

Abstract
Prostasin or human channel-activating protease 1 has been reported to play a critical role in the regulation of extracellular sodium ion transport via its activation of the epithelial cell sodium channel. Here, the structure of the extracellular portion of the membrane associated serine protease has been solved to high resolution in complex with a nonselective d-FFR chloromethyl ketone inhibitor, in an apo form, in a form where the apo crystal has been soaked with the covalent inhibitor camostat and in complex with the protein inhibitor aprotinin. It was also crystallized in the presence of the divalent cation Ca+2. Comparison of the structures with each other and with other members of the trypsin-like serine protease family reveals unique structural features of prostasin and a large degree of conformational variation within specificity determining loops. Of particular interest is the S1 subsite loop which opens and closes in response to basic residues or divalent ions, directly binding Ca+2 cations. This induced fit active site provides a new possible mode of regulation of trypsin-like proteases adapted in particular to extracellular regions with variable ionic concentrations such as the outer membrane layer of the epithelial cell.
Keywords: serine protease, channel activating protein, X-ray structure conformational change, feedback regulation
Introduction
Chymotrypsin-like serine proteases are a diverse set of S1-endoproteases found in all branches of life with the exception of plants and protists.1 Currently, excluding homologous viral proteases, there are sixty seven representative structures of trypsin-like proteases in the protein structure databank and fifteen representative viral proteases.2 Present largely in extracellular environments throughout the body, most are expressed as zymogens, cleaved at a conserved site to generate an active molecule via the rearrangement of the active site. Some such as tissue plasminogen activator (t-PA) display some activity in the zymogen form.3 Plasma proteins are typically mosaic proteins who participate in a number of regulatory networks such as fibrinolysis and blood clotting and the immune system. Regulation of these extracellular networks is carried out by a complex system of protease inhibitors, activators, and the variable number of accessory domains positioned at the N-termini of the proteins. Within the serine protease domain substrate, specificity is generated by diversification of the loops distributed between the secondary structure elements and point mutations around the active site that define the P1-P4 site or the P1′-P4′ substrate binding pockets to induce specificity.4
Recently, many novel membrane associated proteases have been discovered, whose exact role either singly or within a cascade have either yet to be determined or is not fully understood.5 One such protein is prostasin or human channel activating protease (h-CAP), a glycosylphosphatidylinositol-anchored membrane associated serine protease initially found in seminal fluid6 and subsequently in kidneys and airway epithelia where it has been shown to be involved in the matriptase-prostasin, extracellular cascades,7,8 a cell-localized cascade capable of activating the epithelial sodium channel Na+ channels. The activation of epithelial cell sodium channel (ENaC) regulates the transport of sodium ions across the membrane, and thus plays an important role for the regulation of transepithelial fluid movement. ENaC has been demonstrated to control the volume of airway surface liquid in the lungs and to also regulate blood volume through activity in the kidney.8,9 Although it is clear that a number of typsin-like proteases are capable of activating ENaC, prostasin is coexpressed with ENaC and appears to be primarily responsible for the activation; relying initially upon matriptase for its own activation.10,11 Natural protease inhibitors such as aprotinin and bikunin have been shown to regulate protasin and at least one natural protein regulator of prostasin activity has been characterized, the serpin, protease nexin 1 (PN1).12 The primary sequence contains no recognized accessory domains, as do other type II membrane serine proteases such as hepsin, matriptase, and DESC,13–15 and merely consists of a signal sequence at the N-terminal and an ampipathic helix at the C-terminus which is processed to insert as a GPI anchor.16 In addition, prostasin does not auto-activate itself at its zymogen cleavage site (PQAR|ITGG),17 suggesting that regulation of prostasin is controlled in part by upstream proteases. Recently, the work of Krem et al. grouped prostasin into a third class of degradative enzymes18 based on a sequence alignment of the last 50 amino acids corresponding in large to the C-terminal exon of the serine protease domains with a large number of serine protease structures and is closely related by primary sequence to acrosins.
Here, we present structures of the extracellular domain of active prostasin in several forms: inhibited by an irreversible chloromethyl ketone inhibitor (dFFKcmk), in its apo form, complexed with calcium, with the synthetic pan trypsin-like serine protease inhibitor camostat and the natural nonspecific protease inhibitor aprotinin. Conformational changes and the flexibility of specificity pocket loops defining the active site are apparent from these structures. Of particular interest is a conformational change of the S1 subsite relative to other serine protease structures, in its native form this completely closes the subsite to the normal P1 residues arginine or lysine. Upon exposure to substrate via crystal soaking or cocrystallization experiments or via the addition of divalent ions, the S1 subsite opens and adopts a more conventional trypsin S1 conformation. This collapse and opening of the S1 pocket has been observed in an active site mutant of thrombin (D102N), where absence of a sodium ion at a site distant from the substrate binding pocket induces the collapse of the S1 subsite.19,20 This inactive “slow” conformation can be restored via allosteric peptide binding at thrombin exosite I.20 The closed S1 conformation also exists natively in α1-typtase and has only been shown to open by mutation of the key Aspartic acid 216 residue to a glycine.21
This induced fit conformational change of native prostasin in response to ligands or divalent cations at the P1 binding site has not been observed previously at the atomic resolution level and provides a possible explanation for the inhibition of prostasin by divalent cations17 as well as further insight into the regulation of membrane associated proteases by physiological changes specific ion concentrations in areas such as the Airway Surface Liquid or prostate.
Results
Overall structure
By convention, the structure is numbered according to the structural alignment with α-chymotrypsin,22 a convention that is adopted throughout the article. The nomenclature of the active site will follow the convention of Schechter.23 The 32 structurally nonequivalent residues are assigned the number of the previous topologically equivalent residue in α-chymotrypsin supplemented by letters in alphabetical order. These correspond to four loops inserted at residues 58, 144, 172, and 184 with lengths of 8, 9, 13, and 5 amino acids, respectively (Supporting Information 1). Loops are named according to the convention of Perona and Craik4,24 whereby five loops numbered A–E corresponding to residues 34–41, 56–64, 97–103, 143–149, and 74–80, respectively, specify surface loops. Loops numbered 1–3, 185–188, 217–225, and 169–174 specify the definition of the S1 pocket [Fig. 1(a,b) colored magenta and cyan]. The structure in its crystalline form lacks the light chain pro-domain, normally a twenty two residue section covalently attached to the heavy chain by a disulfide bond linked to residue 122 which in this construct has been mutated to a serine.
Figure 1.
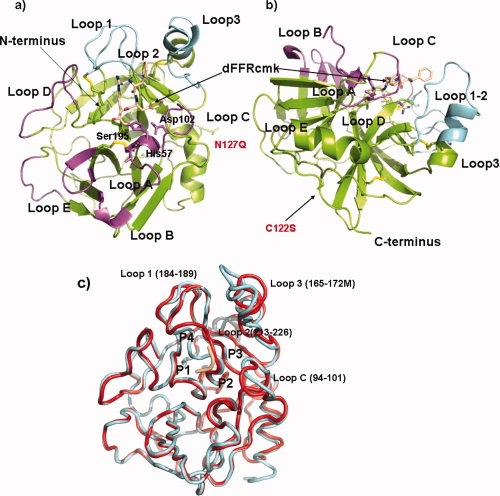
Ribbon diagram of prostasin inhibited and Apo forms produced with Pymol.25 (a) Canonical ribbon representation of dFFR-cmk inhibited structure, ribbon is colored from N and C-terminal via a rainbow representation. Active site residues and atoms of the dFFR inhibitor are portrayed as sticks and colored according to atom. Loops A–E are labeled and shaded magenta whilst those designated at Loops 1–3 are shaded cyan.4,5 Disulfide residues are portrayed as sticks the with sulfur atoms colored yellow. (b) An alternative view of structure in relation to possible membrane associated structure, coloring is identical to that in a), (c) Tube representation of superposition of dFFR-cmk and apo structure, dFFR-cmk inhibited structure in red whilst apo structure in cyan.
Prostasin thus conforms to the general description of an activated trypsin-like serine protease26,27 and consists of an ∼25 Å radius sphere consisting of two sub-domains related by a pseudo two-fold axis.28 Each sub-domain consists of a seven stranded antiparallel β-barrel arranged in a Greek key motif (see Fig. 1). The structure occurs as a monomer in the asymmetric unit within each crystal and as a monomer in the biological unit and is of the type detailed by Sichler whereby residue 190 is an alanine in place of a serine.29 In all crystal structures, the oxianion hole is in a canonical conformation with the salt bridge between Asp194 and the amino terminus of Ile16. As in other chymotrypsin-like serine proteases, the second sub-domain is terminated by an α-helix, in addition a fourteen residue random-coil region follows the helix breaking proline 247 in the construct. Of these fourteen residues, only six could be traced reliably in the electron density for the inhibited structure and only two could be traced for the apo structure; the sequence is clearly flexible as noted by the different conformations in the structures, which would be in accord with the tethering of the molecule to the membrane of the cell via an inserted GPI anchor.16 All other residues could be reliably traced in the electron density. In each of the structures, >96% of residues are in preferred regions of the Ramachandran plot with the exception of one outlier, Asp217 which is in a strained region due to its role in defining the S1 pocket, (vide infra). One cis-proline is present in the structure in the structurally unique Loop D at position 144I.
Comparison with other serine proteases
At the sequence level, prostasin shares ∼40% sequence identity with a number of similar proteases such as human plasma kallikrein β,30 α1-tryptase,31 matriptase13 β-acrosin,32 hepsin,14 and almost all other trypsin like serine proteases. Comparison of representatives from known serine proteases on the structural level indicate that the membrane bound hepsin, α1-tryptase, β-acrosin, and matriptase each have the closest similarity with root mean squared deviations (RMSD) of 0.96E, 1.06 and 1.09, 0.93 Å, respectively, on 211, 210, 211, 221 aligned Cα atoms, all containing a structural core of 210–220 residues with loops decorating the intermittent regions of secondary structure.33 All structures compared contain four structurally conserved disulfide bonds, Cys42-Cys58, Cys136-Cys201, Cys168-Cys182, and Cys191-Cys219. The wild type of prostasin would also contain an additional interchain disulfide between Cys122 and the light chain Cys8; however, the construct for this structure has this position mutated to a serine (see Fig. 1). Figure 2 details the structure alignment of this group of proteases with the dFFR-cmk-inhibited prostasin structure. As would be expected, the core differences in the structure are highlighted in Loops A–E and Loops 1–3, highlighted by the lack of topological equivalent residues between 58A-58E, Loop B, 144A-144E, Loop D 172A-172M, Loop3 183-18E Loop 1 [Fig. 1(a,b)]. Of particular interest is the structurally conserved region before Cys219 downstream of the β-sheet responsible for substrate binding, whereas the Cα's are conserved within this, the rotamers of the disulfide bond are different and the sulfur–sulfur bond points away from the S1 pocket as opposed to toward it in the compared structures, resulting in a slightly large S1 pocket. The ability of this disulfide bond to flip is probably due to the position of a Gly218, situated N-terminal of Cys219, reversed in prostasin relative to other structures (Fig. 2). The region containing the glycosylation mutant N127Q is located at the opposing side of the structure relative to the active site [Fig. 1(a)] and is substituted in all compared structures by small residues and the backbone structure is topologically conserved in this region in all structures with the exception of thrombin which contains an inserted sequence following proline 127. The existence of glycosylation at this site, or the reason for it, is not clear, prostasin retains full activity with the glutamine mutation at this position.
Figure 2.
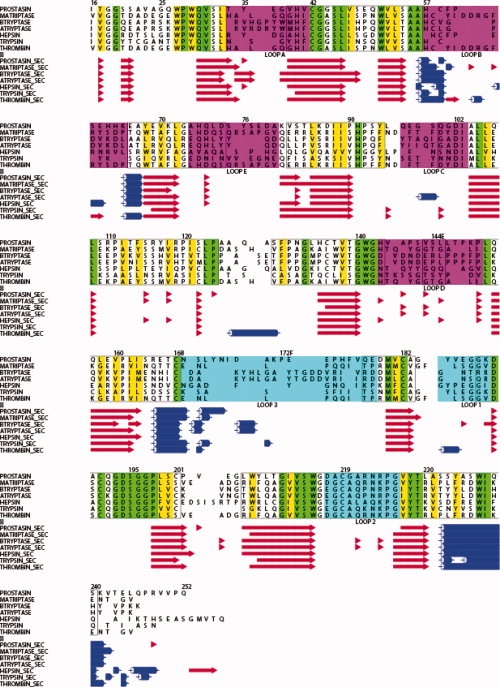
Structure alignment of prostasin with closely related proteases created by MUSTANG,34 STAMP,35 and ALSCRIPT.36 Alignment numbered according to chymotrypsin numbering scheme using coordinate files from, PROSTASIN dFFR, MATRIPTASE, (pdb id 1EAX), BTRYPTASE (pdb id 1A0L), ATRYPTASE (pdb_id 1LTO), HEPSIN (pdb id 1P57) THROMBIN (pdb id 2THF), TRYPSIN (pdb_id 1V2M). Conserved residues are shaded green, conserved hydrophobic residues yellow, conserved charged residues in bold. Loops A–E and 1–3 defined by4,24 are colored magenta and cyan, respectively, and correspond to the colors in Figure 1(a). Secondary structure, for the various sequences are reproduced below at the corresponding sequence number with sheets colored red, helices in blue.
DFFR-chloromethyl ketone-inhibited structure
As in all trypsin-like serine proteases, the S1 subsite is formed by the confluence of three regions designated as Loops 1–3, which collaborate to create a deep pocket containing the negatively charged Asp189 residue at its base. The prostasin structure is no different to this and the backbone of secondary structure elements is largely identical to all homologous structures, with the exception of the substitution of Gly220 for the conventional Ala220, within the pocket downstream of the disulfide bridge Cys191-Cys219.
The high resolution cocrystal of prostasin and dFFR reveals an active site interaction forming a tetrahedral hemiketal structure mimicking a reaction transition state as has been previously characterized.37 The keto-arginine moiety of the inhibitor is covalently linked to Ser195 Oγ via the methyl group and to the imidazole ring nitrogen of His 57 [Fig. 3(a)]. The standard salt-bridge interaction is apparent with the arginine at the base of the P1 pocket and the conserved Asp 189 with the guanidino group separated from the carboxylate by 2.88 Å. The P2 and P3 residues phenylalanine and d-phenylalanine pepides form the canonical anti-parallel β-sheet interaction with Ser214 and Gly216. As would be expected for a generally nonspecific inhibitor, the Phenylalanines at the P2 and P3 positions have very little interaction with main chain or side chains lining their respective pockets. The P2 phenylalanine tucks under a relatively voluminous pocket created by Loop C, whereas the d-Phenylalanine projects into the solvent between the S3 and S4 binding cleft formed by Loop D and Loop 3.
Figure 3.
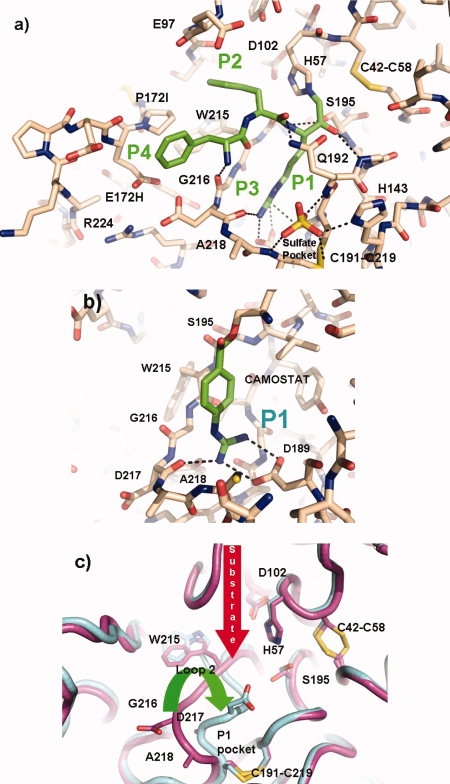
Comparisons of Inhibited and Apo structures, produced with Pymol.25 (a) Stick representation of the active site of the co-crystal of prostasin with dFFR-cmk compound carbon atoms in the protein are colored brown whilst those in the inhibitor are colored green, hydrogen bonds are shown as dashed lines. (b) Stick representation of the region around the S1 pocket in the apo-crystal form soaked with camostat. Carbon atoms in the protein are colored brown while those in the inhibitor are colored green; hydrogen bonds are shown as dashed lines. (c) Representation of the conformational differences of the active site Loop 2 in the apo and camostat inhibited crystal form. Carbon atoms are colored cyan and cyan in the two formes, respectively.
At the bottom of this region below the P3 residue lie two negatively charged residues Asp217 and Glu172H which are unusually close (2.48 Å between the closest carboxyl oxygens). This interaction is probably offset by a compensatory Arg224 interacting with the two residues albeit at a longer distance.
At the base of the S1/S4 pocket, an ordered sulfate anion is nestled in a unique pocket formed from an eight residue insertion positioned after residue 144, Loop D, relative to chymotrypsin and the mutation at position 143 to a histidine. The imidazole ring of His143 forms an electrostatic interaction with one of the sulfate oxygens, in concert with a main chain amide interaction with Ser143C and a further interaction with the main chain nitrogen of Cys219 [Fig. 3(a)]. The presence of this pocket directly adjacent to the P1 and P4 binding site and the uniqueness of the loop that forms this pocket will have important implications in the design of prostasin specific inhibitors. In addition to this, a second sulfate anion decorates the surface of the structure along with three glycerol molecules.
Structure comparison between uninhibited and inhibited — form conformational flexibility in specificity determining regions
Four key conformational changes were apparent in the apo structure relative to the chloromethyl ketone inhibited structure. Loops containing residues 95–100, 168–178, 216–220 and 287–291 (Loop C, Loop3, Loop1 and the C-terminus respectively) all show marked conformational changes when compared [Fig. 1(c)]. With the exception of the latter, all of these moieties are associated with the active site cleft and occur in recognized specificity determining loops.4,24 The S2 subsite loop forms a conformational flexible hood situated above the P2 position and the catalytic His157, this loop is unique to Prostasin on a structural level (see Fig. 2) but in the inhibited form is characterized by poorly defined electron density relative to the rest of the structure. Loop 3 containing residues 168–178 forms the bases of the S4 subsite and marks the largest insertion of residues (numbering 13) relative to chymotrypsin (see Fig. 1). This feature starts at Cys168 forming a helix-turn-sheet motif next to the S2 subsite loop defining the interface between the P2 and P4 binding sites (see Fig. 2). The two motifs are displaced relative by a principally,rigid translation accounting for the large deviations from each other [Fig. 1(c)]. However, both of these loops are bounded by symmetry related molecules, suggesting that the reason for this deviation may be a result of crystal contacts, but does illustrate the flexibility of the region. The Loop 216–219 directly downstream of the β-strand 214–216 that defines the main chain hydrogen bond substrate binding is starkly different to that of the chloromethyl ketone-inhibited form, centered around Asp217. The apo structure shows an S1 pocket that is entirely closed to the solvent relative to the inhibited pocket. This conformational change removes the unfavorable charge–charge interaction between Asp217 and Glu172H and places the residue in the allowed region of the Ramachandran plot relative to the disallowed region of the chloromethyl ketone, suggesting that in the absence of substrate this form is a lower energy conformation. The movement of the loop also configures the rotamer of Asp189 to be in a different nonstandard conformation [Fig. 3(c)]. The final region of diversity is the sequence C-terminal of the final α-helix, the dFFR form terminates its helix one turn early at Thr243. The remaining eight residues then curl back in a kinked β-strand region against the outside of the protein defined by Leu123-Pro131. The helix in the apo region extends to Gln246 before continuing in a completely different direction for a couple of residues. This conformational difference between both crystal forms indicates that this region is flexible as would be expected for a region connecting the GPI anchor to the membrane.
Soaking of crystals with pan specific trypsin inhibitor camostat
The apo structure clearly shows a closed conform of the S1 subsite initiated by the rotation of the Asp217 loop to a lower energy conformation in the absence of substrate. To test whether it was possible to open this site and rescue the active site conformation, the covalent inhibitor camostat was soaked into an apo-crystal for twelve hours before harvesting of the crystal for data collection. Camostat, is a known trypsin inhibitor used in the treatment of pancreatitis38 and a pseudo-irreversible inhibitor. The mechanism-based inhibition is initiated via the nucleophilic attack of Ser195 Oγ on the carbonyl atom of the inhibitor causing the release of a 1-methyl-3-phenyl-pyrazole-5ol group, trapping the residual acyl guanidine-benzoate in the S1 subsite. The crystal structure for trypsin-camostat and thrombin-camostat complexes have been reported,39 but structures to date are not available in public databases. Initial data analysis of the 1.7 Å structure difference Fourier clearly reveals the presence of the covalent intermediate camostat bound to Ser Oγ via the carbonyl atom [Fig. 3(b)]. It also illustrates that the S1 subsite of the structure is completely open to the solvent via the movement of the P1 loop and in an identical conformation to that of the inhibited cocrystal form [Fig. 3(a)]. As the apo and camostat soaked crystals were isomorphous, this indicates that the P1 flexible loop has moved in response to an appropriate P1 substrate thus forming the appropriate S1 subsite via a conformational change and restoring the canonical rotamer of Asp189. The p-guanidino-benzoate nestles in the S1 subsite forming a typical salt bridge interaction between the guanidino and carboxylate groups with a distance of 2.88 Å between opposing nitrogen–oxygen pairs.
Structural explanation for the effect of divalent cations
It has been noted that the addition of cations are inhibitory to prostasin, particularly divalent cations,17 this would insinuate that cations may have an effect on the active site or influence on a distal part of the protein that affects the active site. To test this hypothesis, datasets were collected on crystals that crystallized in the presence of excess calcium and potassium. The calcium-containing form crystallized in an identical crystal form to the apo structure and was indistinguishable to the molecule with the exception of the conformation of the 215–219 loop which contained a Ca+2 ion at the bottom of the S1 site forming an electrostatic interaction with Asp189. The Ca+2 ion is coordinated via 3 waters, Asp 189 and the carbonyl oxygens of Alanine 218 and Alanine 190 making a total coordination number of seven, typical of a Ca+2 ion. The coordination is arranged in a format that the oxygens are arranged in a typical pentagonal bipyrimid around the Ca+2 positions around 2.4 Å giving a typical calcium binding arrangement40 [Fig. 4(a)].
Figure 4.
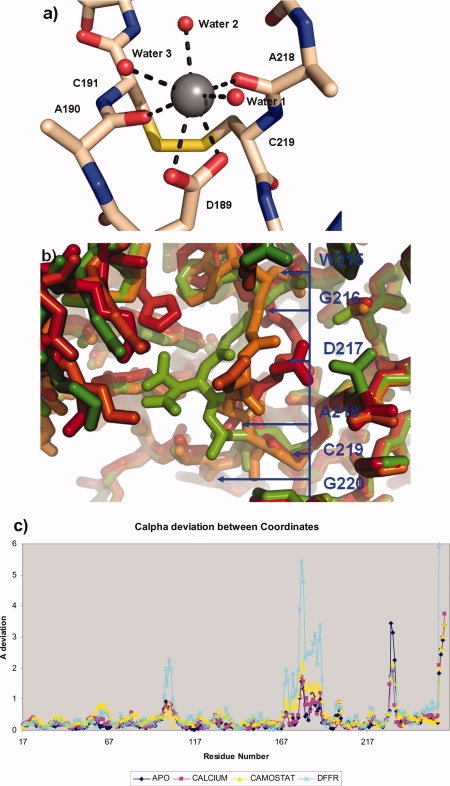
Conformational variability of the P1 binding loop figures produced with Pymol.25 (a) P1 binding site of crystal form of with Ca+2 ion present, detailing the opening of the loop and the coordination of the Calcium ion with Alanines 218 and 190, Aspartic Acid 189 and four water molecules, the seven coordination interactions are represented by dashed lines. (b) The three conformations of the P1 loop. The Apo “closed form” is shaded red, the Ca+2-bound form Amber, and the substrate bound site “open” for green. The blue line represents the position of the residues along the conformationally variant Loop 2. (c) Graph of the deviation of Cα coordinates by residue number for the four representative prostasin structures, numbers relate to the chymotrypsin numbering scheme described in the text.
Although the carboxyl group and carbonyl oxygens necessary for calcium coordination are present in all typsin-like serine proteases, this Ca+2 binding has not previously been observed and may be inhibitory for plasma proteases where the plasma concentration of 1.2 mM for Ca+2 ions is significant and of the same order of magnitude as the binding constant for Ca+2 observed by Shipway17 (Ki of 1.15 mM). The ability to bind calcium appears to arise from the two substitutions around Cys219 relative other serine proteases, the first, the substitution of Ala220 for a glycine and the second the substitution of Gly218 for an alanine (see Fig. 2). This swapping provides some flexibility to the Cys191-Cys219 disulfide bond allowing it to flip to a different rotomer, the alanine insertion at position 218 which is topologically conserved as a glycine in all other trypsin-like serine proteases rigidifies the position allowing the carbonyl oxygen of 218 to be at a convienient bonding distance to the Ca+2.
In comparison to the isomorphous closed and camostat-bound structure (Table I), the position of Loop 2 is in an intermediate position between that of the inhibited and noninhibited form [Fig. 4(b)]. Centering on Asp217 the Cα is 3.75E from the inhibited and 3.25 Å from the apo closed form. The main chain of the high resolution construct is in an intermediate position between the open and closed form of the structure and the density of the high resolution Ca+2 structure revealing the carbonyl oxygen of Ala218 to be in alternate positions, both with and without a bond to the calcium [Fig. 3(c)]. Other work with Kallikrein 441 have shown distinct catalytic effects with zinc, nickel, and cobalt on the protease at sites distal to the active sites, these sites are not conserved within prostasin and we have so far shown no evidence of similar binding in prostasin. An identical experiment carried out with monovalent potassium ion (0.2M) produced a 1.5 Å structure identical to the prostasin closed form, with the exception that residues 219–216 were partially disordered as evident by diffuse electron density indicative of multiple conformations for the loop. As prostasin is of the Pro225 form not the Tyr225, as are the Na+ activated serine proteases, no monovalent ion was present in this P1 subsite or elsewhere in the structure.42 This is in agreement with the observation that the effect of monovalent ions is very weak, and no sodium ions have thus far been located despite the fact that the crystallization drop contains 0.15M sodium chloride.
Table I.
Data and Refinement Statistics
| Protein complexed with | Prostasin dFFKcmk | Prostasin Ca+2 | Prostasin none | Prostasin camostat | Prostasin aprotinin |
|---|---|---|---|---|---|
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| a (Å) | 37.68 | 54.09 | 53.56 | 53.30 | 42.48 |
| b (Å) | 75.88 | 54.07 | 54.07 | 53.80 | 126.93 |
| c (22 Å) | 79.18 | 82.60 | 82.68 | 82.30 | 134.56 |
| Wavelength (Å) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Resolution (Å) | 1.7 | 1.3 | 1.8 | 1.6 | 2.8 |
| Rmerge (%) | 0.088 | 0.061 | 0.063 | 0.063 | 0.152 |
| Unique refs. (observed) | 29,540 (419,031) | 69,317 (337,768) | 29,400 (97,784) | 33,631 (195,979) | 17,082 (108,533) |
| Completeness (%) | 95.8 | 90.6 | 97.5 | 97.6 | 91.2 |
| Rfactor (Rfreea) %b | 0.16 (0.202) | 0.1531 (0.1776) | 0.228 (0.262) | 0.158 (0.192) | 0.225 (0.271) |
| No. protein atoms | 3754 | 3694 | 3670 | 3720 | 4514 |
| No. water atoms | 290 | 367 | 199 | 247 | – |
| No. hetero atoms | 98 | 13 | 0 | 19 | – |
| RMSD bonds (Å) | 0.013 | 0.017 | 0.018 | 0.017 | 0.007 |
| RMSD angles (°) | 1.475 | 1.854 | 1.755 | 1.77 | 1.020 |
| Mean B factor (Å2) | 16.4 | 8.8 | 21.0 | 16.9 | 46.8 |
Rfree = as for Rfactor, but for 5.0% of the total reflections chosen at random and omitted from refinement.
Rfactor = ∑∑|Ii − 〈Ii〉||/∑|Ii| where Ii is the scaled intensity of the ith measurement, and 〈Ii〉 is the mean intensity for that reflection.
P1 conformational flexibility
The inhibited and uninhibited forms of the S1 subsite in prostasin exhibits a large difference in the conformation of Loop 2 (215–219), a region following the β-sheet responsible for the main chain hydrogen bonding stability of the substrate. This closed conformation of the P1 pocket has been previously observed in α1-tryptase43 but the movement of the motif in response to substrate or divalent cation has not been previously recorded, largely because α1-tryptase contains a nonconserved aspartate residue at position 216 locking the protein in the closed position. The α1-tryptase open conformation has only been achieved by the mutation of the Asp216 by a glycine thus allowing the binding of substrate.21 This ability appears to be available in the wild type of prostasin. In relation to wild type α1-tryptase, the P1 loop before Cys 220 is in essentially identical zig-zag conformation with the exception that the indole ring of Trp215 is situated closer to the conventional conformation in prostasin [Fig. 4(b)]. In its open form, the S1 subsite is essentially canonical and forms the identical Arg/Lys specific site recognized in the rest of the serine proteases. The conformation of Asp189 is also very flexible and occurs in a noncanonical position in the apo form and in multiple conformations as in the Ca+2 bound form. Other proteases such as Factor D achieve activation zymogenicity via an induced fit of selectivity of a specific substrate C3bB44,45 via a closing of the site Ser215 and Arg218. A similar effect has been observed recently for thrombin19,20 where absence of a sodium ion usually bound at a site distal to the active site causes the collapse of Trp215 into the P1 pocket generating a so-called “slow” self inhibited thrombin form. The conformation of which can be restored by binding of a fragment of the protease activated receptor PAR1 to exosite I20
Prostasin/aprotinin complex
As expected, the lower resolution (2.8 Å) prostasin aprotinin complex forms a canonical complex in accord with other well studied aprotinin complex such as that with trypsin and matriptase.13,46 In general, the structure of the aprotinin inhibited prostasin has identical conformation to those of the other two small molecule inhibited structures with the exception of the mobile S2 subsite loop (Loop C 95–100), and the S3 subsite loop 168–178 (Loop 3) which is topologically more similar to the chloromethylketone-inhibited form. The reactive site loop of aprotinin forms the expected anti-parallel β-sheet on the scissile bond side 214–217 of prostasin forming carboxyl oxygen to amide nitrogen hydrogen bond from prostasin to aprotinin with Lys215-Ser14, Gly216-Pro13, respectively. The prostasin S2 hood (Loop C, 97–103) adopts an entirely different conformation to each of the other crystal forms and folds back in response to the insertion of aprotinin residues 4–38 to reveal a completely open S2 subsite (see Fig. 5). The S4 subsite containing aprotinin Pro13 in the P4 pocket again adopts a different shape with respect to the other structures, this time His172J forms an interaction with the slightly displaced Glu172H in the apo conformation. The loop 58A–58G inserted after Cys58 does not change conformation as in the Matriptase-Aprotin structure13 but the extended loop does provide additional interactions with the Kunitz domain in the form of an electrostatic interaction of Glu58D with aprotinin Arg20 and Lys46. The S1 subsite of prostasin contains the expected contact with Lys15 of aprotin, but the water-mediated interaction of the ɛ-amino group with Asp189 was not observed, probably due to the low resolution of the data.
Figure 5.
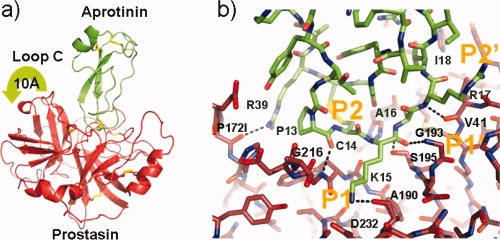
Binding of prostasin to aprotinin, figures produced with Pymol.25 (a) Ribbon representation of the binding of prostasin to aprotinin, prostasin is shaded red whilst aprotinin is colored green. Disulfide bonds are shown in a stick format with sulfur atoms shaded yellow. The movement of Loop C is embodied by a green arrow. (b) Stick representation of the aprotinin/prostasin binding site colored according to Figure 5a. Hydrogen bonds are shown as black dotted line.
Substrate specificity of prostasin
The overall fold of the prostasin catalytic domain is extremely similar to other trypsin-like serine protease, diversity in function/substrate specificity is provided by insertions and substitutions within variable regions of the structure, contained between secondary structure elements.33 Little is known about the natural substrate sequences of prostasin, with the exception that prostasin does not auto-activate itself via cleavage of the Arg15-Ile16 bond. The work by Shipway et al.17 gives a detailed analysis of prostasin substrate specificity based on the release of a coumarin substrate from a synthetic tetrapeptide library.47 This reveals a fairly broad specificity for the substrate binding site with the exception of the P1 being exclusive to basic residues, P2 not favorable to aspartate, glutamine, and glycine residues and, a preference for histidine, arginine, and lysine at the P3 site and P4 preferential to arginine and lysine residues. The unfavorable nature of the acidic P2 residues can easily be accounted for by the presence of acidic residues Glu97 situated directly above the S2 pocket and Asp102, inside the S2 pocket, forming adverse charge–charge interactions.
Although the long broad envaginated and flexible pockets of the S3 and S4 subsite make the positioning of long basic residues within the pocket easy, it seems likely that the positively charge residues are attracted to the pivotal residue Asp217 and Glu172H at the base of the S3 site, whereas the long S4 allows for many water-mediated interactions.
The prime side is a broad pocket with less conformational flexibility than its counterpart on the nonprime side. The structural basis for the P1′ specificity may be partially aided by the aprotinin/prostasin complex structure although interactions within the S2′-S4′ likely do not reflect the natural substrate given that the rigid Kunitz domain quickly veers into the solvent away from the prime pockets and is expected to be different relative to the more flexible native substrate sequences. Shipway17 describes the P1′ specificity to be broad having only major restrictions on isoleucine in this position, consistent with the inability of prostasin to auto-activate itself. From the aprotinin/prostasin structure, it was observed that all standard rotomers of isoleucine would clash with either the disulfide bridge Cys42-Cys58 or the catalytic Histidine 57. The explanation for the isoleucine intolerance of the S2′, S3′, or S4′ subsites observed by Shipway is less apparent from the structure, as are restrictions on the P3′ subsite, which is glutamine, and threonine or the preference at the P4′ for small aliphatic residues. The structures, however, show no contradiction to the previously postulated cleavage sites of prostasin's biological substrate ENaC. If these specificity determinants are applied, then two out of the three subunits (α and γ) contain sequences in the extracellular domains that fulfill these criteria are α201-209 RRARSVAS and γ178-185 RKRKVGGS, respectively,17 sequences that are very likely to be conformationally flexible and easily accommodated within the substrate binding site.
Discussion
The high-resolution structures of prostasin complexed with various inhibitors and ions provides an atomic level view of the protein, a surface associated protease which is an important regulator of the extracellular environment and as such an important therapeutic agent for the treatment of conditions regulated by ENaC such as cystic fibrosis and hypertension. The combination of structural information, describes a detailed mosaic of movements associated with specificity determining loops which will undoubtedly be important in the structure aided design of potential therapeutic agents,48 which for molecules like serine protease are often curtailed by problems of cross-reactivity of compounds with homologous structures.
In all structures, the conformations of the active site loops defined by Perona and Craig4,24 are shown to be structurally unique relative to related serine proteases and variable within different crystal forms (Figs. 1 and 2). Of note is the conformational flexibility of Loop C, defining a unique portion of the S2 subpocket which is variant in all three crystal forms described, from the cocrystal complex with dFFR-cmk which shadows the S2 subsite to the completely unrolled aprotinin-prostasin pocket opening the S2 pocket to the solvent to allow interaction with aprotinin (Figs. 1, 4, and 5). A charged interaction with the acidic residue Glu97 at the apex of this hood could leverage potency and specificity within the P2 binding site. Loop 3 located at the confluence of the S3 and S4 pockets also forms a topologically unique and flexible region which moves in a rigid motion relative to the anchoring residues Arg165-Glu178 in response to crystal contacts. It is possible that the large degree of loop flexibility within the active site loops contributes to the relative inactivity of prostasin on substrates in comparision to more promiscuous proteases such as trypsin or plasmin, but it is more likely that this inactivity is due to prostasin being a more specific inhibitor and our incomplete knowledge of the natural substrate or contributing effects of metals or other proteins. Also of interest is the unique Loop D in the structure which forms a sulfate binding site which via extension of a P4 residue or lateral expansion of a P1 inhibitor may provide a unique specificity determinant.
However, the most remarkable feature is the movement of Loop 2 in response to external stimuli such as ligands or divalent ions [Figs. 3(b) and 4(b)]. In the apo crystal form of the protease, Loop 2 is in a “closed” conformation, and the P1 specificity determining D189 is not accessible to the solvent. In response to the soaking of basic substrates or in the presence of Calcium ions, the loop changes conformation to accommodate binding of the ligand (see Fig. 4). The physiological concentrations of divalent cations in the environments where prostasin is expressed can far exceed the binding constants for these ions with prostasin particularly in tissues such as the prostate where calcium concentrations can reach 9 mM. This closure of the S1 pocket has been observed in the trypsin-like serine proteases in the closed zig-zag confirmation of the α1-tryptase structure43 and opening of the pocket could be induced by a simple mutation of Asp 216 to a glycine. This has also been recently demonstrated with an active site (D102N) mutant of thrombin.19,20 Here, the collapse of the pocket is observed in the absence of Na+ and is influenced allosterically by substrate binding in thrombin exosites. Prostasin does not appear to be activated or inactivated allosterically by interactions at distal sites, instead the S1 pocket appears to open and close in direct response to the presence or absence of ligands or divalent cations, a phenomenon not previously observed in trypsin like serine proteases. It is clear that in the inhibited form this loop is in a strained conformation indicated by the nonconventional φ-psi angles of Asp217. It seems likely that this is a molecular mechanism for negative feedback inhibition and points the way to a novel and subtle method of protease regulation in response to the surrounding environment. Although no structural effect are observed in the presence of sodium or potassium ions, calcium ions have been implicated as regulators of ENaC49 and are shown to bind in the S1 pocket of prostasin in an inhibitory manner unseen in previously solved trypsin-like structures.17 One plausible scenario consistent with previous observations50–53 is that raising intracellular Na+ levels by inhibition of the Na+ pump causes a decrease in extracellular Ca+2 ions, whereas Ca+2 ions have also been shown to effect sodium channels directly.50,51,53 ENaC activation may also be influenced by the increasing activity of Prostasin in a Ca+2 depleted environment, suggesting that in an environment with constantly changing ionic concentrations a subtle feedback mechanism is available to regulate the control of the sodium channel in response to the negative feedback provided by the concentration of calcium ions.
Experimental Procedures
Construct and cloning
A construct was designed on the basis of other active chymotrypsin-like serine proteases lacking the N-terminal light chain sequence and with the heavy chain disulfide 122 mutated to a serine to prevent miscellaneous disulfide bonding due to the lack of the light chain. In addition a “spare” cysteine residue 170 was mutated to a Ser to prevent intermolecular disulfide formation as well as a putative glycosylation site mutate N127Q. The hydrophobic C-terminal sequence was also truncated by 38 residues ending at residue 262 (chymotrypsin numbering).
Expression, purification crystallization and soaking
Prostasin was expressed in the baculovirus system in SF9 cells using a honey-bee melittin signal sequence and C-terminal 10 Histidine tag, expression was low (∼800 μg/L) so large volumes of cells were needed to produce enough material for crystallization (∼50 L). Primary purification was performed with a Fast Flow Sepharose IMAC column (Pharmacia). Secondary purification was carried out with a Resource Q followed by buffer exchange into 20 mM Tris pH 8.0, 150 mM sodium chloride, and 1 mMtris(2-carboxyethyl)phosphine (TCEP) before concentrating to 20 mg/mL.
All crystallization experiments were carried out in 96 well low profile Greiner crystallization plates in a sitting drop vapor diffusion format with 480 conditions screens performed at both 4 and 20°C.54 Equal volumes of protein concentrated at 20 mg/mL were added to the reservoir solutions to create a total drop volume of 500 nL. Crystals of the dFFRcmk (soaked for 8 h with 1:3 molar excess of chloromethyl ketone) compound were produced from a crystallization solution containing 1.8M ammonium sulfate buffered with a 0.1M solution of Phosphate/Citrate at pH 4.7, whereas those of the apo form were produced from 30% PEG-6000 buffered with a 0.1M Mes buffer at pH 6.0, those with Ca+2 at 0.2M Calcium Acetate, 20% Peg-3000 buffered with 0.1M Tris at pH 7.0, potassium crystals 0.2M potassium dihydrogen phosphate with 20% Peg-3350.
Screening for the aprotin/prostasin was carried out in an identical manner after mixing purified aprotinin with prostasin in a 1:1 molar ratio of aprotinin the crystallized in 1.6M potassium hydrogen phosphate, 0.4M sodium dihydrogen phosphate, 0.2M sodium chloride in an imidazole buffer at pH 8.0.
All crystals were mounted using 20% glycerol as a cryoprotectant before cooling to 100 degrees K. Soaking of camostat was carried out by adding camostat dissolved at a concentration of 1 mM to the crystal drop before a final concentration of 1 mM in the crystal drop for 12 h before mounting.
Data collection and structure solution
All data were collected at beamline 5.0.3 of the ALS and were processed with the HKL2000 package.55 Refinement and building was performed with Refmac5 and Phenix56,57 and coot58 all other crystallographic manipulations were carried out with the CCP4 package.59
The structure was solved with Molrep60 using the structure of β-tryptase (PDB code 1A0L)31 as a search probe. All data between 50.0 and 4.0 Å resolution were used for the rotation and translation search the final correlation coefficient and R-factor after placement of the one molecule were 0.321 and 0.521, respectively. After this, the model a model constructed from the core of the probe model clearly revealed the dFFKchloromethyl inhibitor in a Sigmaa61 weighted electron density map. After which iterative cycles of manual rebuilding with Coot, Refmac5, and Phenix56–58 were used to complete the structure. The geometry of the structures is excellent and all residues are in allowed regions of the Ramachandran plot with the exception of one residue, Asp217 in the inhibited structures.
All other nonisomorphous forms were solved and refined using identical procedures with the exception that the inhibited prostasin structure was used as the molecular replacement search probe (Table I).
Acknowledgments
The authors would like to thank Andreas Kreusch and Chris Lee for help with data collection, Andrew Schumacher for useful discussion, and Peter Schultz for continued support. The work in this paper is based on experiments conducted at beamline 5.0.3 of the advanced light source (ALS). The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, Material Sciences Division of the U.S. Department of Energy under contract No. DEAC03-76SF00098 at Lawrence Berkeley National Laboratory.
Coordinates
The Structures have been deposited in the protein databank. The apo structure, PDB ID 3E1X, the dFFRcmk complex, PDB ID 3EON, the camostat complex, PDB ID 3FVF, the Calcium complex, 3GYL and the aprotinin complex 3GYM.
Glossary
Abbreviations:
- ENaC
endothelial cell sodium channel
- h-CAP1
human channel activating prostease
- PEG
polyethylene glycol.
References
- 1.Rojas A, Doolittle RF. The occurrence of type S1A serine proteases in sponge and jellyfish. J Mol Evol. 2006;55:790–794. doi: 10.1007/s00239-002-2361-x. [DOI] [PubMed] [Google Scholar]
- 2.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 3.Tachias K, Madison EL. Converting tissue-type plasminogen activator into a zymogen. J Biol Chem. 1996;271:28749–28752. doi: 10.1074/jbc.271.46.28749. [DOI] [PubMed] [Google Scholar]
- 4.Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 5.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, Antalis TM. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22:237–258. doi: 10.1023/a:1023003616848. [DOI] [PubMed] [Google Scholar]
- 6.Chao L, Chao J, Yu JX. Prostasin is a novel human serine proteinase from seminal fluid. Purification, tissue distribution, and localization in prostate gland. J Biol Chem. 1995;270:13483–13489. [PubMed] [Google Scholar]
- 7.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 8.Planes C, Caughey GH. Regulation of the epithelial Na+ channel by peptidases. Curr Top Dev Biol. 2007;78:23–46. doi: 10.1016/S0070-2153(06)78002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong Z, Illek B, Bhagwandin VJ, Verghese GM, Caughey GH. Prostasin, a membrane-anchored serine peptidase, regulates sodium currents in JME/CF15 cells, a cystic fibrosis airway epithelial cell line. Am J Physiol Lung Cell Mol Physiol. 2004;287:L928–L935. doi: 10.1152/ajplung.00160.2004. [DOI] [PubMed] [Google Scholar]
- 10.Netzel-Arnett S, Currie BM, Szabo R, Lin CY, Chen LM, Chai KX, Antalis TM, Bugge TH, List K. Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J Biol Chem. 2006;281:32941–32945. doi: 10.1074/jbc.C600208200. [DOI] [PubMed] [Google Scholar]
- 11.List K, Hobson JP, Molinolo A, Bugge TH. Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J Cell Physiol. 2007;213:237–245. doi: 10.1002/jcp.21115. [DOI] [PubMed] [Google Scholar]
- 12.Chen LM, Zhang X, Chai KX. Regulation of prostasin expression and function in the prostate. Prostate. 2004;59:1–12. doi: 10.1002/pros.10346. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich R, Fuentes-Prior P, Ong E, Coombs G, Hunter M, Oehler R, Pierson D, Gonzalez R, Huber R, Bode W, et al. Catalytic domain structures of MT-SP1/matriptase, a matrix-degrading transmembrane serine proteinase. J Biol Chem. 2002;277:2160–2168. doi: 10.1074/jbc.M109830200. [DOI] [PubMed] [Google Scholar]
- 14.Somoza JR, Ho JD, Luong C, Ghate M, Sprengeler PA, Mortara K, Shrader WD, Sperandio D, Chan H, McGrath ME, et al. The structure of the extracellular region of human hepsin reveals a serine protease domain and a novel scavenger receptor cysteine-rich (SRCR) domain. Structure. 2003;11:1123–1131. doi: 10.1016/s0969-2126(03)00148-5. [DOI] [PubMed] [Google Scholar]
- 15.Kyrieleis OJ, Huber R, Ong E, Oehler R, Hunter M, Madison EL, Jacob U. Crystal structure of the catalytic domain of DESC1, a new member of the type II transmembrane serine proteinase family. FEBS J. 2007;274:2148–2160. doi: 10.1111/j.1742-4658.2007.05756.x. [DOI] [PubMed] [Google Scholar]
- 16.Chen LM, Skinner ML, Kauffman SW, Chao J, Chao L, Thaler CD, Chai KX. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J Biol Chem. 2001;276:21434–21442. doi: 10.1074/jbc.M011423200. [DOI] [PubMed] [Google Scholar]
- 17.Shipway A, Danahay H, Williams JA, Tully DC, Backes BJ, Harris JL. Biochemical characterization of prostasin, a channel activating protease. Biochem Biophys Res Commun. 2004;324:953–963. doi: 10.1016/j.bbrc.2004.09.123. [DOI] [PubMed] [Google Scholar]
- 18.Krem MM, Rose T, Di Cera E. Sequence determinants of function and evolution in serine proteases. Trends Cardiovasc Med. 2000;10:171–176. doi: 10.1016/s1050-1738(00)00068-2. [DOI] [PubMed] [Google Scholar]
- 19.Pineda AO, Chen ZW, Bah A, Garvey LC, Mathews FS, Di Cera E. Crystal structure of thrombin in a self-inhibited conformation. J Biol Chem. 2006;281:32922–32928. doi: 10.1074/jbc.M605530200. [DOI] [PubMed] [Google Scholar]
- 20.Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Natl Acad Sci USA. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohr KB, Selwood T, Marquardt U, Huber R, Schechter NM, Bode W, Than ME. X-ray structures of free and leupeptin-complexed human alphaI-tryptase mutants: indication for an alpha–>beta-tryptase transition. J Mol Biol. 2006;357:195–209. doi: 10.1016/j.jmb.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 22.Meloun B, Kluh I, Kostka V, Moravek L, Prusik Z, Vanecek J, Keil B, Sorm F. Covalent structure of bovine chymotrypsinogen A. Biochim Biophys Acta. 1966;130:543–546. doi: 10.1016/0304-4165(66)90258-3. [DOI] [PubMed] [Google Scholar]
- 23.Schechter I, Berger A. On the size of the active site in proteases. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 24.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLano WL. The Pymol Molecular Graphics System. San Carlos, CA, USA: 2002. http://www.pymol.org. DeLano Scientific LLC. [Google Scholar]
- 26.Kossiakoff AA, Chambers JL, Kay LM, Stroud RM. Structure of bovine trypsinogen at 1.9 A resolution. Biochemistry. 1977;16:654–664. doi: 10.1021/bi00623a016. [DOI] [PubMed] [Google Scholar]
- 27.Kraut J. Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 28.McLachlan AD. Gene duplications in the structural evolution of chymotrypsin. J Mol Biol. 1979;128:49–79. doi: 10.1016/0022-2836(79)90308-5. [DOI] [PubMed] [Google Scholar]
- 29.Sichler K, Hopfner KP, Kopetzki E, Huber R, Bode W, Brandstetter H. The influence of residue 190 in the S1 site of trypsin-like serine proteases on substrate selectivity is universally conserved. FEBS Lett. 2002;530:220–224. doi: 10.1016/s0014-5793(02)03495-6. [DOI] [PubMed] [Google Scholar]
- 30.Tang J, Yu CL, Williams SR, Springman E, Jeffery D, Sprengeler PA, Estevez A, Sampang J, Shrader W, Spencer J, et al. Expression, crystallization, and three-dimensional structure of the catalytic domain of human plasma kallikrein. J Biol Chem. 2005;280:41077–41089. doi: 10.1074/jbc.M506766200. [DOI] [PubMed] [Google Scholar]
- 31.Pereira PJ, Bergner A, Macedo-Ribeiro S, Huber R, Matschiner G, Fritz H, Sommerhoff CP, Bode W. Human beta-tryptase is a ring-like tetramer with active sites facing a central pore. Nature. 1998;392:306–311. doi: 10.1038/32703. [DOI] [PubMed] [Google Scholar]
- 32.Tranter R, Read JA, Jones R, Brady RL. Effector sites in the three-dimensional structure of mammalian sperm beta-acrosin. Structure. 2000;8:1179–1188. doi: 10.1016/s0969-2126(00)00523-2. [DOI] [PubMed] [Google Scholar]
- 33.Greer J. Comparative modeling methods: application to the family of the mammalian serine proteases. Proteins. 1990;7:317–334. doi: 10.1002/prot.340070404. [DOI] [PubMed] [Google Scholar]
- 34.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: a multiple structural alignment algorithm. Proteins. 2006;64:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- 35.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 36.Barton GJ. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 37.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombstructure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otsuki M, Tani S, Okabayashi Y, Fuji M, Nakamura T, Fujisawa T, Itoh H. Beneficial effects of the synthetic trypsin inhibitor camostate in cerulein-induced acute pancreatitis in rats. Dig Dis Sci. 1990;35:242–250. doi: 10.1007/BF01536770. [DOI] [PubMed] [Google Scholar]
- 39.Kubodera H, Sasaki C, Okumura C, Matsuzaki T. Binding Mechanisms of thrombin inhibitors and trypsin inhibitors revealed by X-ray crystallography. Anal Sci. 1991;7:857–860. [Google Scholar]
- 40.Kaufman Katz A, Glusker JP, Beebe SA, Bock CW. Calcium ion coordination: a comparison with that of beryllium, magnesium and zinc. J Am Chem Soc. 1996;118:5752–5763. [Google Scholar]
- 41.Debela M, Magdolen V, Grimminger V, Sommerhoff C, Messerschmidt A, Huber R, Friedrich R, Bode W, Goettig P. Crystal structures of human tissue kallikrein 4: activity modulation by a specific zinc binding site. J Mol Biol. 2006;362:1094–1107. doi: 10.1016/j.jmb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 43.Marquardt U, Zettl F, Huber R, Bode W, Sommerhoff C. The crystal structure of human alpha1-tryptase reveals a blocked substrate-binding region. J Mol Biol. 2002;321:491–502. doi: 10.1016/s0022-2836(02)00625-3. [DOI] [PubMed] [Google Scholar]
- 44.Narayana SV, Carson M, el-Kabbani O, Kilpatrick JM, Moore D, Chen X, Bugg CE, Volanakis JE, DeLucas LJ. Structure of human factor D. A complement system protein at 2.0 A resolution. J Mol Biol. 1994;235:695–708. doi: 10.1006/jmbi.1994.1021. [DOI] [PubMed] [Google Scholar]
- 45.Volanakis JE, Narayana SV. Complement factor D, a novel serine protease. Protein Sci. 1996;5:553–564. doi: 10.1002/pro.5560050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huber R, Kukla D, Bode W, Schwager P, Bartels K, Deisenhofer J, Steigemann W. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor II. Crystallographic refinement at 1.9 A resolution. J Mol Biol. 1974;89:73–101. doi: 10.1016/0022-2836(74)90163-6. [DOI] [PubMed] [Google Scholar]
- 47.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci USA. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tully DC, Vidal A, Chatterjee AK, Williams JA, Roberts MJ, Petrassi HM, Spraggon G, Bursulaya B, Pacoma R, Shipway A, et al. Discovery of inhibitors of the channel-activating protease prostasin (CAP1/PRSS8) utilizing structure-based design. Bioorg Med Chem Lett. 2008;18:5895–5899. doi: 10.1016/j.bmcl.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 49.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 50.Garty H, Lindemann B. Feedback inhibition of sodium uptake in K+-depolarized toad urinary bladders. Biochim Biophys Acta. 1984;771:89–98. doi: 10.1016/0005-2736(84)90114-7. [DOI] [PubMed] [Google Scholar]
- 51.Frindt G, Windhager EE. Ca2(+)-dependent inhibition of sodium transport in rabbit cortical collecting tubules. Am J Physiol. 1990;258:F568–F582. doi: 10.1152/ajprenal.1990.258.3.F568. [DOI] [PubMed] [Google Scholar]
- 52.Silver RB, Frindt G, Windhager EE, Palmer LG. Feedback regulation of Na channels in rat CCT. I. Effects of inhibition of Na pump. Am J Physiol. 1993;264:F557–F564. doi: 10.1152/ajprenal.1993.264.3.F557. [DOI] [PubMed] [Google Scholar]
- 53.Champigny G, Voilley N, Lingueglia E, Friend V, Barbry P, Lazdunski M. Regulation of expression of the lung amiloride-sensitive Na+ channel by steroid hormones. EMBO J. 1994;13:2177–2181. doi: 10.1002/j.1460-2075.1994.tb06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Page R, Grzechnik SK, Canaves JM, Spraggon G, Kreusch A, Kuhn P, Stevens RC, Lesley SA. Shotgun crystallization strategy for structural genomics: an optimized two-tiered crystallization screen against the Thermotoga maritima proteome. Acta Crystallogr Section D Biol Crystallogr. 2003;59:1028–1037. doi: 10.1107/s0907444903007790. [DOI] [PubMed] [Google Scholar]
- 55.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 56.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 57.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 59.Collaborative, Computing, Project, and Number 4. The CCP4 suite: programs for protein crystallography, Version 3.1. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 60.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 61.Read RJ. Improved fourier coefficients for maps using phases from partial strucutres with errors. Acta Crystallogr. 1986;A42:140–149. [Google Scholar]