terminal synthesis
For the synthesis of AcTSP-1(412–443)-COSR, a number of increased-mass adducts were also observed, resulting in low overall yields. While the addition of EDT might be expected to improve yields of this segment in the same way as the C-terminal segment, thiol-based HF scavengers complicate the workup of thioester peptides. Other HF scavengers including p-cresol and indole15 were tested, but none improved yields significantly.
AcTSP-1(412–443)-COSR contains three tryptophan residues, which were initially incorporated as the side-chain unprotected Boc-Trp-OH. To investigate whether the adducts observed are forming on Trp residues, AcTSP-1(412–443)-COSR was synthesized incorporating either Trp(hoc) or Trp(formyl). While the use of Trp(hoc) was not an improvement over the use of unprotected tryptophan, the use of Trp(formyl) improved yields from ∼10% to ∼30% by analytical HPLC [Fig. 2(A,B)]. As the hoc group is removed upon treatment with HF, while the formyl group is stable to HF, these results suggest that modifications to Trp are occurring during HF-mediated deprotection, rather than during the process of SPPS. Despite the observed improvement in crude yield, the use of Trp(formyl) was not chosen as a synthetic strategy because the use of the formyl group on the thioester-containing N-terminal fragment necessitates protection of internal cysteines in both ligation fragments due to formyl migration during ligation.
Figure 2.

HPLC traces of N-terminal synthetic products varying in Trp protection and resin. Peaks with a “*” correspond to the +194 Da adduct. Gradient is 22.5 to 58.5% ACN in H2O, 0.1% TFA over 30 min. A: N-terminal on MBHA resin with Trp(hoc). B: N-terminal on MBHA resin with Trp(formyl). C: N-terminal on ChemMATRIX resin with unprotected Trp.
Synthesis of AcTSP-1(412–443)-COSR was attempted on two different resins, the standard MBHA-1% crosslinked polystyrene resin and ChemMATRIX, a PEG-based resin. ChemMATRIX has been used frequently in Fmoc synthesis,16 but has not been previously applied to Boc synthesis. It was observed that couplings on ChemMATRIX are significantly slower than those on standard MBHA, with double couplings (2 × 15 min) being required to obtain quantitative chain assembly. Interestingly, this resin also swells significantly more in TFA than it does in DMF or DCM, swelling to ∼2–3 times its volume in DMF as TFA is added for the Boc-removal step. As a result, ChemMATRIX resin requires more TFA than an equivalent volume of polystyrene resin. Thus, the handling of ChemMATRIX resin during a synthesis differs from the handling of polystyrene resins. However, upon optimization of the coupling cycles, synthesis on ChemMATRIX resin was highly successful: Using double couplings and unprotected tryptophan, synthesis on ChemMATRIX resin resulted in product that was ∼40% pure by HPLC [Fig. 2(C)], whereas the identical synthesis on MBHA-polystyrene resin of similar loading (ChemMATRIX, 0.62 mmol/g; MBHA, 0.59 mmol/g) yielded a product that was ∼10% pure. Most observed side products were greater in molecular weight than the desired product. We postulate that the increased resin swelling observed in TFA might also occur in HF, which could improve the removal and scavenging of protecting groups from the peptide-resin.
Despite the improvement in the overall purity of the synthesis, neither the use of Trp(formyl) nor ChemMATRIX resin affected an abundant side product. In both cases, a >10% impurity with observed masses of +91 ± 1 Da and +194 ± 1 Da remained. In an attempt to characterize this impurity, a truncated peptide lacking the Trp-rich sequence, 429Cys-TSP-1(430–443)-COSR, was synthesized; however, the same +91 ± 1 Da and +194 ± 1 Da peaks were observed [Fig. 3(A)]. Similarly, the +194 adduct was observed in a sequence lacking Cys429 (data not shown). Looking at the linear polypeptide sequence, 430Ser(Bzl)-Val-432Thr(Bzl)-433 Cys(4-MeBzl)-Gly-Asp(OcHex)-Gly-Val-Ile-Thr(Bzl)-Arg(Tos)-Ile-Arg(Tos)-443Leu-Mpa-Lys-MBHA, we noted that Cys433(4-MeBzl) and Thr432(Bzl) are adjacent to each other, and 194 Da corresponds to the combined mass of their protecting groups. In light of recent work showing that intramolecular reactions between adjacent Cys residues can yield disulfide bonded products during HF-mediated cleavage,17 we reasoned that the +194 adduct could be the result of Bzl group migration between Thr433 and Cys432 (see Fig. 4). To test this hypothesis, we synthesized 429Cys-TSP-1(430–443)-COSR with unprotected Thr(OH) instead of the Thr(Bzl) utilized in the initial synthesis. It was found that replacement of Thr(Bzl) with unprotected Thr eliminates the observed mass adducts in the 429Cys-TSP-1(430–443)-COSR peptide [Fig. 3(B)].
Figure 3.
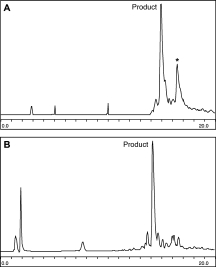
HPLC traces of the synthesis of 429Cys-TSP-1(430–443)-COSR with a gradient of 18 to 54% ACN in H2O, 0.1% TFA over 20 min. A: Thr (Bzl). B: Thr lacking side-chain protection.
Figure 4.

Proposed mechanism for the formation of a +194 Da adduct in the synthesis of 429Cys-TSP-1(430–443)-COSR (left) and potential mechanism for the formation of a +90 Da adduct (right).
Although most benzylation-based side products arising during HF cleavage are thought to result from reaction with free benzyl cations, the sequence-dependent nature of this side reaction strongly suggests an intramolecular mechanism. Before HF-mediated deprotection, when Thr(Bzl) is used, this peptide includes a 432Thr(Bzl)-Cys(4-MeBzl)433 sequence. The deprotection of Thr(Bzl) is expected to be significantly faster than Cys(4-MeBzl) and to occur by an SN1 mechanism, thus generating the benzyl cation.18 This cation could, in principle, react with the nearby Cys(4-MeBzl) before it diffuses away. However, we propose that this adduct could also result from an SN2 mechanism, in analogy to low-HF conditions that use a high concentration of the thioether dimethylsulfide (Fig. 4, left).19 Simultaneous removal of the benzyl group from threonine and attachment to the protected thioether of cysteine would be facilitated by the intramolecular nature of the reaction. The resulting sulfonium moiety on the Cys would be stable to acidolysis, but it could react as an electrophile with another unprotected Cys on the peptide20 (Fig. 4, right). The resulting Cys(4-MeBzl) would undergo the expected acidolysis, but as acidolysis of Bzl from cysteine is significantly slower,21 some Cys(Bzl) would remain and account for the observed +90 adducts. These studies suggest that such intramolecular side reactions could be a more common problem than is generally appreciated.
Purification and ligation
The polypeptides corresponding to Ac-TSP-1(412–443)-COSR, Ac-TSP-1(412–428)-COSR, Ac-TSP-1(412–432)-COSR, 444Cys-TSP-1(445–473)-CONH2, 429Cys-TSP-1(430–473)-CONH2, and 433Cys-TSP-1(434–473)-CONH2 were purified by reverse-phase HPLC on a 10–70 mg scale. After purification, equimolar amounts of each segment were reacted under standard native ligation conditions13,22 to give the desired full length, reduced TSR2 product. Chemical ligation of the N-terminal and C-terminal fragments for all ligation sites is complete after 18 h and the major HPLC peak corresponds to the desired full-length product (see Fig. 5). Products were purified by reverse-phase HPLC.
Figure 5.

HPLC trace of completed ligation reaction, with a gradient of 18 to 54% ACN in H2O, 0.1% TFA over 30 min.
Folding and structural characterization
The TSR2 domain has six cysteines that form three disulfide bonds. Recombinant TSR2 domains have been shown to maintain activity after reduction and refolding in vitro.23 To fold the synthetic domain correctly, a number of conditions for oxidative refolding were tested. Folding of the domain was analyzed by analytical HPLC, with two assumptions: first, that the properly folded product would show a shift in retention time from both unfolded product and incorrectly folded product, and second, that the native disulfide bonding pattern corresponds to the thermodynamic minimum, such that if disulfide exchange is allowed to reach equilibrium, the native disulfide bonding pattern will be the result.24 Mass spectrometry was used to confirm that the folded product corresponded to the formation of three disulfide bonds: the folded product had an observed mass of 6788 Da, while the unfolded, reduced ligation product had a mass of 6794 Da. It was found that folding equilibriums were most readily achieved in a highly oxidizing environment. While most protein folding conditions use a 10:1 ratio of reduced cysteine to oxidized cystine, for the TSR2 domain it was found that a 1:2 ratio of reduced cysteine to oxidized cystine in degassed Tris buffer, pH 7.5, produces folded protein after 4 h of stirring under argon at 4°C (see Fig. 6). The highly oxidizing conditions required for refolding suggest that this domain is comparably unstable to reduction relative to other disulfide-rich proteins.
Figure 6.
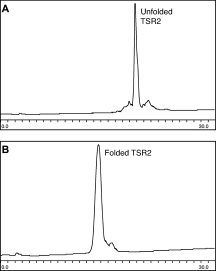
Oxidative folding of TSR2: HPLC traces with a gradient of 18 to 54% ACN in H2O, 0.1% TFA over 30 min. A: Before folding. B: At completion of folding.
TSR domains have a unique CD signal that is diagnostic of the folded structure. The characteristic π-stacking conformation of the TSR domain results in an unusual strong, positive CD signal from 205 to 240 nm with maxima at 212 and 230 nm. To confirm the folding of the TSR2 domain, a CD spectra was taken and compared to the published spectra of a recombinant TSR2 domain,25 as well as the published spectra for the TSR3 domain and TSR123, a protein consisting of all three TSR domains of TSP-1.26 The observed far-UV CD spectra obtained from the folded TSR domain (see Fig. 7) indicates the presence of the expected TSR fold.
Figure 7.
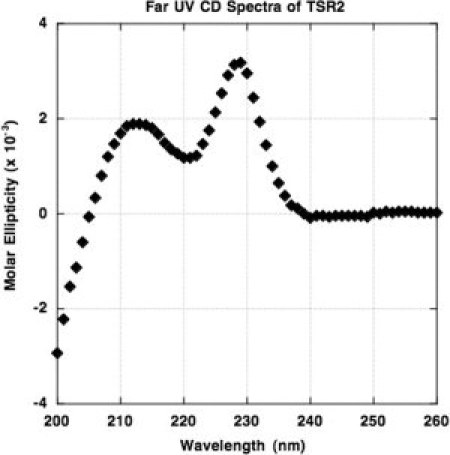
Circular dichroism spectra of folded, “native-like” TSR2.
Design, synthesis, and folding of biotinylated TSR2 analogs
The introduction of a new functionality on the surface of a protein that does not interfere with the folding of that protein can be challenging. To introduce a biotin moiety on the TSR2 domain, first a site of attachment had to be identified. In principle, any surface-exposed residue could be selected. However, in practice it was found that changing the surface charge has a significant impact on the folding of the TSR2 domain. Initially, Lys 454 and Lys 467 were chosen as attachment sites, but it was found that incorporating biotin at these positions abolished proper folding of the domain. It is possible that eliminating a surface charge from lysine interferes with solvation of the domain, leading to aggregation rather than folding during the refolding process. Varying the ratio of cysteine to cystine during the refolding process did little to improve yields of folded material. To investigate other potential attachment sites, a scan was performed, substituting Lys(Biotin) individually at each of the positions 450, 451, and 452. This loop is not thought to be critical to the folding of the domain, as the side chains of these three residues are not well-conserved across TSRs.5 Additionally, the side chains of these residues are uncharged yet solvent-exposed in the crystal structure. We found that Lys(Biotin) variants yielded folded material when biotin-lysine was incorporated at position 450 or 452, but not at the 451 position. An additional variant was produced that incorporates a miniPEG linker between the lysine side chain and the biotin (see Fig. 8), as the inclusion of a linker between biotin and a protein has been shown to improve binding to avidin. TSR2 variants with biotin-miniPEG-Lys fold similarly to biotin-Lys variants. Circular dichroism spectra of the folded 450Lys(biotin-miniPEG)TSR2 is comparable to that of unmodified TSR2 (data not shown), and both demonstrate a similar melting temperature upon thermal denaturation monitored by CD (see Fig. 9). Biotinylated TSR2 will be useful in assays for detection of interactions with other proteins, and can be used for imaging through conjugation with fluorescent avidin.
Figure 8.
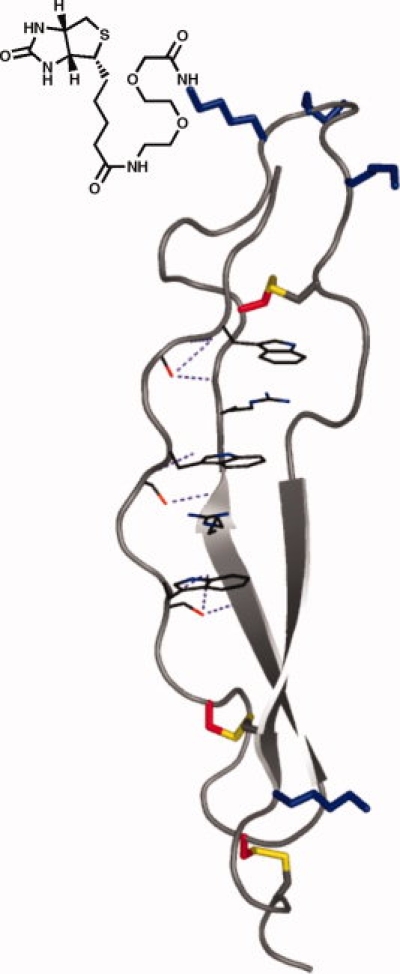
Schematic of biotin-miniPEG-TSR2, with biotin-PEG incorporated at position 450. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 9.

Thermal unfolding of “native-like” TSR2 and biotin-miniPEG(450)-TSR2 monitored by circular dichroism. The data were fit to a two-state transition.
Materials and Methods
Materials
Most Nα-Boc amino acids were purchased from CS-Bio, with the exception of Boc-Trp(Hoc)-OH, which was obtained from Peptides International, Boc-Lys(Fmoc)-OH, obtained from Midwest Biotech, and Boc-Thr-OH, obtained from NovaBiochem. Boc-Lys-PAM was from NeoMPS. S-trityl-β-mercaptopropionic acid, Fmoc-miniPEG-OH, p-methylbenzylhydrazine resin (0.59 meq/g), and HCTU were purchased from Peptides International. ChemMATRIX resin (0.6 meq/g) was obtained from Matrix Innovation. Dimethylformamide (DMF) and trifluoroacetic acid (TFA) were from EMD Biosciences. Acetonitrile was from J. T. Baker. Dichloromethane (DCM), Tris, and NaCl were from Fisher. N,N-diisopropylethylamine, anisole, p-cresol, ethane dithiol, triisopropyl silane, cysteine, and cystine were from Sigma. Thiophenol and benzyl mercaptan were from Aldrich. Hydrofluoric acid (HF) was from Matheson Trigas. Ninhydrin reagents were from Applied Biosystems.
Solid-phase peptide synthesis
All peptides were synthesized by manual SPPS using standard Boc protection and HCTU activation with in situ neutralization.12 Except where otherwise noted, side chain protecting groups used were Arg(Tos), Asn (Xan), Asp(OcHex), Cys(4-MeBzl), Glu(OcHex), Gln(Xan), His(Dnp), Lys(ClZ), Ser(Bzl), and Thr(Bzl). Tryptophan was used without a side chain protecting group. C-terminal peptides (429Cys-TSP-1(430–473)-CONH2, 433Cys-TSP-1(434–473)-CONH2, 444Cys-TSP-1(445–473)-CONH2, and variants thereof incorporating Lys(Fmoc)) were synthesized directly on MBHA resin. Following chain elongation, the N-terminal Boc group was removed and the resins were dried under vacuum. N-terminal thioester peptides (Ac-TSP-1(412–443)-COSR, Ac-TSP-1(412–428)-COSR, and Ac-TSP-1(412–432)-COSR) were synthesized on S-trityl-mercaptopropionic acid-Lys-MBHA (MpaK-MBHA) resin. MpaK-MBHA resin was synthesized on a large scale utilizing standard Boc synthesis procedures to couple Lys and then S-trityl-β-mercaptopropionic acid onto MBHA resin. This resin is known to generate a thioester upon cleavage with HF.13 To begin each synthesis, the S-trityl group was removed using a 2.5% TIS, 2.5% H2O, 95% TFA cocktail. Coupling of the next amino acid was allowed to proceed for 40 min instead of the usual 20. For the synthesis of Ac-TSP-1(412–443)-COSR on ChemMATRIX resin, first, a Lys-PAM linker was incorporated using standard HCTU activation. Then, the S-trityl-β-mercaptopropionic acid thioester linker was introduced as earlier, and double couplings were used throughout. Following incorporation of the first tryptophan on either resin, 5% anisole, 95% TFA was used for each subsequent deprotection step, to limit the formation of tBu adducts on the unprotected tryptophans. The N-terminal peptides were acetylated by incubation of neutralized resin in 0.5M acetic anhydride in DMF for 10 min, followed by washing with DCM and drying in vacuo.
HF cleavage
In a typical HF cleavage reaction, 300 mg of resin are stirred in a vessel containing 1 mL of scavenger (anisole or p-cresol) in 10 mL of HF. The reaction is allowed to proceed at 0°C for 1 h, and then the HF is distilled off under vacuum. The peptide-resin mixture is washed three times with ice-cold diethyl ether and filtered. The peptide is then dissolved in 30% B (27% ACN, 73% H2O, 0.1%TFA) and lyophilized. For C-terminal peptides, 200 μL of ethane dithiol is included in the HF cleavage reaction along with 1 mL of p-cresol.
Reverse-phase HPLC analysis and purification
HPLC buffer A is 99.9% H2O, 0.1% TFA and buffer B is 90% ACN, 9.9% H2O, 0.1%TFA. Analytical HPLCs were run using a Phenomenex Jupiter 4 μ Proteo 90 Å column at 0–67% B over 30 min (peptides) or a Phenomenex Jupiter 5 μ C18 300 Å column at 20–60% B over 30 min (ligations and foldings). Preparative scale HPLC was run on a Waters DeltaPrep4000 instrument using a 21.2 mm Jupiter 10 μ Proteo 90 Å column for peptides and a 10 mm Jupiter 10 μ C18 300 Å column for ligation products and folding. Gradients were adjusted as needed depending on the elution profile of each peptide.
Native chemical ligation
In a typical ligation reaction, equimolar amounts of corresponding N- and C-terminal peptides were dissolved in 6M GnHCl, 200 mM NaPhosphate buffer, pH 8.0, to a total concentration of ∼20 mg/mL. 1% thiophenol and 1% benzylmercaptan were then added to the ligation mixture, the mixture was shaken, and was then allowed to react for ∼18 h at 37°C. Before preparative HPLC of the ligation product, thiols were extracted in diethylether. Pure fractions were identified by ESI-MS, combined, and lyophilized.
Oxidative folding
Purified, lyophilized protein was dissolved at 0.1 mg/mL in freshly degassed TBS buffer (10 mM Tris, 150 mM NaCl, pH 7.5) containing 1.5 mM cysteine and 3 mM cystine (note: cystine is not fully soluble at 3 mM at pH 7.5, so actual concentration in solution is lower). The solution was stirred at 4°C for 2–4 h. Folded protein was purified by RP-HPLC as earlier.
Synthesis of biotinlyated variants of TSR2
Peptides were synthesized as described earlier with Boc-Lys(Fmoc) incorporated at the desired modification site. After chain elongation was complete, but before final N-terminal deprotection, the Fmoc group was cleaved with 20% piperidine in DMF (2 × 15 min). Biotin was coupled using DIC/HOBt activation in DMSO:DMF:DCM (1:5:5). The reaction was allowed to go for 1 h at RT and then monitored by ninhydrin. Double and sometimes triple couplings were required for quantitative biotinylation. For biotin-PEG variants, Fmoc-miniPEG-OH was coupled onto the Lys(-NH2) after piperidine treatment. A twofold molar excess of Fmoc-miniPEG was activated by the addition of 1 eq. HATU and 1.5 eq. DIEA in DMF and coupled for 1 h at RT. A ninhydrin reaction indicated that the coupling was not quite complete, so a second coupling was done using a single equivalent (to resin) of Fmoc-miniPEG. Then, the Fmoc was removed and biotin was coupled as described earlier. Finally, the N-terminal Boc group was removed and peptides were cleaved and purified as described earlier. The conditions for ligation and folding were identical for biotinylated and nonbiotinylated TSR2 variants.
Circular dichroism spectroscopy
CD spectra were acquired on an Aviv 202 CD Spectrophotometer. For far UV spectra, 28 μM of protein was dissolved in 10 mM phosphate, 100 mM NaCl, pH 7.5 buffer (native), or TBS (PEG-biotin) and run in a 1 mm cuvette with 3 s integration times. For thermal denaturation studies, 20–25 μM of protein was dissolved in TBS buffer and heated. The CD signal at 228 nm was recorded every 1.5°C with 3 min equilibration times. Thermal denaturation curves were fit in Prism assuming a two-state transition with ΔCp = 0 and linear baselines.27
Conclusions
The TSR2 domain of human TSP-1 is a promising target for use in antitumorogenic therapies as an antiangiogenic agent. While recombinant TSRs have shown some therapeutic promise,3 synthetic access to the domain provides more flexibility for directed modifications to improve either activity or stability in vivo. Despite advances in peptide and protein chemistry, establishing synthetic access to a new protein domain can require significant troubleshooting and optimization. TSR2 is a challenging target because it is functionally rich and its folding requires the formation of three disulfide bonds. We have demonstrated the synthesis of the TSR2 domain from human TSP-1 and variants utilizing a two-piece ligation strategy followed by oxidative folding. Although the TSR2 domain might be synthetically accessible by SPPS alone (62 residues), a ligation strategy was chosen to facilitate modifications in each half of the protein and to better ensure the purity of the synthetic product.
Once established, synthetic access to the TSR2 domain can be used to create chemically modified variants. Here, we have synthesized several biotinylated variants of TSR2, incorporating either biotin or biotin and a PEG-based linker at several positions. As the biological activity of TSR2 is complex and poorly understood, having a site-specifically biotinylated variant of the protein might help to deconvolute the multiple interactions involved in the regulation of tumorogenesis by TSR2. The biotinylated variants we have synthesized will be used to elucidate the interaction network of the domain. In addition, the total synthesis of the TSR2 domain enables access to a large number of variants that can then be used to further our understanding of the molecular basis of the unique fold and thrombospondin's biochemical interaction network. These ongoing studies will provide insights into the structure and function of this important antitumorogenic domain.
Glossary
Abbreviations:
- 4-MeBzl
4-methylbenzyl
- ACN
acetonitrile
- Bzl
benzyl
- DCM
dichloromethane
- DIC
diisopropylcarbodiimide
- DIEA
diisopropylethylamine
- DMF
dimethylformamide
- EDT
ethanedithiol
- GnHCl
guanidine hydrochloride
- HATU
2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HCTU
2-(6-Chloro-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HF
hydrofluoric acid
- HOBt
N-hydroxybenzotriazole
- Mpa
mercaptopropionic acid
- PEG
polyethylene glycol
- RP-HPLC
reversed-phase high-performance liquid chromotography
- SPPS
solid-phase peptide synthesis
- TBS
tris-buffered saline
- TFA
trifluoroacetic acid
- TIS
triisopropyl silane
- TSP-1
thrombospondin-1
- TSR
thrombospondin-1 type 1 repeat
- TSR1
TSR2, TSR3, first, second, and third type 1 repeats from thrombospondin-1.
References
- 1.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA. 1990;87:6624–6628. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang XF, Galardi E, Duquette M, Lawler J, Parangi S. Antiangiogenic treatment with three thrombospondin-1 type 1 repeats versus gemcitabine in an orthotopic human pancreatic cancer model. Clin Cancer Res. 2005;11:5622–5630. doi: 10.1158/1078-0432.CCR-05-0459. [DOI] [PubMed] [Google Scholar]
- 4.Miao WM, Seng WL, Duquette M, Lawler P, Laus C, Lawler J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-beta-dependent and -independent mechanisms. Cancer Res. 2001;61:7830–7839. [PubMed] [Google Scholar]
- 5.Tucker RP. The thrombospondin type 1 repeat superfamily. Int J Biochem Cell Biol. 2004;36:969–974. doi: 10.1016/j.biocel.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang JH. Crystal structure of the tsp-1 type 1 repeats: a novel layered fold and its biological implication. J Cell Biol. 2002;159:373–382. doi: 10.1083/jcb.200206062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J. C-mannosylation and o-fucosylation of the thrombospondin type 1 module. J Biol Chem. 2001;276:6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 8.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 9.Silverstein RL. The face of tsr revealed: an extracellular signaling domain is exposed. J Cell Biol. 2002;159:203–206. doi: 10.1083/jcb.200209138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultzcherry S, Lawler J, Murphyullrich JE. The type-1 repeats of thrombospondin-1 activate latent transforming growth-factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 11.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262:785–794. [PubMed] [Google Scholar]
- 12.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB. In situ neutralization in boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Res Ther. 2007;13:31–44. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 13.Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc Natl Acad Sci USA. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakakibara S. Chemical synthesis of proteins in solution. Biopolymers. 1999;51:279–296. doi: 10.1002/(SICI)1097-0282(1999)51:4<279::AID-BIP4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 15.Pennington MW. HF cleavage and deprotection procedures for peptides synthesized using a boc/bzl strategy. Methods Mol Biol. 1994;35:41–62. doi: 10.1385/0-89603-273-6:41. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Martin F, Quintanar-Audelo M, Garcia-Ramos Y, Cruz LJ, Gravel C, Furic R, Cote S, Tulla-Puche J, Albericio F. Chemmatrix, a poly(ethylene glycol)-based support for the solid-phase synthesis of complex peptides. J Comb Chem. 2006;8:213–220. doi: 10.1021/cc0600019. [DOI] [PubMed] [Google Scholar]
- 17.Harris KM, Flemer S, Jr, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J Pept Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson BW, Merrifield RB. Acid stability of several benzylic protecting groups used in solid-phase peptide synthesis. Rearrangement of o-benzyltyrosine to 3-benzyltyrosine. J Am Chem Soc. 1973;95:3750–3756. doi: 10.1021/ja00792a046. [DOI] [PubMed] [Google Scholar]
- 19.Tam JP, Heath WF, Merrifield RB. Sn 1 and sn 2 mechanisms for the deprotection of synthetic peptides by hydrogen fluoride. Studies to minimize the tyrosine alkylation side reaction. Int J Pept Protein Res. 1983;21:57–65. doi: 10.1111/j.1399-3011.1983.tb03078.x. [DOI] [PubMed] [Google Scholar]
- 20.Heath WF, Tam JP, Merrifield RB. Improved deprotection of cysteine-containing peptides in HF. Int J Pept Protein Res. 1986;28:498–507. [Google Scholar]
- 21.Hallinan EA. Formation of a dehydroalanyl residue from s-benzylcysteine upon HF cleavage of a [sar1, cys8]-angiotensin II peptide resin. Int J Pept Protein Res. 1991;38:601–602. doi: 10.1111/j.1399-3011.1991.tb01546.x. [DOI] [PubMed] [Google Scholar]
- 22.Dawson PE. Synthesis of chemokines by native chemical ligation. Chemokines. 1997;287:34–45. doi: 10.1016/s0076-6879(97)87005-x. [DOI] [PubMed] [Google Scholar]
- 23.Adams JC, Lawler J. Cell-type specific adhesive interactions of skeletal myoblasts with thrombospondin-1. Mol Biol Cell. 1994;5:423–437. doi: 10.1091/mbc.5.4.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price-Carter M, Gray WR, Goldenberg DP. Folding of α-conotoxins. I. Efficient disulfide-coupled folding of mature sequences in vitro. Biochemistry. 1996;35:15537–15546. doi: 10.1021/bi961574c. [DOI] [PubMed] [Google Scholar]
- 25.Roszmusz E, Patthy A, Trexler M, Patthy L. Structural characterization of the second tsp1-module of human thrombospondin. Biochem Biophys Res Commun. 2002;296:156–160. doi: 10.1016/s0006-291x(02)00826-4. [DOI] [PubMed] [Google Scholar]
- 26.Huwiler KG, Vestling MM, Annis DS, Mosher DF. Biophysical characterization, including disulfide bond assignments, of the anti-angiogenic type 1 domains of human thrombospondin-1. Biochemistry. 2002;41:14329–14339. doi: 10.1021/bi026463u. [DOI] [PubMed] [Google Scholar]
- 27.Greenfield NJ. Analysis of circular dichroism data. Methods Enzymol. 2004;383:282–317. doi: 10.1016/S0076-6879(04)83012-X. [DOI] [PubMed] [Google Scholar]