

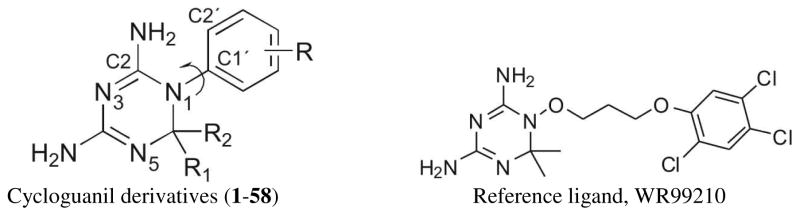
Abstract
Drug resistance acquired by Plasmodium falciparum (Pf) is a major problem in the treatment and control of malaria. One of the major examples of drug resistance is that caused by mutations in the active site of dihydrofolate reductase (DHFR) of Pf (PfDHFR-TS). A double mutation, A16V+S108T, is specific for resistance to the marketed drug cycloguanil. In this study, we used 58 cycloguanil (2,4-diamino-1,6-dihydro-1,3,5-triazine) derivatives to explore the relationship between various physico-chemical properties and reported binding affinity data on wild type and A16V+S108T mutant type. Using the Hansch 2D-QSAR method, we obtained a parabolic relationship of hydrophobicity of substituents at the N1-phenyl ring with the wild type binding affinity data. Hydrophobicity being a key property for wild type binding affinity data, we found steric factors to be crucial for A16V+S108T mutant resistance. We investigated FlexX, GOLD, Glide and Molegro virtual docking programs and 13 different scoring functions on 10 of the cycloguanil derivatives to evaluate which program was best for reproducing the experimental binding mode and correlating the docking scores with the reported binding affinity data. We identified GOLD using its GoldScore fitness function as the most accurate docking program for predicting binding affinity data of cycloguanil derivatives to DHFR and Molegro virtual docker with its template docking algorithm and MolDock [GRID] scoring function as most accurate for reproducing the experimental binding mode of a reference ligand that is structurally similar to the cycloguanil derivatives studied. We also report an interaction index which best describes the structure-activity relationships exhibited by these analogs in terms of PfDHFR-TS active site interactions.
Introduction
Classified as one of the leading causes of death in the world, malaria is a major threat for public health, especially in the tropical and sub-tropical countries. Malaria has been declared endemic particularly in the sub-Sahara African countries, Asia, Oceania and Latin America.1 Malaria is caused by Plasmodium parasites, of which the most lethal is Plasmodium falciparum (Pf). The biology of this parasite is well understood with the help of genomics2 and proteomics projects3 and has aided identification of new molecular targets that could be used to aid rational design of drugs or vaccines. Drug resistance acquired by the malarial parasites is a major problem in the treatment and control of malaria. Dihydrofolate reductase (DHFR) in Pf (PfDHFR-TS; TS refers to thymidylate synthase, bound to DHFR in Pf), which catalyzes the reduction of dihydrofolate to tetrahydrofolate, is one of the most widely studied enzymes in antimalarial drug design due to its potential role in DNA synthesis.4, 5 DHFR is also considered to be a good target for other protozoal diseases like leishmaniasis, trypanosomiasis and Chagas’ diseases.6 Resistance of the parasite to antifolates has been observed and is mainly caused by mutations around the enzyme active site.7–9 Some of the resistance-causing mutations include single S108N, double C59R+S108N, triple N51I+C59R+S108N, C59R+S108N+I164L and quadruple N51I+C59R+S108N+I164L. A double mutation, A16V+S108T, is specific for resistance to the marketed drug cycloguanil. Over the past few years, several computational techniques have been applied to better understand PfDHFR-TS-ligand interactions.10 Currently there are four crystal structures of protozoal DHFR available in the Protein Data Bank, including the wild type PfDHFR-TS complexed with triazine inhibitor WR99210 (1j3i),11 a double mutant PfDHFR-TS complexed with pyrimethamine (1j3j),11 a quadruple mutant PfDHFR-TS complexed with WR99210 (1j3k)11 and wild type Plasmodium vivax DHFR-TS complexed with pyrimethamine (2bl9).12 Before the release of PfDHFR-TS crystal structures, researchers developed homology models to study the resistance mechanism of commonly used antifolate drugs.13 Recently, Fogel et al. performed molecular modeling studies including docking studies with the GOLD program on pyrimethamine derivatives into the 1j3k DHFR structure, evaluated several scoring functions, including those from GOLD, Molegro virtual docker, Discovery Studio and MOE and found Molegro virtual docker’s Protein-Ligand interaction score to show good correlation with the reported binding affinity data.14 The same authors performed docking studies of pyrimethamine derivatives into both wild type and mutant enzymes.15 QSAR studies using neural networks on this class of compounds have also been performed by the same authors.16 4D-QSAR studies for some of the antifolates have been published.17 A 3D pharmacophore model has been prepared using Catalyst software on a diverse set of PfDHFR-TS inhibitors, including cycloguanil and pyrimethamine derivatives.18 The Autodock program has also been shown to predict the correct binding modes of folic acid-competitive DHFR antagonists identified by Plouffe et al. through their cell-based high-throughput screening.19
In summary, previous modeling studies focused on studying the interactions of various small molecule inhibitors of DHFR with the active site residues of wild and mutant types using different arbitrarily chosen docking programs and QSAR studies. However there is need for new research on the importance of physico-chemical properties such as the linear and/or bilinear relationship of hydrophobicity with binding affinity data and on systematic attempts to find a good docking program/scoring function to study molecular interactions accurately for this highly interesting and therapeutically important class of compounds. Hence our report has two main objectives: (1) to gain new insights into the structure activity relationship (SAR) among the cycloguanil derivatives binding to wild and mutant type PfDHFR-TS and (2) to find an optimal docking program/scoring function combination to study the binding interactions between these compounds and wild type PfDHFR-TS that are not revealed by the SAR studies. For objective 1, we made use of QSAR to study a series of 58 cycloguanil (2,4-diamino-1,6-dihydro-1,3,5-triazine) derivatives20–22 which all originated from the same laboratory, since well-defined binding constants against wild and mutant types of PfDHFRTS have been reported for them. We chose to use traditional Hansch 2D-QSAR analysis for this series of compounds for two main reasons. First, 48 of the 58 compounds had an undefined stereogenic center at C6 of the triazine ring of cycloguanil, which rendered the data unsuitable for 3D-QSAR studies. Secondly, Hansch 2D-QSAR is well known for its simplicity and ease of mechanistic interpretation. For the docking studies, we used the only 10 compounds with well-defined stereochemistry to study interactions with the wild type PfDHFR-TS that could not be delineated from our 2D-QSAR studies. For objective 2, we evaluated FlexX, GOLD, Glide and Molegro virtual docking programs. We then investigated in detail the interactions of the analogs with the active site of PfDHFR-TS, using what we found to be the most suitable docking program for this class of protein-ligand interactions. The two objectives of this work fit together well since the methods of 2D-QSAR and docking give complementary information on the structure-activity relationship.
Materials and Method
1. Hansch 2D-QSAR
2D-QSAR models were constructed using the data set of 58 compounds.20–22 The compounds used for the present analysis along with their observed affinities for wild and mutant type PfDHFR-TS are listed in Table 1. We used two types of standard descriptors: indicator variables and physico-chemical constants. A complete list of all the descriptors used in 2D model generation is given in Table 2. An indicator variable designated as I with a relevant subscript was set to 1 if a particular substituent is present and to 0 if absent. R1 and R2 are both equivalently attached at C6, so to make sure the indicator variables were well defined, we ordered the R1/R2 substituents so that for any analog the less bulky substituent is at R1. Physico-chemical constants—hydrophobic (π); electronic (σ), including resonance effect (R) or field/inductive effect (f); molar refractivity (MR); and hydrogen bond donor (HDR) or acceptor (HAR)—were taken from the literature.23, 24 The substituent hydrophobicity constant (π) which we used from the literature is in fact measured experimentally. The hydrophobicity constant (πX) for any substituent X is calculated according to the equation, πX = logPX − logPH, where PX and PH are the partition coefficients of the reference compound with and without substituent, respectively. The Hammett electronic parameter (σ) is calculated based on the influence of substituent X on the ionization of benzoic acid. The molar refractivity (MR), defined in the Lorenz-Lorentz equation as , where n is the refractive index, MW is the molecular weight and d is the density, is considered to be a measure of the volume occupied by an atom or group of atoms according to Hansch and co-workers, since usually there is only very slight variation in the refractive index between a set of analogs.25 A correlation matrix was used to correlate the biological activity with physico-chemical and structural predictor variables. Descriptors with inter-correlation |r| > 0.6 were not included in the same QSAR model. Predictor variables with p > 0.05 were eliminated whilst deriving the QSAR models, in order to assure statistical reliability. Linear regression analysis was performed using Systat Version 11. The Student’s t-distribution was used to assess the significance of individual regression terms. Forward stepping regression was used to build the QSAR models. This method initially generates a QSAR model containing only one variable, which is chosen to be the one with the highest t-statistic, and subsequent variables are added based on their relative importance, also as determined by t-statistics. The QSAR models were evaluated using standard statistical parameters including the correlation coefficient (r), coefficient of determination (r2), standard error of estimate (s) and Fisher F-value. The figures within the parentheses following the coefficient terms are the standard error of regression terms and constants. A data point was considered as an outlier if its residual value exceeded twice the standard error of estimate of the model. Self-consistency of the derived models was verified using the leave-one-out (loo) process, and the predictability of each model was assessed using cross-validated r2, called q2. The best measure of reliability of a QSAR model is a high q2, not just a high r2 which could be a result of over-fitting to data. In general, a value of q2 > 0.5 is considered acceptable.26, 27
Table 1.
Compoundsa and their observed binding affinities.
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R | Kiwild (nM) | Kimut (nM) |
| 1 | H | Me | H | 113 | 456 |
| 2 | H | Me | 4-Cl | 4.1 | 127 |
| 3 | H | Me | 4-Me | 23.4 | 186 |
| 4 | H | Me | 3,4-Cl2 | 1.4 | 17.8 |
| 5 | Me | nC6H13 | H | 1.5 | 180 |
| 6 | Me | nC6H13 | 4-Cl | 0.7 | 564 |
| 7 | Me | nC6H13 | 4-Br | 3 | 2186 |
| 8 | Me | nC6H13 | 4-Et | 1.1 | 552 |
| 9 | Me | nC6H13 | 3,4-Cl2 | 1.4 | 107 |
| 10 | H | nC6H13 | 4-Me | 0.8 | 823 |
| 11 | H | Ph | H | 49.1 | 61.8 |
| 12 | H | Me | 4-Et | 11 | 322 |
| 13 | H | Ph | 4-Br | 2.9 | 90.3 |
| 14 | H | Ph | 4-Me | 7.7 | 170 |
| 15 | H | Ph | 4-Et | 12.1 | 86.6 |
| 16 | H | Ph | 3,4-Cl2 | 1.6 | 11 |
| 17b | Me | Me | 4-Cl | 1.5 | 1314 |
| 18b | H | H | 4-Cl | 24.4 | 646 |
| 19 | H | Et | 4-Cl | 3.6 | 189 |
| 20 | H | nPr | 4-Cl | 4.6 | 107 |
| 21 | H | nBu | 4-Cl | 3.7 | 167 |
| 22 | H | tBu | 4-Cl | 3838 | 82721 |
| 23b | Me | Me | 4-Br | 1.1 | 1947 |
| 24 | H | nPr | 4-Br | 2.6 | 127 |
| 25 | H | iPr | 4-Br | 30 | 1195 |
| 26 | H | Et | 4-Me | 5.9 | 128 |
| 27 | H | nPr | 4-Me | 13.7 | 188 |
| 28 | H | iPr | 4-Me | 167 | 1460 |
| 29b | Me | Me | H | 20 | 1375 |
| 30b | Me | Me | 4-F | 4.6 | 1633 |
| 31b | H | H | H | 329 | 585 |
| 32b | Me | Me | 3-Cl | 3.7 | 340 |
| 33b | Me | Me | 3,4-Cl2 | 1.1 | 130.7 |
| 34 | H | Me | 3-Cl | 10.2 | 38.7 |
| 35 | H | Me | 4-Br | 5.7 | 202 |
| 36 | H | iPr | 4-Cl | 60.5 | 1538 |
| 37 | H | Et | 4-Br | 2.7 | 99 |
| 38b | Me | Me | 4-Me | 1.8 | 1584 |
| 39b | H | H | 4-F | 270 | 469 |
| 40 | Me | iPr | 3-Cl | 25.5 | 2177 |
| 41 | H | Ph | 4-Cl | 4.5 | 49.3 |
| 42 | H | Ph | 3-Cl | 11.7 | 10.5 |
| 43 | Me | nPr | 4-Cl | 3.5 | 9229 |
| 44 | Me | iPr | 4-Cl | 36.5 | 44791 |
| 45 | Me | nPr | 3-Cl | 4.6 | 471 |
| 46 | Me | nC6H13 | 3-Cl | 2.4 | 101 |
| 47 | H | 4-C6H5OC6H4 | 4-Cl | 0.4 | 3.8 |
| 48 | H | 4-C6H5OC6H4 | 3-Cl | 0.7 | 2.7 |
| 49 | H | 3-C6H5OC6H4 | 3-Cl | 1.1 | 2.5 |
| 50 | H | 3-C6H5CH2OC6H4 | 4-Cl | 0.7 | 6.2 |
| 51 | H | 3-C6H5CH2OC6H4 | 3-Cl | 2.3 | 3.2 |
| 52 | H | 3-(4-ClC6H4O)C6H4 | 3-Cl | 1.3 | 3.5 |
| 53 | H | nC7H15 | 3-Cl | 2.7 | 3.8 |
| 54 | H | 4-PrOC6H4 | 3-Cl | 3.3 | 9.3 |
| 55 | H | 3-[2,4,5-Cl3C6H2O(CH2)3O)C6H4 | 3-Cl | 4.0 | 2.6 |
| 56 | H | 3-C6H5OC6H4 | 4-Cl | 0.5 | 2.7 |
| 57 | H | 3-(4-ClC6H4O)C6H4 | 4-Cl | 1.4 | 7.5 |
| 58 | H | 3-(3,5-Cl2C6H3O)C6H4 | 3-Cl | 1.8 | 4.7 |
Table 2.
Descriptors used in the 2D-QSAR analysis.
| No. | Descriptor | Family | Description |
|---|---|---|---|
| 1 | I1 | Structural | Presence of hydrogen atom at R1 |
| 2 | I2 | Structural | Presence of hydrogen atom at R2 |
| 3 | I3 | Structural | Presence of methyl group at R1 |
| 4 | I4 | Structural | Presence of iso-propyl group at R2 |
| 5 | I5 | Structural | Presence of 3-phenoxy phenyl group at R2 |
| 6 | I6 | Structural | Presence of 3-benzyloxy phenyl group at R2 |
| 7 | I7 | Structural | Presence of 4-phenoxy phenyl group at R2 |
| 8 | I8 | Structural | Presence of 3-(4-Chlorophenoxy)phenyl group at R2 |
| 9 | I9 | Structural | Presence of methyl group at R2 |
| 10 | I10 | Structural | Presence of n-hexyl group at R2 |
| 11 | I11 | Structural | Presence of phenyl group at R2 |
| 12 | I12 | Structural | Presence of ethyl group at R2 |
| 13 | I13 | Structural | Presence of n-propyl group at R2 |
| 14 | πpara(R) | Hydrophobic | Hydrophobicity of para-substituents around R |
| 15 | ΣπR | Hydrophobic | Hydrophobicity of para- and meta-substituents around R |
| 16 | MRp | Steric | Molar refractivity of para-substituents around R |
| 17 | ΣMR | Steric | Molar refractivity of para- and meta-substituents around R |
| 18 | f | Electronic | Field (inductive) effect of para- and meta-substituents around R |
| 19 | R | Electronic | Resonance effect of para- and meta-substituents around R |
| 20 | σp | Electronic | Hammett constant for para-substituents around R |
| 21 | Σσ | Electronic | Hammett constant for para- and meta-substituents around R |
| 22 | Espara | Steric | Taft’s steric constant for para-substituents around R |
| 23 | Lp | Steric | Sterimol parameter, length of para-substituents around R |
| 24 | B1para(R) | Steric | Sterimol parameter, minimal width of para-substituents around R |
| 25 | B5para | Steric | Sterimol parameter, newly defined maximal width of parasubstituents around R |
2. Docking
For docking studies, we used 10 of the 58 compounds (indicated in Table 1) to study the interaction of these compounds with the active site residues of PfDHFR-TS, information not uncovered from our QSAR studies. The 10 were the only compounds with well defined stereochemistry at C6 of the triazine ring among the 58 cycloguanil derivatives used in the QSAR studies. For all the docking studies we used wild type PfDHFR-TS complexed with WR99210 (1j3i).11 The structure of this reference ligand is shown in Table 1. The whole tetrameric protein was used. All water molecules and the co-crystallized ligand, WR99210, were removed before docking. All the docking analyses were confined to chain A of 1j3i. Hydrogen atoms were added to the protein for each type of docking. The protein was optimized to remove any bad contacts only for Glide docking (it is a part of the standard protein preparation in the Glide program). The 10 ligands were each constructed starting from an overlay with the bioactive conformation of WR99210 and then minimized using MMFF94 in Sybyl 7.2. We used two types of measures to evaluate the accuracy of the docking programs. First, we considered the ability of the docking program to reproduce the experimental binding mode of WR99210. For this we used the minimized structure of WR99210 and docked it along with the 10 other ligands during each docking run and then compared the RMSD between the predicted binding mode and the experimental structure of WR99210. We used an all-atom-in-place superposition using Schrödinger 8.5 suite of programs to calculate the RMSD. The second measure we used was R2 for correlation between the docking scores and the experimental binding affinity data.
2.1 FlexX
FlexX Release 2.0 implemented in Sybyl 7.228 was used as one of the four docking programs investigated. We prepared an active site comprising of all complete amino acids containing any atom within 6.5 Å of any ligand atom of WR99210. We considered four scoring functions, DScore,29 GScore,30, 31 CScore,32 PMF Score33 and the composite FlexX Total scores. 30 poses for each ligand were generated and all were considered for the RMSD calculation, whereas the scores from only the top-most pose were used to correlate with the reported binding affinity.
2.2 Glide
We used the Glide program from Schrödinger 7.5 software, which is well known for its speed and accuracy.34–36 We used the standard precision (SP) module of Glide. The Glide GScore and EModel which are based on the ChemScore function of Eldridge et al.32 were considered. The protein was prepared using the protein preparation and refinement tool in Glide. For the active site, a grid box centered at the ligand, WR99210, was used, set to accommodate a maximum ligand length of 15 Å. We docked the ligands flexibly, allowing for the flip of 5- and 6-membered rings, writing out a maximum of 30 poses per ligand and also enabling the post-docking minimization of the ligands. We also evaluated the performance of Ruvinsky’s correction to GScore and EModel.37, 38 This uses a number of docked poses within each energy well and calculates a refined docking score. For Ruvinsky’s correction treatment, we generated 100 poses per ligand using the SP mode of Glide. These poses were then clustered and the entropy was calculated for each cluster. This was accomplished using the pose_entropy script (revision 1.8) available within the Schrödinger suite of programs.
2.3 Molegro virtual docker
We used Molegro virtual docker, which has been recently introduced and gained attention among medicinal chemists.39, 40 Due to the availability of the bioactive conformation of a structurally-related compound, WR99210, we used the template docking available in Molegro virtual docker and evaluated MolDock, rerank and protein-ligand interaction scores from MolDock and MolDock [GRID] options. Template docking is based on extracting the chemical properties like the pharmacophore elements of a ligand bound in the active site and using that information for docking structurally similar analogs. We used W99210 from 1j3i as the template. We used the default settings, including a grid resolution of 0.30 Å for grid generation and a 15 Å radius from the template as the binding site. We used the MolDock optimizer as a search algorithm, and the number of runs was set to 10. A population size of 50, maximum iteration of 2000, scaling factor of 0.50, crossover rate of 0.90 and a variation-based termination scheme for parameter settings were used. The maximum number of poses to generate was increased to 10 from a default value of 5. Since the Molegro virtual docker works by an evolutionary algorithm, consecutive docking runs do not give exactly the same fitness scores. To address this issue of inherent randomness, we used five consecutive runs and then used the top ranked score from each run to calculate the average score for each ligand to correlate with the experimental binding
2.4 GOLD
We used the GOLD 4.0 docking program.41 The active site was defined as any atom that lies within a 15 Å radius of the ligand, WR99210. Since GOLD, like Molegro, uses a genetic algorithm to dock ligands, we performed five consecutive docking runs to obtain an average docking score for each ligand. Default GA settings that ensure 100% search efficiency were used for the genetic algorithm (GA). An early termination of the number of GA runs was allowed when the RMSDs of the top three GA solutions were within 0.5 Å. We evaluated GoldScore, ChemScore and Astex Statistical Potential (ASP) scoring functions.42 ASP is an atom-atom potential that is based on a database of protein-ligand complexes. Its accuracy is comparable to GoldScore and ChemScore.42
Results and discussion
1. Hansch 2D-QSAR
Hansch 2D-QSAR models are listed in Table 3, and the corresponding statistical parameters of the regression equations are given in Table 4. Models 1–2a were developed by considering the observed binding affinity data on the wild type enzyme as a dependent variable (pKi(wt)) and the derived physico-chemical and indicator variables as independent variables. Model 1 explains 80.2% variance in observed binding affinity data on wild type. We found 22 to be a serious outlier when deriving Model 1. Model 1a, prepared without 22, had significantly improved statistical parameters such as much higher r2 compared to Model 1, supporting that 22 is an outlier. Model 1a explains 90.4% variance in the observed binding affinity. The coefficients for the descriptors are nearly the same for Model 1a as those for Model 1, so we analyze in detail Model 1a. None of the descriptors used in Model 1a are auto-correlated with each other. Model 1a gave the order of importance of the variables as I4 > I7 > I2 > I5 > Σ;πR > I6 > I8 > I3. It is quite obvious that other than hydrophobicity, which is a very common feature for compounds acting on DHFR, certain structural features of these analogs are crucial for PfDHFR-TS inhibition. Published results show that substituents at the C6 position interact with Ala16 in the wild type enzyme.9 For example, one of the C6-methyl groups of cycloguanil showed hydrophobic interactions with this residue.9 The negative regression coefficient of the indicator variable I2 in Model 1a indicates that a hydrogen atom at both C6 positions on the triazine ring eliminates the attractive hydrophobic interactions which other substituents can make with the adjacent alanine side chain. The negative contribution of I4, showing the untoward effect of the isopropyl group at the R2 position, is probably due to a steric clash of this bulky hydrophobic group with the alanine side chain. On the other hand, the positive values of the regression coefficients for other descriptors such as I3 (Me at R1) or I5, I6, I7 and I8 [for 3-phenoxyphenyl, 3-benzyloxyphenyl, 4-phenoxyphenyl and 3-(4-chlorophenoxy)phenyl at R2, respectively] indicate that some bulky substituents increase the binding affinity of the ligands towards the wild type. This in part may be because the compounds in the data set with bulky aryloxy R2 substituents have only a hydrogen atom at the R1 position and have either 3- or 4-chloro substitution at the N1-aryl ring (R position) (note that cycloguanil possesses a 4-chloro group at this position).
Table 3.
Regression coefficients and intercept (with standard errors in parentheses) of Hansch 2D-QSAR models.
| Model | I1 | I2 | I3 | I4 | I5 | I6 | I7 | I8 | ΣπR | (ΣπR)2 | πPara(R) | B1Para(R) | Intercept |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | – | −0.868 (0.321) | 0.563 (0.151) | −1.022 (0.237) | 1.023 (0.368) | 0.788 (0.368) | 1.168 (0.368) | 0.763 (0.368) | 0.828 (0.219) | – | – | – | 7.520 (0.187) |
| 1aa | – | −0.976 (0.203) | 0.466 (0.096) | −1.087 (0.150) | 0.919 (0.233) | 0.684 (0.233) | 1.064 (0.233) | 0.659 (0.233) | 0.816 (0.139) | – | – | – | 7.631 (0.119) |
| 2 | – | −0.814 (0.327) | 0.582 (0.153) | −1.061 (0.242) | 0.989 (0.370) | 0.754 (0.370) | 1.134 (0.370) | 0.729 (0.370) | 1.303 (0.573) | −0.349 (0.389) | – | – | 7.392 (0.236) |
| 2ab | – | −0.797 (0.180) | 0.447 (0.084) | −1.139 (0.131) | 0.854 (0.201) | 0.619 (0.201) | 0.999 (0.201) | 0.594 (0.201) | 1.945 (0.333) | −0.721 (0.219) | – | – | 7.259 (0.138) |
| 3 | 0.962 (0.208) | – | – | −1.127 (0.336) | 1.400 (0.519) | 1.165 (0.519) | 1.310 (0.519) | – | – | – | – | −0.735 (0.253) | 7.252 (0.409) |
| 3aa | 1.042 (0.174) | – | – | −1.198 (0.280) | 1.321 (0.432) | 1.086 (0.432) | 1.231 (0.432) | – | – | – | – | −0.625 (0.212) | 7.097 (0.342) |
Model obtained without compound 22;
Model obtained without compounds 5 and 22
Table 4.
Nonvalidated and validated statistical parameters for Hansch 2D-QSAR models.
| Model | n | r2 | F | s | q2 |
|---|---|---|---|---|---|
| 1 | 58 | 0.802 | 11.04 | 0.502 | 0.572 |
| 1a | 57 | 0.904 | 26.68 | 0.317 | 0.743 |
| 2 | 58 | 0.806 | 9.86 | 0.503 | 0.649 |
| 2a | 56 | 0.932 | 33.92 | 0.272 | 0.869 |
| 3 | 58 | 0.778 | 13.03 | 0.711 | 0.541 |
| 3a | 57 | 0.834 | 18.98 | 0.592 | 0.640 |
The positive coefficient of the ΣπR term of Model 1a shows that hydrophobic substituents at R increase the binding affinity of the ligands to the wild type. The importance of the hydrophobicity of these anti-folates is well documented in the literature and has previously been studied using Hansch 2D-QSAR.43–45 We then tested for any non-linear relationship between hydrophobicity and the binding affinity data on the wild type. Model 2, developed using the square of ΣπR, shows that adding in this new descriptor is not statistically acceptable, since the standard error of the regression term for this descriptor is more than the regression coefficient, making a low t (two-tail) value considerably lower than the tabulated value at a 95% confidence interval. An explanation for this is that Model 2 suffers from the presence of 2 outliers, namely compounds 5 and 22. Model 2a was developed after eliminating 5 and 22 as outliers. Model 2a shows that the addition of the (ΣπR)2 term is statistically acceptable at greater than 95% confidence interval. The occurrence of a non-linear relationship is not very surprising as several bilinear relationships between hydrophobicity (using π) and DHFR binding affinity data for various species have already been reported in the literature.44, 45 The optimum π values for meta substituents of the N1-aryl ring of cycloguanil analogs towards chicken, human, rat, murine, E. coli, Leishmania casei, L. major and P. carinii were 5.00, 2.10, 5.00, 1.76, 3.00, 4.31, 4.54 and 4.00, respectively. According to our literature search, there is no report about optimal hydrophobicity requirements for cycloguanil analogs against PfDHFR-TS binding affinity. Hence, we calculated the optimum value of the hydrophobicity of substituents at R for better binding affinity towards the wild type using simple differentiation of the parabolic equation (Model 2a) with respect to ΣπR, treating contributions from all other indicator variables as constant, and found d[pKi(wt)]/d[ΣπR] = −0.349*2(ΣπR) + 1.303 = 0.61 as the optimum πR value. Most of the potent compounds possess either a 4-chloro or 3-chloro substituent (π = 0.71 for Cl); however most of the compounds containing di-chloro substituents are inactive (π = 1.42 for di-Cl because of the additive effect of hydrophobicity). Neither a methyl substituent (π = 0.56, which is low) nor the ethyl substituent (π = 1.02, which is too high) are optimal for binding affinity. Substituents like CF3, CH2Br, SCH3, SeCH3, CH=CH2, SCOC2H5, NHC3H7 with π values 0.88, 0.79, 0.61, 0.74, 0.62, 0.82, 0.64, respectively, would be some better choices, depending upon the synthetic feasibility of these analogs. Some thioalkyl, selenium and CF3 containing-derivatives have already been reported to show promising activity on Pneumocystis carinii and human DHFR binding affinity.44, 45 The low coefficient of this π term shows that there exists a total desolvation of substituents in the PfDHFR binding site. Since this descriptor is not a whole molecular property, it is difficult to say whether the hydrophobic contributions of the C6-position substituents are important or not; but certainly the hydrophobic contribution from the N1–aryl ring substituents are additive and an optimal value is required for enhanced binding affinity to the wild type.
Model 3 was obtained by considering binding affinity data reported on the A16V+S108T mutant form of the enzyme, pKi(mut), as the dependent variable and the physico-chemical and indicator variables as independent variables. Model 3a was obtained by removing 22 as an outlier from Model 3. The positive contribution of the indicator variable I1 in Model 3a suggests that having the R1 position unsubstituted is favorable for enhancing binding to the mutant form of the enzyme. This is consistent with the SAR observations of Vilaivan et al.,21 who showed that removal of one of the dimethyl groups of cycloguanil at the C6 position results in better binding affinity to the mutant form. The reason, as pointed out by that research group, is that one of the methyl groups experiences a steric clash with the Val16 side chain in the A16V+S108T mutant (referred to as the “steric constraint hypothesis”9). Analogous to our observation for the wild type, a bulky isopropyl group (reflected by the negative contribution of the indicator variable, I4) is detrimental for binding to the mutant form. The positive contributions of the indicator variables I5, I6, and I7 show that 3-phenoxyphenyl, 3-benzyloxyphenyl and 4-phenoxyphenyl at R2 are favorable for enhanced binding affinity. However the strong binding of these analogs is in part due to the presence of H as the other substituent (at the R1 position) of C6, a fact that was already described above for Model 1a. B1Para(R) is a measure of the width of the first atom of the R substituents. Its negative coefficient indicates that bulkier para substituents around the N1-aryl ring (R) are detrimental for binding to this mutant type. This is again because of steric constraints imposed by Ser108. Most of the active compounds for this mutant type possess, instead, meta substituents.
Compound 22, an outlier in Models 1–3, is the least active among the series, and possesses a bulky t-butyl group at the R2 position. Interestingly, a compound with a t-butyl group at the meta position of the N1-aryl ring of cycloguanil was noted as an outlier in the QSAR study of cycloguanil analogs for Pneumocystis carinii DHFR inhibition by Marlowe et al.45 It was pointed out by those authors that the t-butyl group’s interaction with Pneumocystis carinii or Leishmania major DHFR is more highly twisted than in bacterial and mammalian DHFRs. The probable reason suggested for such behavior is that certain bulky amino acids like Trp, Ile or Phe restrict the entry of cycloguanil analogs to the hydrophobic binding pocket of DHFR in the former species; the inactivity of 22 suggests that this also occurs for Pf DHFR. Since compound 5 does not have any unusual substitution pattern and its binding affinity can be captured adequately by the descriptors used in Model 2, the outlying behavior of this compound may be due to experimental error. The reported binding affinity data (Ki) for these compounds were an average of triplicated assay results and there was a relatively high standard error reported for compound 5 (Ki = 1.5±0.8) compared to for other compounds 9 (Ki = 1.4±0.2), 16 (Ki = 1.6±0.2), 17 (Ki = 1.5±0.3) and 57 (Ki = 1.4±0.3) with Ki’s in a similar range to that for compound 5. The major results derived from the Hansch 2D-QSAR models are summarized in Figure 1.
Figure 1.

Summary of Hansch 2D-QSAR results for wild type and mutant binding affinity data.
Docking
A summary of all the average RMSDs and the R2 between docking scores and reported binding affinities is given in Table 5. Here we present the results for the 4 docking algorithms.
Table 5.
Summary of average RMSD and the R2 between docking scores and reported binding affinities for the 4 docking programs.
| Program | Scoring function | R2 | Average RMSDa |
|---|---|---|---|
| FlexX | Total score | 0.116 | – |
| DScore | 0.276 | – | |
| PMF Score | 0.039 | – | |
| GScore | 0.134 | – | |
| CScore | 0.008 | 4.97 | |
| Glide-SP | GScore | 0.466 | – |
| EModel | 0.450 | 3.40 | |
| Ruvinsky-GScore | 0.167 | – | |
| Ruvinsky-EModel | 0.196 | – | |
| Molegro virtual docker | MolDock | 0.741 | 2.14 |
| Rerank | 0.740 | – | |
| Protein-ligand | 0.740 | – | |
| GOLD | GoldScore | 0.911 | 2.99 |
| ChemScore | 0.855 | 3.88 | |
| ASP | 0.626 | 1.96 |
In Å.
1.1 FlexX
FlexX docking scores, including the Total score, GScore, ChemScore, PMF Score and DScore, all resulted in a poor correlation of R2 < 0.3 with the reported binding affinity data. FlexX predicted consistent binding modes for 9 of the 10 cycloguanil derivatives. Compound 39, which is unsubstituted at R1 and R2 and with a 4-F substitution at R, was predicted to adopt a different binding mode than the other 9 compounds. In contrast, the only other compound with such unsubstituted R1 and R2 and with a 4-Cl substitution at R was predicted to bind in a similar way to the other 9 compounds. This shows that the predictions of FlexX are inconsistent because such a big change in the binding mode is not expected to be caused by the small steric change from the F to the Cl atom at the R position, considering the space available in the active site. The average RMSD between the 30 docked poses from FlexX and the crystal structure of W99210 was 4.971 Å. The RMSD between the top-ranked of the 30 poses and the experimental structure was 3.3 Å. This high RMSD shows that FlexX was not capable of reproducing the experimental structure. Together with the performance of the docking scores, it can be concluded that FlexX as implemented in Sybyl 7.2 is not an accurate docking program to study this class of protein-ligand interactions. The binding modes of the 10 cycloguanil derivatives and the reference ligand, W99210, are given in Supplementary Figure S1. The individual docking scores for the 10 cycloguanil derivatives from FlexX and the RMSD values are given in Supplementary Tables S1 and S2, respectively.
1.2 Glide
The Glide SP module predicted consistent binding modes for all 10 compounds, matching the reference ligand binding mode. Ruvinsky’s treatment of the docking scores also resulted in similar binding modes of 9 compounds, but one compound, 17, for no apparent reason failed to refine through this procedure. The correlation R2 between the Glide GScore and the reported binding affinity data for the 10 compounds was 0.416. The correlation R2 between the Glide EModel and the reported binding affinity data was 0.426. As can be seen from the correlation coefficient values, neither the Glide GScore nor the EModel was satisfactorily able to predict the binding affinities of these analogs. The Glide GScore and EModel refined using Ruvinsky’s correction were still worse than the corresponding unprocessed GScore and EModel from Glide. R2 between the reported binding affinity data and the Ruvinsky refined Glide GScore and EModel for the nine compounds were 0.167 and 0.196, respectively. The average RMSD between the experimental and 30 predicted binding poses of W99210 was 3.399 Å. But the RMSD of the top-ranked pose among the 30 poses of the reference ligand and the experimental structure was only 1.734 Å, and hence, it can be concluded that the top-ranked pose from Glide can be confidently used for further calculations, since RMSD < 2 Å. The top-ranked pose of each ligand and the reference molecule from a Glide SP run before and after Ruvinsky’s treatment are given in Supplementary Figure S2. The individual docking scores for the 10 cycloguanil derivatives from Glide SP and the RMSD values are given in Supplementary Tables S3 and S4, respectively.
2.3 Molegro virtual docker
The chemical properties used in the bound ligand, W99210, which was used for template docking using Molegro virtual docker are given in Figure 2. Four hydrogen bond donor atoms (purple), three hydrogen bond acceptors (green) and 12 ring atoms (yellow) of the reference ligand, as shown in Figure 2, were used to dock other compounds. Molegro predicted consistent binding modes for all 10 compounds, matching the binding mode of the reference ligand, just as was found with Glide. The predicted binding modes are given in Figure 3. Three different scores can be obtained using two different scoring methods, MolDock and MolDock [GRID], so a total of six sets of scores were investigated using Molegro, and the results are given in Supplementary Table S5. Each score in the table is an average of five consecutive runs. The correlation R2 between the reported binding affinity data of the 10 cycloguanil derivatives and the MolDock, rerank and Protein-Ligand interaction scores from the MolDock scoring method were 0.741, 0.740 and 0.740, respectively. The correlation R2 between the reported binding affinity data of the 10 cycloguanil derivatives and the MolDock, rerank and the Protein-Ligand interaction scores from the MolDock [GRID] scoring method were 0.735, 0.703 and 0.739, respectively. This shows that Molegro docking scores were approximately equally successful in predicting the binding affinity, and that for docking scores Molegro performed better than FlexX or Glide. Also, since hydrogen bonding interactions do not influence the variation in the binding affinity data exerted by these analogs (discussed in detail below, under active site interactions), the correlation obtained with the two scoring functions used, MolDock and MolDock [GRID], which differ only by taking hydrogen bond directionality into account, was similar. The RMSDs obtained using the two different scoring functions are given in Supplementary Table S6. The average RMSDs were 2.14 and 2.18 Å, respectively. We found an impressively low RMSD = 1.02 Å between the top-ranked pose and the experimental structure of W99210 using the MolDock [GRID] scoring function. This clearly demonstrates that the Molegro virtual docker was very accurate in reproducing the experimental binding mode and the software also gave a reasonably accurate prediction of binding affinity of the cycloguanil derivatives.
Figure 2.
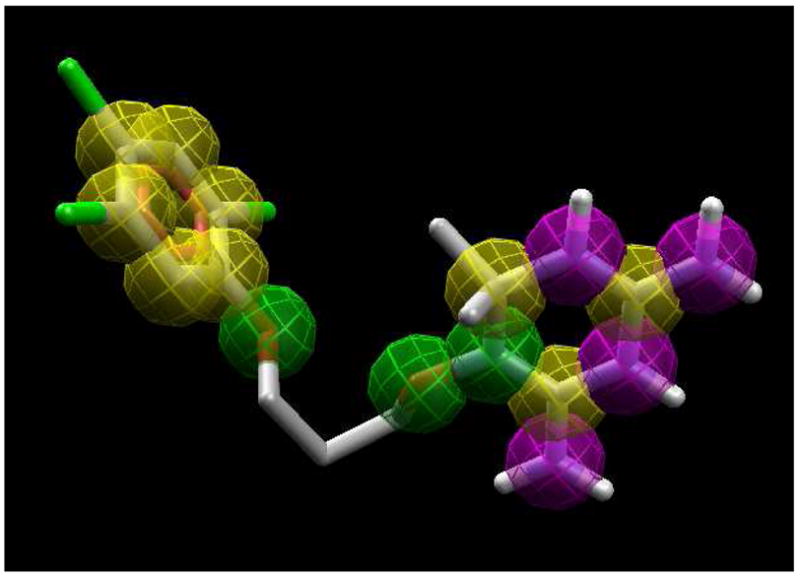
Template used for Molegro template docking. The chemical properties are also shown. Yellow: ring atoms; green: hydrogen bond acceptors; purple: hydrogen bond donors.
Figure 3.

Predicted binding modes of 10 cycloguanil analogs and the reference ligand using Molegro.
2.4 GOLD
Using GOLD software, among the three available docking scores investigated, only GoldScore predicted a consistent binding mode for all 10 cycloguanil analogs which was similar to that of the reference ligand. The correlation R2 between the reported binding affinity and the GoldScore, ChemScore and ASP were 0.911, 0.855 and 0.626, respectively. Hence, the order of accuracy of the scoring functions for predicting the binding affinity was GScore > ChemScore > ASP. The average RMSD between the predicted binding modes of W99210 and the experimental structure using GoldScore, ChemScore and ASP were 2.99, 3.88 and 1.96 Å, respectively. The order of accuracy for reproducing the experimental binding mode was ASP > GoldScore > ChemScore, and this order was maintained when the RMSDs of only the top-ranked poses were considered. This data demonstrates that the GoldScore scoring function predicted the binding affinity of the cycloguanil analogs very well, whereas the ASP was most accurate for reproducing the experimental binding mode. The ChemScore performance was not satisfactory for predicting either the binding mode or the binding affinity when compared to the other two scoring functions. Since the GoldScore yielded consistent binding modes and also an excellent correlation with the experimental binding affinity data, we selected the top-ranked binding pose of each ligand from GoldScore to study in detail the interaction of these derivatives with the active site residues. The predicted binding modes from GoldScore are given in Figure 4, whereas results from the other two scoring functions, ChemScore and the ASP, are given in Supplementary Figure S3.
Figure 4.

Binding modes of 8 cycloguanil derivatives, showing the key polar and non-polar interactions predicted obtained using GOLD (with GoldScore).
2.5 Active site interactions
We analyzed the top-ranked poses of the 10 cycloguanil ligands from GOLD (GoldScore) for important hydrogen bond and hydrophobic interactions (Figure 4). Two polar interactions are conserved in all the 10 cycloguanil derivatives: the 4-amino group forms a hydrogen bond with the side chain carboxylic acid of Asp54, and the 2-amino group forms a hydrogen bond with the backbone carbonyl of Ile14. Except for 31 and 39, which are unsubstituted at the C6 position, all the compounds showed an additional hydrogen bond with Asp54, between the carboxylic acid OH of Asp54 and N5 of the triazine ring of the ligand.
C6 of the triazine ring of cycloguanil derivatives lies within 4.5–5.0 Å of the methyl of Ala16 (Dist1 in Table 6). C4′ of the N1 phenyl of the triazine ring lies within 4.8–5.3 Å of the side chain methyl of Ser108 (Dist2 in Table 6), allowing favorable hydrophobic interactions. Both of these hydrophobic interactions are roughly equally important for these analogs as we can see from the correlations, R2 = 0.793 and 0.725 between the reported wild type binding affinity and Dist1 and Dist2, respectively. To make one interaction index which would account for both these interactions we used the average of the two distances = (Dist1+Dist2)/2, which showed an excellent correlation of R2 = 0.874 with the binding affinity data (Figure 5). We also calculated a key dihedral angle C2-N1-C1′-C2′ (atom labels given in Table 1) involved in the various substitution patterns in these analogs, using the docked poses, and found it to show a good correlation of R2 = 0.873 with binding affinity data (Figure 5). This clearly demonstrates that hydrophobic interactions of these analogs with the active site of PfDHFR-TS and the C2-N1-C1′-C2′ dihedral angle are crucial for their binding affinity in the wild type enzyme.
Table 6.
Interaction index and key dihedral angle which are correlated to the wild type activity.
| Compd. | pIC50 | Dist1a | Dist2a | Dihedralb | Indexc |
|---|---|---|---|---|---|
| 17 | 8.82 | 4.533 | 4.825 | −99.4 | 4.68 |
| 18 | 7.61 | 4.564 | 5.136 | −108.1 | 4.85 |
| 23 | 8.96 | 4.471 | 4.988 | −94.3 | 4.73 |
| 29 | 7.70 | 4.552 | 5.098 | −98.2 | 4.83 |
| 30 | 8.34 | 4.545 | 4.833 | −97.2 | 4.69 |
| 31 | 6.48 | 4.958 | 5.245 | −114.3 | 5.10 |
| 32 | 8.43 | 4.654 | 4.876 | −96.4 | 4.77 |
| 33 | 8.96 | 4.519 | 5.002 | −93.7 | 4.76 |
| 38 | 8.74 | 4.449 | 4.974 | −96.0 | 4.71 |
| 39 | 6.57 | 5.004 | 5.326 | −116.6 | 5.17 |
In Å;
C2-N1-C1′-C2′, in °;
Index = (Dist1+Dist2)/2
Figure 5.

Correlation of binding affinity with the C2-N1-C1′-C2′ torsion angle (upper graph) and with the distance between the cycloguanil derivatives and active site residues (lower graph).
Since the X-ray structure is unavailable for the double mutant relevant to these cycloguanil compounds, A16V + S108T, we did not proceed with docking into the mutant protein. It would be possible to mutate the wild type protein or another mutant X-ray structure and then perform docking but this would be less reliable than working directly from a crystal structure, as we were able to do for the wild type.
Conclusions
In the modern scenario of the drug design program, in addition to designing potent and safer drug candidates against malaria, necessary care has to be taken in addressing the emergence of resistance to anti-malarial drugs. In order to understand the importance of various physico-chemical and structural properties of cycloguanil derivatives towards their wild and mutant type PfDHFR-TS binding and to identify and use an optimal docking program to study the molecular interactions, we reported herein our extensive 2D-QSAR investigations followed by comparative docking analysis. Our 2D-QSAR investigations suggested that several structural features of the chosen ligands and the hydrophobicity of N1-phenyl substituents of 2,4-diamino-1,6-dihydro-1,3,5-triazine scaffold are crucial for effective binding with the wild type of the enzyme. This observation was analogous to our previously published results on a different antimalarial enzyme target which have shown the importance of ligands’ hydrophobicity to be crucial for their in vitro antimalarial activities.46 For binding to the A16V+S108T mutant form of the same enzyme, we determined that several structural variables such as the presence of 3-phenoxyphenyl, 3-benzyloxyphenyl and 4-phenoxyphenyl at the C6 position of the triazine ring of cycloguanil derivatives influence binding, and also found a negative steric tolerance for para-substituents on the N1-phenyl ring.
We studied the binding mode of the cycloguanil derivatives with DHFR-TS by focusing on the 10 reported analogs which had well-defined stereochemistry. Among the four different docking programs and 13 different scoring functions analyzed, we obtained the best correlation R2 = 0.911 between reported binding affinity data and docking scores for the GOLD program using the GoldScore fitness function. Also, among the programs and scoring functions analyzed, we found the Molegro virtual docker program, with its template docking algorithm and MolDock [GRID] score, to be very accurate for reproducing the experimental binding mode of ligand WR99210, with an RMSD of 1.02 Å. The average of two key hydrophobic distances and a key dihedral angle were found to be well correlated to the wild type activity exhibited by the 10 cycloguanil derivatives. Hence a combination of 2D-QSAR and docking analysis provided fresh insights into the existing SAR of cycloguanil derivatives towards their PfDHFR-TS inhibition.
Supplementary Material
Acknowledgments
This research was supported in part by funding from the University of Mississippi, including from its Faculty Research Program and the Office of Research and Sponsored Programs; from the National Center for Zoonotic, Vector-borne, and Enteric Diseases (CK) of the Centers for Disease Control and Prevention (U01/CI000211); from the National Science Foundation (EPS-0556308); and from the National Institute of Health’s National Center for Research Resources (P20 RR021929). SP is a Natural Products Neuroscience Fellow. PNT was a Summer Research Institute for Undergraduates participant. This investigation was conducted in a facility constructed with support from research facilities improvement program C06 RR-14503-01 from the NIH National Center for Research Resources.
Footnotes
Supporting Information: A complete list of different docking scores from FlexX, Glide, Molegro and GOLD and predicted binding modes using FlexX, Glide and GOLD for all 10 compounds used in the docking analysis are given in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434(7030):214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu YM, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419(6906):520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 4.Gregson A, Plowe CV. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol Rev. 2005;57(1):117–145. doi: 10.1124/pr.57.1.4. [DOI] [PubMed] [Google Scholar]
- 5.Kompis IM, Islam K, Then RL. DNA and RNA synthesis: Antifolates. Chem Rev. 2005;105(2):593–620. doi: 10.1021/cr0301144. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert IH. Inhibitors of dihydrofolate reductase in leishmania and trypanosomes. Biochem Biophys Acta-Mol Basis Dis. 2002;1587(2–3):249–257. doi: 10.1016/s0925-4439(02)00088-1. [DOI] [PubMed] [Google Scholar]
- 7.Anderson AC. Targeting DHFR in parasitic protozoa. Drug Discov Today. 2005;10(2):121–128. doi: 10.1016/S1359-6446(04)03308-2. [DOI] [PubMed] [Google Scholar]
- 8.Hyde JE. The dihydrofolate reductase-thymidylate synthetase gene in the drug resistance of malaria parasites. Pharmacol Ther. 1990;48(1):45–59. doi: 10.1016/0163-7258(90)90017-v. [DOI] [PubMed] [Google Scholar]
- 9.Yuthavong Y, Yuvaniyama J, Chitnumsub P, Vanichtanankul J, Chusacultanachai S, Tarnchompoo B, Vilaivan T, Kamchonwongpaisan S. Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: structural basis for antifolate resistance and development of effective inhibitors. Parasitology. 2005;130:249–259. doi: 10.1017/s003118200400664x. [DOI] [PubMed] [Google Scholar]
- 10.Adane L, Bharatam PV. Modelling and informatics in the analysis of P-falciparum DHFR enzyme inhibitors. Curr Med Chem. 2008;15(16):1552–1569. doi: 10.2174/092986708784911551. [DOI] [PubMed] [Google Scholar]
- 11.Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD, Yuthavong Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol. 2003;10(5):357–365. doi: 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- 12.Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchompoo B, Walkinshaw MD, Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: Pyrimethamine displacement linked with mutation-induced resistance. PNAS. 2005;102(37):13046–13051. doi: 10.1073/pnas.0501747102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delfino RT, Santos OA, Figueroa-Villar JD. Molecular modeling of wild-type and antifolate resistant mutant Plasmodium falciparum DHFR. Biophys Chem. 2002;98(3):287–300. doi: 10.1016/s0301-4622(02)00077-7. [DOI] [PubMed] [Google Scholar]
- 14.Fogel GB, Cheung M, Pittman E, Hecht D. Modeling the inhibition of quadruple mutant Plasmodium falciparum dihydrofolate reductase by pyrimethamine derivatives. J Comput-Aided Mol Des. 2008;22(1):29–38. doi: 10.1007/s10822-007-9152-9. [DOI] [PubMed] [Google Scholar]
- 15.Fogel GB, Cheung M, Pittman E, Hecht D. In silico screening against wild-type and mutant Plasmodium falciparum dihydrofolate reductase. J Mol Graph Model. 2008;26:1145–1152. doi: 10.1016/j.jmgm.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Hecht D, Cheung M, Fogel GB. QSAR using evolved neural networks for the inhibition of mutant PfDHFR by pyrimethamine derivatives. Biosystems. 2008;92(1):10–15. doi: 10.1016/j.biosystems.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Santos OA, Hopfinger AJ. A search for sources of drug resistance by the 4D-QSAR analysis of a set of antimalarial dihydrofolate reductase inhibitors. J Comput-Aided Mol Des. 2001;15(1):1–12. doi: 10.1023/a:1011152818340. [DOI] [PubMed] [Google Scholar]
- 18.Parenti MD, Pacchioni S, Ferrari AM, Rastelli G. Three-dimensional quantitative structure-activity relationship analysis of a set of Plasmodium falciparum dihydrofolate reductase inhibitors using a pharmacophore generation approach. J Med Chem. 2004;47(17):4258–4267. doi: 10.1021/jm040769c. [DOI] [PubMed] [Google Scholar]
- 19.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. PNAS. 2008;105(26):9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamchonwongpaisan S, Quarrell R, Charoensetakul N, Ponsinet R, Vilaivan T, Vanichtanankul J, Tarnchompoo B, Sirawaraporn W, Lowe G, Yuthavong Y. Inhibitors of multiple mutants of Plasmodium falciparum dihydrofolate reductase and their antimalarial activities. J Med Chem. 2004;47(3):673–680. doi: 10.1021/jm030165t. [DOI] [PubMed] [Google Scholar]
- 21.Vilaivan T, Saesaengseerung N, Jarprung D, Kamchonwongpaisan S, Sirawaraporn W, Yuthavong Y. Synthesis of solution-phase combinatorial library of 4,6-diamino-1,2-dihydro-1,3,5-triazine and identification of new leads against A16V+S108T mutant dihydrofolate reductase of . Plasmodium falciparum Bioorg Med Chem. 2003;11(2):217–224. doi: 10.1016/s0968-0896(02)00344-9. [DOI] [PubMed] [Google Scholar]
- 22.Yuthavong Y, Vilaivan T, Chareonsethakul N, Kamchonwongpaisan S, Sirawaraporn W, Quarrell R, Lowe G. Development of a lead inhibitor for the A16V+S108T mutant of dihydrofolate reductase from the cycloguanil-resistant strain (T9/94) of Plasmodium falciparum. J Med Chem. 2000;43(14):2738–2744. doi: 10.1021/jm0009181. [DOI] [PubMed] [Google Scholar]
- 23.Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. Wiley-Interscience: New York; 1979. pp. 48–63. [Google Scholar]
- 24.Verloop A, Hoogenstraaten W, Tipker J. Development and application of new steric substituent parameters in drug design. In: Ariens EJ, editor. Drug Design. Vol. 7. Academic Press; New York: 1976. pp. 165–207. [Google Scholar]
- 25.Hansch C, Garg R, Kurup A. Searching for allosteric effects via QSARs. Bioorg Med Chem. 2001;9(2):283–289. doi: 10.1016/s0968-0896(00)00248-0. [DOI] [PubMed] [Google Scholar]
- 26.Doweyko AM. 3D-QSAR illusions. J Comput-Aided Mol Des. 2004;18(7–9):587–596. doi: 10.1007/s10822-004-4068-0. [DOI] [PubMed] [Google Scholar]
- 27.Ponce YM, Garit JAC, Torrens F, Zaldivar VR, Castro EA. Atom, atomtype, and total linear indices of the “molecular pseudograph’s atom adjacency matrix”: Application to QSPR/QSAR studies of organic compounds. Molecules. 2004;9(12):1100–1123. doi: 10.3390/91201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sybyl. Vol. 7.2. Tripos; St. Louis, MO: 2006. [Google Scholar]
- 29.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J Comput Chem. 1992;13(4):505–524. [Google Scholar]
- 30.Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245(1):43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 31.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 32.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions. 1. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput-Aided Mol Des. 1997;11(5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 33.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42(5):791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 34.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid accurate docking and scoring 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 35.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A new approach for rapid accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 36.Kontoyianni M, McClellan LM, Sokol GS. Evaluation of docking performance: Comparative data on docking algorithms. J Med Chem. 2004;47(3):558–565. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- 37.Ruvinsky AM. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11 scoring functions. J Comput Chem. 2007;28(8):1364–1372. doi: 10.1002/jcc.20580. [DOI] [PubMed] [Google Scholar]
- 38.Ruvinsky AM, Kozintsev AV. New and fast statistical-thermodynamic method for computation of protein-ligand binding entropy substantially improves docking accuracy. J Comput Chem. 2005;26(11):1089–1095. doi: 10.1002/jcc.20246. [DOI] [PubMed] [Google Scholar]
- 39.Thomsen R, Christensen MH. MolDock: A new technique for high-accuracy molecular docking. Journal of Medicinal Chemistry. 2006;49(11):3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 40.Molegro ApS. Molegro Virtual Docker. Vol. 2.4. Aarhus C; Denmark: 2008. [Google Scholar]
- 41.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins-Structure Function and Genetics. 2003;52(4):609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 42.Mooij WTM, Verdonk ML. General and targeted statistical potentials for protein-ligand interactions. Proteins-Structure Function and Bioinformatics. 2005;61(2):272–287. doi: 10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- 43.Hansch C, Hathaway BA, Guo ZR, Selassie CD, Dietrich SW, Blaney JM, Langridge R, Volz KW, Kaufman BT. Crystallography, quantitative structureactivity relationships, and molecular graphics in a comparative analysis of the inhibition of dihydrofolate reductase from chicken liver and Lactobacillus casei by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(substituted-phenyl)-s-triazines. J Med Chem. 1984;27(2):129–143. doi: 10.1021/jm00368a006. [DOI] [PubMed] [Google Scholar]
- 44.Hathaway BA, Guo ZR, Hansch C, Delcamp TJ, Susten SS, Freisheim JH. Inhibition of human dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(substituted-phenyl)-s-triazine s. A quantitative structure-activity relationship analysis. J Med Chem. 1984;27(2):144–149. doi: 10.1021/jm00368a007. [DOI] [PubMed] [Google Scholar]
- 45.Marlowe CK, Selassie CD, Santi DV. Quantitative structure-activity relationships of the inhibition of Pneumocystis carinii dihydrofolate reductase by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(X-phenyl)-s-triazines. J Med Chem. 1995;38(6):967–972. doi: 10.1021/jm00006a016. [DOI] [PubMed] [Google Scholar]
- 46.Xie A, Sivaprakasam P, Doerksen RJ. 3D-QSAR analysis of antimalarial farnesyltransferase inhibitors based on a 2,5-diaminobenzophenone scaffold. Bioorg Med Chem. 2006;14(21):7311–7323. doi: 10.1016/j.bmc.2006.06.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.