

Abstract
Plasmodium vivax invasion of human erythrocytes requires that the ligand domain of the Duffy-binding protein (DBP) recognize its cognate erythrocyte receptor, making DBP a potential target for therapy. The recently determined crystal structure of the orthologous DBP ligand domain of the closely related simian malaria parasite Plasmodium knowlesi provides insight into the molecular basis for receptor recognition and raises important questions about the mechanism of immune evasion employed by the malaria parasite.
Plasmodium merozoite invasion is a multi-step process
Invasion of erythrocytes by malaria parasites is a multi-step process involving discrete parasite ligands and host cell receptors [1]. Duffy binding protein (DBP), the first such ligand identified in micronemes of invasive malaria merozoites [2], is absolutely vital for the invasion process of Plasmodium vivax [3]. Cysteine-rich region II of the DBP comprises the prototypical Duffy binding like (DBL) ligand domain [4, 5], which is also found in other erythrocyte binding proteins (EBA-175, BAEBL, JESEBL) and in cytoadherence proteins (PfEMP-1) [6]. Although the putative ligand domains of these paralogues have <30% sequence identity, these cysteine-rich regions share a core set of conserved residues (e.g., cysteines and aromatic amino acids) believed to be structurally and functionally important. DBL domains of both the human parasite P. vivax DBP and simian parasite P. knowlesi DBPα interact with the Duffy antigen receptor for chemokines (DARC) [7] on the erythrocyte surface, leading to formation of a tight junction necessary for invasion. The crystal structure of the P. knowlesi DBPα DBL domain recently reported by Singh et al provides exciting insights into the functional character of the P. vivax DBP [8].
Plasmodium knowlesi α DBL structure
The overall structure of the P. knowlesi DBPα DBL is similar to that of the F1 and F2 DBL domains of EBA-175 [9]. All twelve conserved cysteines of the P. knowlesi DBPα DBL domain are involved in intradomain disulfide bridges that delimit three DBL subdomains in the backbone, which forms a ‘boomerang-shaped unit’. The pattern of disulfide bonding is identical between the P. knowlesi DBPα DBL and the F1 and F2 DBLs of P. falciparum EBA-175, although the F2 has an additional disulfide bridge. Subdomains 1, 2, and 3 have two, one and three disulfide bonds, respectively, and are comprised of twelve alpha helices (Fig. 1). Residues 15-52 form a random-coil stretch that makes up subdomain 1. The region between subdomains 1 and 2 (residues 53-63) is disordered and missing from the crystal structure, but is predicted to form a flexible linker. The ‘β finger’ motifs that facilitate dimerization of the P. falciparum EBA-175 F1/F2 DBL [9] appear functionally present in subdomain 1, although their role is unclear, as P. knowlesi DBPα DBL is not known to dimerize. Subdomain 2 (residues 64-180) and subdomain 3 (residues 186-307) each contain six alpha helices and are attached by a short linker segment. Subdomain 3 forms a large loop stabilized by three disulfide bridges with alpha helix 8 atop alpha helices 7 and 9; however, the functional role of the subdomain 3 structure is unclear.
Figure 1. Subdomain structure of the P. knowlesi DBPα DBL domain is defined by disulfide bonding.
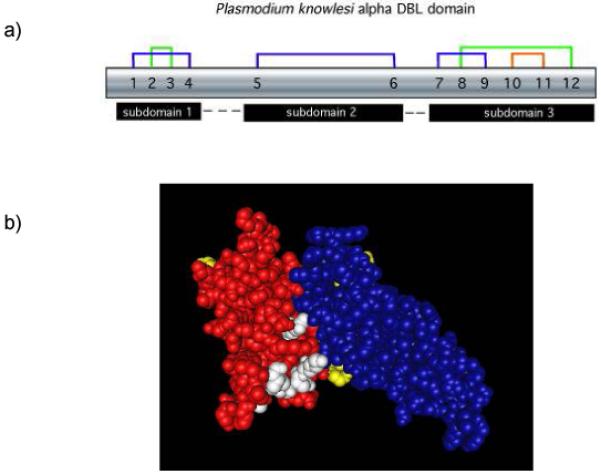
(A) Disulfide bonds, numbered sequentially 1-12, create three subdomains within the DBL ligand domain. (B) The subdomains highlighted by separate colors are connected by flexible linkers and weak hydrophobic forces maintain the subdomain configuration. In this model subdomain 1 is yellow, subdomain 2 is red, subdomain 3 is blue, and residues of the predicted binding pocket for receptor recognition are white.
Proposed DARC Recognition Site
The model proposed by Singh et al places the DARC binding site in a solvent accessible groove on a fairly flat surface atop subdomain 2. Based on previous mutational analysis [10-12], key residues for DARC recognition were identified as a cluster of nonpolar residues (Y94, L168, I175) grouped adjacent to basic residues (K96, K100, R103, K177) on the subdomain 2 surface to promote interaction with the sulfated Y41 of DARC, a critical element for receptor recognition identified in in vitro assays [13]. Major conformational changes to the P. knowlesi DBPα DBL structure are not predicted for DBL-DARC interaction, although this interaction is thought to bring the subdomain 3 loop into close contact with the host cell surface possibly to stabilize the ligand-receptor interaction or lead to a subsequent event in invasion. Unlike EBA175-GPA interaction, sugar side chains on the erythrocyte receptor have no apparent role in promoting the specificity of the DBP-DARC ligand-receptor interaction [9, 14].
Analysis of site-directed mutagenesis data suggests that additional residues, other than those identified above, are involved in the DARC binding site or have a role in receptor recognition [11, 12]. Mutations that completely abrogated P. vivax DBP binding to the DARC receptor map to multiple locations on the DBL structure outside of the proposed binding groove and a number of those residues cluster together on the outer surface of the DBL structure, including residues in unstructured exposed regions (e.g., PkDBPα DBL H59, S60). The dispersed pattern of these functionally important residues on the surface of the DBL suggests some involvement in recognition of the host receptor or in subsequent molecular changes or interactions that stabilize the ligand-receptor complex. Other mutated residues that exhibited loss of function are buried or on the surfaces of the DBL subdomains and their mutation may create significant structural changes.
Immune Evasion Mechanisms
Presentation of the DBP onto the merozoite surface must occur if the parasite is to invade an erythrocyte. Therefore, Plasmodium must be able to evade the host immune responses targeted against the functionally important parts of the DBP in order for this ligand to effectively recognize the erythrocyte receptor and for invasion to proceed. Various mechanisms of immune evasion have been described for other non-homologous ligands that have similar roles in other microbial pathogens. One of the best characterized examples is the influenza molecule hemagglutinin, which has a high degree of polymorphism surrounding its binding domain [15], suggesting that variants are selected by immune surveillance. In such a mechanism of immune escape polymorphic residues near the binding site elude binding of inhibitory antibody thus protecting the critical functional site on the ligand domain. Previously, this type of mechanism similar to influenza hemagglutinin was proposed for the P. vivax DBP [16-18] and recent structural data suggests that polymorphisms of another Plasmodium microneme ligand, apical membrane antigen 1 (AMA1), also serve to evade immune antibody inhibition [19] .
Implications of the structure of DBPα DBL for Immune Evasion
Singh et al. propose an alternative immune evasion model, termed ‘just in time’ release, based on their analysis that the most highly polymorphic residues cluster on the opposite side of the DBL from the predicted critical DARC binding site of subdomain 2. Invasive merozoites are believed to sequester microneme proteins until merozoites contact the target erythrocyte, presumably as a mechanism to reduce exposure of the DBP to immune inhibition [2]. Singh et al. propose that DBP release from the micronemes is followed by very rapid interaction with DARC and that this is the primary mechanism of evading immune inhibition. Residues critical for receptor recognition are protected from immune inhibition by swift binding to the cognate receptor, so that the residues under the strongest immune selective pressure are those that remain exposed on the protein surface opposite the binding domain. Supporting evidence depicted selected polymorphic residues identified from P. vivax field isolates mapped onto the P. knowlesi DBPα DBL structure localized to knobby protrusions on the face of the molecule opposite from the proposed DARC recognition site. However, P. vivax DBL polymorphisms [16-18, 20-22] are more extensive than those depicted in the Singh et al. paper (Fig. 2), and are widely dispersed over the DBL domain. Of particular interest are polymorphisms in the P. vivax DBL corresponding to residue K171 and W191 of the P. knowlesi DBPα DBL (Fig. 2). Variant residues at these locations in the P. vivax DBP DBL domain appear to represent part of a linked haplotype common among P. vivax DBP alleles, which was shown to collectively alter antigenic character and significantly change sensitivity to inhibitory antibodies [23]. Some of the other scattered polymorphisms may be random neutral mutations, but it is likely that DARC has a significantly larger binding footprint than that identified so far. Therefore, some polymorphisms distal to the proposed binding site may represent residues under weaker immune selective pressure or possibly alter affinity to DARC at other residues of the minimal 35 amino acid receptor in a way to enhance invasion efficiency. The position of polymorphisms relative to the complete DARC binding site will affect their potential contribution to antigenic character, sensitivity to inhibitory antibodies, and affinity of the DBL domain for the DARC receptor. Additional data are needed, such as co-crystallization of DBP-DARC, to resolve the importance of polymorphisms in the DBL domain to help determine the role of these two models in immune evasion and the importance of allelic diversity for anti-DBP immune therapies.
Figure 2. Three-dimensional structure of the P. knowlesi DBPα DBL.

The deduced 3D structure of the DBL ligand domain is depicted as a space-filling model (left side) and the ‘worm’ model (right side) rotated in each representation by 90°. The space filling models show the dimorphic residues as green, polymorphic residues as yellow, residues of the predicted binding pocket for the receptor recognition as white, and residues known to change antigenic character and sensitivity to antibody inhibition in red (K171, W191). Dimorphic and polymorphic residues were determined as those that were dimorphic or polymorphic from Sal I DBP in at least two other strains [16-18, 20-22]. The worm model depicts the amino acid backbone and highlights the alpha helices of the second and third domains. Subdomain 1 is yellow, subdomain 2 is red and subdomain 3 is blue.
Concluding remarks
Recognition of the tertiary structure of P. knowlesi DBPα DBL is an important advance in the field of malaria research. Although DBL domains are found across the Plasmodium genus and have important implications for erythrocyte invasion and cytoadherence, their role in P. vivax merozoite invasion is especially important. Knowledge of the structure of the P. knowlesi DBPα DBL domain, along with the P. falciparum EBA-175 F1/F2 DBL domain structure, will greatly enhance research towards effectively blocking merozoite invasion of erythrocytes. Co-crystallization of the DBL domain with its receptor will certainly lend clarity to the process of erythrocyte invasion as well as the mechanism of immune evasion employed by the P. vivax merozoite.
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health R01 AI33656 to JHA and University of Notre Dame Schmitt Fellowship to AMM.
References
- 1.Chitnis CE, Blackman MJ. Host cell invasion by malaria parasites. Parasitol Today. 2000;16:411–415. doi: 10.1016/s0169-4758(00)01756-7. [DOI] [PubMed] [Google Scholar]
- 2.Adams JH, et al. The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell. 1990;63:141–153. doi: 10.1016/0092-8674(90)90295-p. [DOI] [PubMed] [Google Scholar]
- 3.Miller LH, et al. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med. 1976;295:302–304. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 4.Adams JH, et al. A family of erythrocyte binding proteins of malaria parasites. Proceedings of the National Academy of Sciences USA. 1992;89:7085–7089. doi: 10.1073/pnas.89.15.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson DS, et al. Isolation of multiple sequences from the Plasmodium falciparum genome that encode conserved domains homologous to those in erythrocyte-binding proteins. Proc Natl Acad Sci U S A. 1995;92:7100–7104. doi: 10.1073/pnas.92.15.7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michon P, et al. Evolutionary relationships of conserved cysteine-rich motifs in adhesive molecules of malaria parasites. Mol Biol Evol. 2002;19:1128–1142. doi: 10.1093/oxfordjournals.molbev.a004171. [DOI] [PubMed] [Google Scholar]
- 7.Chitnis CE, Miller LH. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J Exp Med. 1994;180:497–506. doi: 10.1084/jem.180.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SK, et al. Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature. 2006;439:741–744. doi: 10.1038/nature04443. [DOI] [PubMed] [Google Scholar]
- 9.Tolia NH, et al. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005;122:183–193. doi: 10.1016/j.cell.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 10.Singh SK, et al. Definition of structural elements in Plasmodium vivax and P. knowlesi Duffy-binding domains necessary for erythrocyte invasion. Biochem J. 2003;374:193–198. doi: 10.1042/BJ20030622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hans D, et al. Mapping binding residues in the Plasmodium vivax domain that binds Duffy antigen during red cell invasion. Mol Microbiol. 2005;55:1423–1434. doi: 10.1111/j.1365-2958.2005.04484.x. [DOI] [PubMed] [Google Scholar]
- 12.Vanbuskirk KM, et al. Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc Natl Acad Sci U S A. 2004;101:15754–15759. doi: 10.1073/pnas.0405421101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choe H, et al. Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC) Mol Microbiol. 2005;55:1413–1422. doi: 10.1111/j.1365-2958.2004.04478.x. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay D, et al. The structure of the Plasmodium falciparum EBA175 ligand domain and the molecular basis of host specificity. Trends Parasitol. 2006;22:143–145. doi: 10.1016/j.pt.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- 16.Baum J, et al. Evidence for Diversifying Selection on Erythrocyte-Binding Antigens of Plasmodium falciparum and P. vivax. Genetics. 2003;163:1327–1336. doi: 10.1093/genetics/163.4.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole-Tobian J, King CL. Diversity and natural selection in Plasmodium vivax Duffy binding protein gene. Mol Biochem Parasitol. 2003;127:121–132. doi: 10.1016/s0166-6851(02)00327-4. [DOI] [PubMed] [Google Scholar]
- 18.Tsuboi T, et al. Natural variation within the principal adhesion domain of the Plasmodium vivax duffy binding protein. Infect Immun. 1994;62:5581–5586. doi: 10.1128/iai.62.12.5581-5586.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai T, et al. Structure of AMA1 from Plasmodium falciparum reveals a clustering of polymorphisms that surround a conserved hydrophobic pocket. Proc Natl Acad Sci U S A. 2005;102:12736–12741. doi: 10.1073/pnas.0501808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ampudia E, et al. Genetic polymorphism of the Duffy receptor binding domain of Plasmodium vivax in Colombian wild isolates. Mol. Biochem. Parasitol. 1996;78:269–272. doi: 10.1016/s0166-6851(96)02611-4. [DOI] [PubMed] [Google Scholar]
- 21.Xainli J, et al. The erythrocyte binding motif of Plasmodium vivax Duffy binding protein is highly polymorphic and functionally conserved in isolates from Papua New Guinea. Mol Biochem Parasitol. 2000;111:253–260. doi: 10.1016/s0166-6851(00)00315-7. [DOI] [PubMed] [Google Scholar]
- 22.Kho WG, et al. Analysis of polymorphic regions of Plasmodium vivax Duffy binding protein of Korean isolates. Korean J Parasitol. 2001;39:143–150. doi: 10.3347/kjp.2001.39.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanbuskirk KM, et al. Antigenic Drift in the Ligand Domain of Plasmodium vivax Duffy Binding Protein Confers Resistance to Inhibitory Antibodies. J Infect Dis. 2004;190:1556–1562. doi: 10.1086/424852. [DOI] [PubMed] [Google Scholar]