

Abstract
The ability to manipulate mitochondrial DNA (mtDNA) heteroplasmy would provide a powerful tool to treat mitochondrial diseases. Recent studies showed that mitochondria-targeted restriction endonucleases can modify mtDNA heteroplasmy in a predictable and efficient manner if it recognizes a single site in the mutant mtDNA. However, the applicability of such model is limited to mutations that create a novel cleavage site, not present in the wild-type mtDNA. We attempted to extend this approach to a ‘differential multiple cleavage site’ model, where an mtDNA mutation creates an extra restriction site to the ones normally present in the wild-type mtDNA. Taking advantage of a heteroplasmic mouse model harboring two haplotypes of mtDNA (NZB/BALB) and using adenovirus as a gene vector, we delivered a mitochondria-targeted Scal restriction endonuclease to different mouse tissues. Scal recognizes five sites in the NZB mtDNA but only three in BALB mtDNA. Our results showed that changes in mtDNA heteroplasmy were obtained by the expression of mitochondria-targeted ScaI in both liver, after intravenous injection, and in skeletal muscle, after intramuscular injection. Although mtDNA depletion was an undesirable side effect, our data suggest that under a regulated expression system, mtDNA depletion could be minimized and restriction endonucleases recognizing multiple sites could have a potential for therapeutic use.
Keywords: mitochondria, mtDNA heteroplasmy, mitochondrial diseases, restriction endonuclease, adenovirus
Introduction
Normal expression of the mitochondrial genome (mtDNA) is essential for the biogenesis of the oxidative phosphorylation system (OXPHOS) that is required by essentially all cells for respiration. Mutations in mtDNA have been associated with a wide variety of human disorders and have a prevalence of at least 1:5000.1,2 Potential genetic approaches to the treatment of diseases caused by mtDNA mutations are distinct from conventional gene therapy strategies, as the genetic features of mtDNA differ substantially from those of the nuclear genome.
Cells contain multiple copies of mtDNA; thus mutant loci within the mtDNA population of a cell or tissue commonly exist in a heteroplasmic state. This means that mtDNA populations contain variable proportions of mutant and wild-type mtDNA.3 Disease phenotypes are only observed once the proportion of wild-type mtDNA drops below a threshold level4,5 and so increasing the proportion of wild-type mtDNA in affected tissues is seen as a viable therapeutic strategy for the treatment of diseases caused by mtDNA mutations. This has been achieved to variable extents in muscle by inducing the incorporation of satellite cells containing predominantly wild-type mtDNA through either exercise training,6 tissue damage and regeneration,7 or pharmacological ablation.8 In cultured cells, selection against mutant mtDNA has been achieved using the OXPHOS inhibitor oligomycin.9 Peptide nucleic acid oligomers, homologous to mtDNA mutations, can also preferentially inhibit replication of specific mtDNA haplotypes in vitro, but this has showed limitations in the context of intact cells.10,11
An important feature of mitochondrial genetics is that cells tend to maintain a fixed abundance of mtDNA12 such that if mtDNA levels are depleted artificially, they will repopulate to original levels once the ‘depletion pressure’ is removed.13,14 We and other groups have taken advantage of this phenomenon to manipulate mtDNA heteroplasmy by delivering restriction endonucleases (RE) to mitochondria that target specific mtDNA sub-populations. Cleaved mtDNA is rapidly degraded allowing repopulation with residual mtDNA.15
Srivastava et al.16 expressed PstI targeted to mitochondria in a heteroplasmic murine cell line, containing one mtDNA haplotype with two sites for PstI and another haplotype having none, causing a significant shift in heteroplasmy. This approach was also used by Tanaka et al.,17 who used SmaI that recognizes the pathogenic 8993T→G mutation in MT-ATP6. Elimination of the mutant mtDNA was observed, which resulted in restoration of both the normal intracellular ATP levels and mitochondrial membrane potential. Bayona-Bafaluy et al.15 developed a system to modify mtDNA heteroplasmy by expressing a mitochondria-targeted form of ApaLI in cells and tissues from heteroplasmic mice that carry the BALB/NZB mtDNA haplotypes, only BALB mtDNA containing an ApaLI site. They showed both in cultured cells and in vivo (brain and muscle) that mitochondria-targeted ApaLI could directionally alter mtDNA heteroplasmy.
All of the above examples relied on the existence of unique RE sites in a sub-population of mtDNA in heteroplasmic cells or tissues. Unfortunately, clinically relevant mtDNA mutations do not always create unique RE sites. A good example is the 3243A→G transition in the mitochondrial tRNALeu(UUR) gene (MT-TL1) present in patients with MELAS syndrome (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes). This mutation creates an extra ApaI site in addition to the five sites present in wild-type mtDNA. Thus we were interested in testing the effectiveness of a mitochondriatargeted RE that recognized a different number of sites in two sub-populations of mtDNA, a so-called ‘differential multiple cleavage site’ model. To do this, we used heteroplasmic mice carrying two normal mtDNA genotypes, BALB/cByJ (BALB) and NZB/BINJ (NZB), and chose ScaI as the model RE as it recognizes three sites in BALB mtDNA and five sites in NZB mtDNA (Figure 1a). Recombinant adenovirus (rAd) was used to deliver a mitochondria-targeted form of the enzyme expressed under a cytomegalovirus (CMV) promoter.
Figure 1.
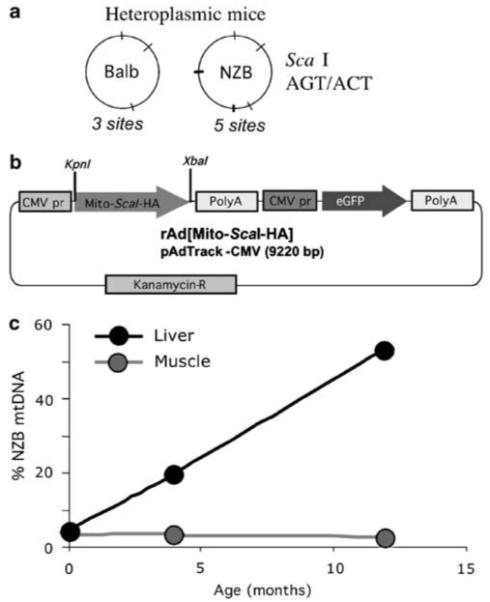
Production and characterization of the tools and reagents used to test mitochondria-targeted restriction endonucleases. (a) ScaI restriction endonuclease can digest both BALB and NZB mtDNA, recognizing three sites in the BALB mtDNA and five sites in the NZB mtDNA. ScaI was chosen because it resembles a human disease-related mutation model, namely the creation of an extra ApaI restriction endonuclease site in MELAS syndrome mtDNA, in addition to sites present in the wild-type human mtDNA. (b) The recombinant rAd[Mito-ScaI-HA] contained a mitochondrial targeting sequence (cytochrome oxidase subunit 8 (Cox8)), a gene coding for Streptomyces caespitosus ScaI restriction endonuclease synthesized in vitro and an HA tag for immunological detection. The construct was cloned in a recombinant adenovirus vector also encoding the enhanced green fluorescence protein (eGFP) under the control of cytomegalovirus promoter (CMV). (c) The mouse model utilized (NZB/BALB mtDNA) shows an age-related shift in mtDNA heteroplasmy in liver, where the NZB haplotype increases with age. On the other hand, mtDNA heteroplasmy in tissues such as muscle remains unchanged.20 mtDNA, mitochondrial DNA; MELAS, mitochondrial encephalopathy, lactic acidosis and stroke-like episodes; NZB, New Zealand Black.
Results
A recombinant ScaI protein, Mito-ScaI-HA, was created by the addition of an N-terminal mitochondrial targeting pre-sequence and a C-terminal hemagglutinin (HA) tag (Figure 1b). The efficiency of targeting Mito-ScaI-HA to mitochondria was analyzed by immunocytochemistry in murine hepatocyte-derived cells infected with rAd[Mito-ScaI-HA]. HA expression colocalized with the mitochondrial dye Mitotracker (Figure 2a). The rAd vector used in these studies was derived from adenovirus type-5 and contained an enhanced green fluorescence protein (eGFP) gene under an independent CMV promoter to enable visualization of infection (Figure 1b). An identical vector containing only eGFP under the CMV promoter (rAd[eGFP]) was used throughout as a control.
Figure 2.
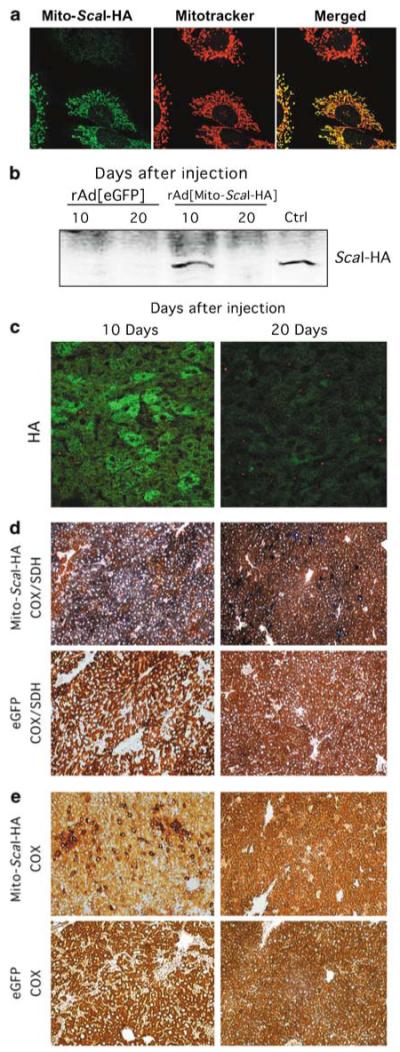
Strong expression of rAd[Mito-ScaI-HA] in liver leads to COX deficiency. (a) The mitochondrial localization of ScaI was confirmed by confocal microscopy of mouse cell line (hepatocyte-derived) infected with rAd[Mito-ScaI-HA]. Colocalization of Mito-ScaI-HA and Mitotracker was evident in transduced cells. (b) Steady-state levels of Mito-ScaI-HA were detected by western blot (anti-HA) of liver samples after injection. Strong protein expression was observed in a liver sample transduced with rAd[Mito-ScaI-HA] 10 days after jugular injection. No HA expression was detected at 20 days after rAd[Mito-ScaI-HA] or after rAd[eGFP] injection. Cultured cells transduced with rAd[Mito-ScaI-HA] were used as positive controls. (c) The same pattern of expression was observed by immunocytochemistry analysis with an anti-HA antibody (green). COX (e) and COX/SDH (d) activities in liver samples at 10 or 20 days after injection. COX-negative regions with increased SDH staining were observed in rAd[Mito-ScaI-HA]-transduced liver 10 days after injection. These areas were reduced 20 days after systemic gene delivery. rAd[eGFP]-injected mice showed no change in COX or COX/SDH staining (d and e). COX, cytochrome c oxidase; eGFP, enhanced green fluorescence protein; HA, hemag-glutinin; SDH, succinate dehydrogenase.
Liver studies
Taking advantage of the tropism of adenovirus 5 (Ad 5) for liver,18,19 right external jugular vein injections of rAd[Mito-ScaI-HA] or rAd[eGFP] were performed in two groups of mice: 1-year-old mice, which harbor high proportions of NZB mtDNA, and 2.5- to 4-month-old mice with lower and more variable proportions of NZB mtDNA. Previous characterization of mtDNA haplotype distribution in this mouse model showed that the segregation is uneven in liver, but not in muscle (Figure 1c).20 For unknown reasons, liver cells tend to accumulate the NZB haplotype. Liver biopsies, obtained immediately before and at various time points following injection, were analyzed for Mito-ScaI-HA expression and changes in mtDNA levels and heteroplasmy.
Mito-ScaI-HA expression in liver
We analyzed the expression of the transgenes after injection of rAd[Mito-ScaI-HA] (two animals) or rAd[eGFP] (two animals). For both groups, eGFP expression could be detected for up to 2 weeks in liver (not shown). Western blots of liver homogenates showed high levels of HA expression in a sample obtained 10 days after injection of rAd[Mito-ScaI-HA] but no detectable expression in a sample obtained 20 days after injection (Figure 2b). Immunohistochemical studies confirmed this pattern of expression (Figure 2c).
rAd [Mito-ScaI-HA] causes cytochrome c oxidase deficiency and mtDNA depletion in liver
Liver sections were evaluated histochemically for cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) activities at 10 or 20 days after injection. COX-deficient areas were observed 10 days after injection of rAd[Mito-ScaI-HA]. This deficiency was less pronounced, but still detectable at 20 days after injection (Figures 2d-e). No COX deficiency was detected in liver sections after rAd[eGFP] injection (Figures 2d-e). At 7 or 14 days after injection, COX activity in liver homogenates was decreased in samples from mice injected with rAd [Mito-ScaI-HA] when compared to those injected with rAd[eGFP] or sham animals (Figure 3a).
Figure 3.
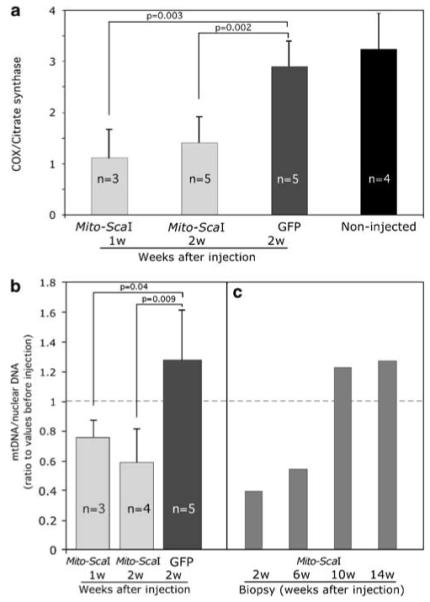
Transduction of rAd[Mito-ScaI-HA] in liver leads to a partial COX deficiency and mtDNA depletion. (a) Enzymatic activity measured spectrophotometrically in liver homogenates of rAd-injected mice. Specific COX activity is expressed as ratios to citrate synthase activity. rAd[Mito-ScaI-HA]-infected liver samples showed decreased COX activity when compared to rAd[eGFP] samples or uninjected controls. (b) mtDNA levels were analyzed by Southern blot. rAd[Mito-ScaI-HA]-infected samples from old-mice group showed decreased ratios of mtDNA/nuclear DNA when compared to the respective pre-injection biopsy samples. (c) Serial liver biopsies were analyzed for mtDNA content in 4-month-old mice. Each bar represents the liver biopsy performed in the same animal analyzed at different times after rAd[Mito-ScaI-HA] delivery. Error bars represent standard deviation. Significance was assessed by the Student’s t-test (individual comparisons to the GFP-treated group). COX, cytochrome c oxidase; eGFP, enhanced green fluorescence protein; mtDNA, mitochondrial DNA.
mtDNA levels were quantified in liver samples at different time points after injection of either vector by Southern blotting. When normalized to a nuclear DNA signal (18S rDNA),21 liver samples from the older mice showed a decrease in mtDNA content at 7 or 14 days after injection of rAd[Mito-ScaI-HA] that was not apparent in samples from mice injected with rAd[eGFP] (Figure 3b). Serial liver biopsies performed in the 2.5- to 4-month-old mice showed the same pattern of rAd [Mito-ScaI-HA]-associated mtDNA depletion for up to 6 weeks after injection, followed by recovery at 10 and 14 weeks after injection (Figure 3c). rAd[eGFP]-injected 2.5- to 4-month-old mice did not show any decrease in mtDNA content (data not shown).
rAd[Mito-ScaI-HA] induces a rapid shift in mtDNA heteroplasmy in liver
We tested the ability of rAd[Mito-ScaI-HA] to promote a shift in mtDNA heteroplasmy. As shown in Figure 4a, the old-mice group liver samples were analyzed 1 or 2 weeks after intravenous injections. The initial proportion of NZB mtDNA ranged from 70 to 90%. There was a decrease in NZB mtDNA that ranged from 7 to 15% in all the samples analyzed after rAd[Mito-ScaI-HA] injection. rAd[eGFP]-injected mice showed an increase in NZB mtDNA due to the age-related segregation as previously described20 (Figure 1c).
Figure 4.
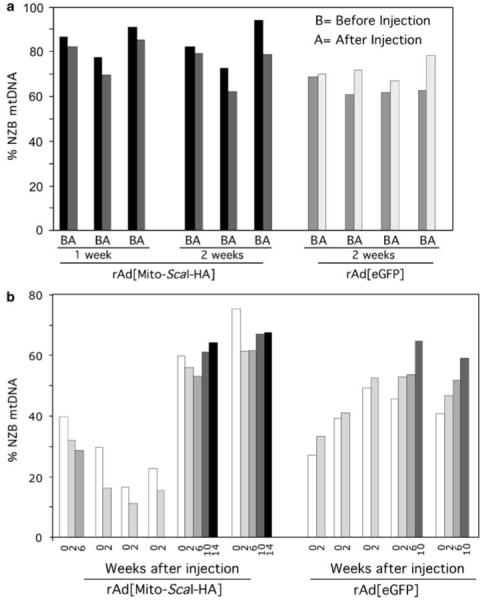
Transduction of rAd[Mito-ScaI-HA] in liver leads to a shift in mtDNA heteroplasmy. (a) The percentage of NZB mtDNA genotype was quantified by the last-cycle hot PCR/RFLP analysis. NZB mtDNA was decreased in 1-year-old mice 1 or 2 weeks after injection of rAd[Mito-ScaI-HA], and increased as expected after rAd[eGFP] administration. Bars represent the percentage of NZB mtDNA in liver samples before and after adenovirus injection. The decrease of NZB mtDNA in the rAd[Mito-ScaI-HA]-injected liver was 7.2±2.4% (1 week, n = 3) and 11.5±7.1% (2 weeks, n = 3). In contrast, the rAd[eGFP]-injected livers showed an increase in NZB mtDNA (13.5±10.3%, n = 4). The difference in heteroplasmy shift between the 2 weeks injected animals (rAd[Mito-ScaI-HA] vs rAd[eGFP]) was significant (P 0.015). (b) Serial liver biopsies were performed after rAd[Mito-ScaI-HA] or rAd[eGFP] injection. Each cluster of bars represents data from one animal, which was analyzed for different periods. Each bar corresponds to liver biopsies analyzed at different times (1-14 weeks) after adenoviral injection. eGFP, enhanced green fluorescence protein; mtDNA, mitochondrial DNA; NZB, New Zealand Black.
Serial liver biopsies were performed on the 2.5- to 4-month-old group after rAd[Mito-ScaI-HA] or rAd[eGFP] injection to investigate the time frame of changes in mtDNA heteroplasmy. Figure 4b shows the changes in the proportion of NZB mtDNA in different mice from 0 to 14 weeks after injection. We detected a decrease in the proportion of NZB mtDNA that was maintained for 6 weeks after injection of rAd[Mito-ScaI-HA]. Beyond this time, the relative amount of NZB mtDNA tended to return to its natural upward trend. When the mice were injected with rAd[eGFP], only the expected age-related increase in the proportion of NZB mtDNA was observed (Figure 4b). No overt health problems were detected in the injected animals.
Muscle studies
Because skeletal muscle is commonly affected in mitochondrial diseases, we tested if rAd[Mito-ScaI-HA] also could promote a shift in mtDNA heteroplasmy after intramuscular injection.
rAd [Mito-ScaI-HA] and rAd [eGFP] expression in muscle fibers
Intramuscular injections of rAd[Mito-ScaI-HA] in the right femoris muscle of 5-day-old mice led to local expression of the transgenes as shown by the eGFP fluorescence, which was not present in the uninjected muscle (Supplementary Figure S1A). To follow the expression of rAd[Mito-ScaI-HA] in muscle fibers, HA immunoreactive signal was followed at 1, 2, 3 and 4 weeks after rAd delivery. HA expression was observed up to 3 weeks after injection, a pattern that correlated with the eGFP expression. Both HA and eGFP levels were reduced 4 weeks after injection (Supplementary Figure S1B). To identify the site of injection beyond this time, we coinjected rAd[Mito-ScaI-HA] with a 0.25% solution of fluorescent latex microspheres. Microspheres were consistently detected in the areas surrounding injection sites (Supplementary Figure S2). By using this procedure, we could analyze the injection sites for up to 12 weeks after injection.
rAd [Mito-ScaI-HA] generates COX-deficient muscle fibers in the injected areas
Serial sections of the right femoris muscle of injected mice were evaluated histochemically for COX and SDH activities. Figure 5a (fourth column) shows a typical COX/SDH dual stain pattern where normal fibers stain dark brown for COX activity and pale blue for SDH activity. A COX deficiency was observed in some fibers of the animals injected with rAd[Mito-ScaI-HA]. Increased SDH staining was also seen in COX-deficient fibers, suggesting mitochondrial proliferation.22 Immunohistochemistry using monoclonal antibodies against subunit MT-CO1 of COX (Figure 5a, first column) showed a decrease in some of the injected fibers, which also had increased SDHA staining (Figure 5a, second and third columns). No reductions in COX activity or MT-CO1 expression were observed in uninjected muscle (Figure 5a) or muscle injected with rAd[eGFP] (data not shown).
Figure 5.
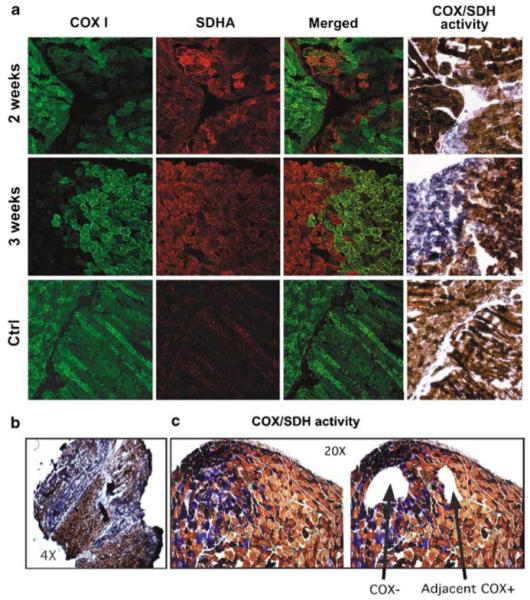
Expression of mitochondrial ScaI in muscle leads to COX deficiency in injected areas. (a) Immunostaining of muscle samples at different time points after focal injection with rAd[Mito-ScaI-HA] with MT-CO1-Alexa-conjugated monoclonal antibody shows a decrease in CO1. Mitochondrial proliferation can be inferred from the increased staining with the SDHA antibody. CO1-deficient areas also showed decreased COX activity with increased SDH staining (fourth column). (b) Muscle sections stained for the COX/SDH activity showed a pattern of COX-deficient areas (at the sites of focal injections) that coexists with normal COX areas (as shown with different magnifications). (c) Laser capture microscope microdissected areas of COX-negative (COX-) areas and COX-positive (COX+) areas adjacent to the deficient areas were collected for analyses. COX, cytochrome c oxidase; SDH, succinate dehydrogenase.
rAd [Mito-ScaI-HA] induces a rapid shift in mtDNA heteroplasmy in muscle
We analyzed changes in mtDNA heteroplasmy after injecting rAd[Mito-ScaI-HA] or rAd[eGFP] into the right femoris muscle. To determine the proportion of NZB mtDNA in injected muscle fibers, we microdissected fiber segments from sections stained for COX or COX/SDH activity. Figure 5b shows that injections of rAd[Mito-ScaI-HA] produced areas of COX-deficient fibers. COX-negative fibers as well as COX-positive fibers adjacent to the COX-negative areas were microdissected and analyzed (Figure 5c). Tissue samples were collected from mice killed at 1, 2, 3, 4, 8 and 12 weeks after injection and analyzed using ‘last-cycle hot’ PCR/restriction fragment length polymorphism (RFLP). The mtDNA heteroplasmy showed a rapid, small, but significant shift in mtDNA heteroplasmy with a decrease in the proportion of the NZB mtDNA haplotype. This shift in heteroplasmy was followed in animals up to 12 weeks after injection, when all animals were killed (Figures 6a and b). When muscle samples were obtained from COX-positive areas that were adjacent to the COX-negative areas (Figure 5c), similar shifts in heteroplasmy were obtained (Figure 6c). No changes in the proportion of the NZB mtDNA haplotype were seen in muscle fibers dissected from muscle injected with rAd[eGFP]. No morphological evidence of necrotic or apoptotic tissuewas observed in muscle injected with either vector. Similar to the liver experiments, no obvious changes in the muscle appearance were noted.
Figure 6.
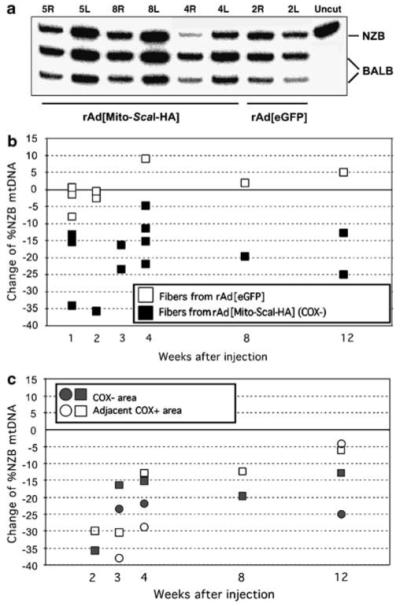
Shift in mtDNA heteroplasmy after rAd[Mito-ScaI-HA] transduction in muscle. (a) Representative PAGE analysis showing the percentage of NZB mtDNA of injected samples (R = right) after DNA extraction and last-cycle hot PCR-RFLP procedures. Each sample was a pool of muscle fiber segments (10-20) from individual mice. Samples 5, 8 and 4 right (R) from rAd[Mito-ScaI-HA]-injected muscle at 1 week after virus administration showed a decrease in the relative ratio of NZB mtDNA, when compared to the uninjected leg (L = left). Sample 2R (leg injected with rAd[eGFP]) did not show a shift in mtDNA heteroplasmy. ‘Uncut’, PCR fragment non-digested. NZB or BALB indicates each of the two mtDNA haplotypes after PCR/RFLP analysis. The original percentage of NZB mtDNA ranged from 25 to 40% of the total mtDNA. (b) Differences in the mtDNA haplotype in LCM-microdissected muscle samples. Each square represents the difference in the %NZB haplotype between injected and uninjected hind limbs of single rAd[Mito-ScaI-HA] (black squares)- or rAd[eGFP] (white squares)-injected mice. (c) Differences in the proportion of NZB mtDNA between injected and uninjected femoris COX-positive (COX+) fibers located adjacent to COX-negative (COX-) fibers were microdissected and %NZB mtDNA quantified. Both COX-areas (gray squares or circles) and adjacent COX+ areas (corresponding white squares or circles) showed decreased NZB mtDNA at all time points. COX, cytochrome c oxidase; LCM, laser capture microscope; mtDNA, mitochondrial DNA; NZB, New Zealand Black.
Discussion
Extending the usefulness of mitochondria-targeted REs to promote a shift in mtDNA heteroplasmy
REs targeted to mitochondria specific for restriction sites created by mtDNA mutations have been shown to selectively degrade mutant mtDNA.15-17,23 In its simplest form (a single and unique cleavage site created by a mutation, such as the SmaI site created by the 8993T→G NARP mutation), this approach was effective, but very limited. Our study is the first attempt to expand the usefulness of mitochondria-targeted RE to a ‘differential multiple cleavage site’ model where restriction sites are present on both the mutant and wild-type mtDNA (for example the additional ApaI site created by the 3243A→G MELAS mutation).
As mentioned previously, adenoviral vectors are particularly attractive in this approach as they may avoid the issues related to the integration of viral or other foreign sequences in the host genome. The transient expression of adenovirus has the potential to promote an adequate change in mtDNA heteroplasmy. Although we used young animals for the muscle studies, to avoid a neutralizing immunological response, there are many approaches being developed to circumvent this problem in adult mice, including immunosuppression, immuno-modulation, serotype switching, use of targeted Ad vectors, microencapsulation of Ad vectors, use of helper-dependent Ad vectors, and development of non-human Ad vectors.24
Shifts in mtDNA heteroplasmy in liver
rAd vectors are widely used in research owing to their extremely high transduction efficiency and wide tropism.25 In rodents and non-human primates, systemic administration of rAd 5 results in approximately 95% of measurable transduction in hepatocytes18,19,26 with little toxic effect.27,28 However, rAd vectors do illicit an immunogenic response and as such are rapidly cleared from the host leading to transient transgene expression.29 In the case of mitochondria-targeted RE strategies, transient expression can be considered an advantage as it may be sufficient to promote long-lasting changes in the balance of mtDNA haplotypes.15 Liver was an interesting organ to study in this model because NZB mtDNA is preferentially maintained in the aging liver of NZB/BALB heteroplasmic mice. The molecular basis for this natural, non-random segregation remains unclear. It does not appear to be based on OXPHOS function at the cellular or organelle level, nor to a replicative advantage of the NZB haplotype.20,30 Recently, Moreno-Loshuertos et al.31 found evidence that non-random segregation may be a consequence of an increased generation of reactive oxygen species and upregulation of mitochondrial biogenesis.
We were able to revert the natural segregation of heteroplasmic NZB/BALB mtDNA in liver towards an increasing proportion of NZB mtDNA haplotype after intravenous injection of rAd[Mito-ScaI-HA]. This heteroplasmy shift persisted for 6 weeks after injection, concurrent with a partial COX deficiency and mtDNA depletion, despite the fact that expression of rAd [Mito-ScaI-HA] was not detectable beyond 3 weeks. At time points beyond 6 weeks, mtDNA levels returned to normal and mtDNA segregation appeared to regain its natural segregation pattern in which the NZB haplotype increases with age.20 Nevertheless, for a specific age, the % NZB mtDNA was reduced from the initial trend by the action of Mito-ScaI.
Our results indicate that the COX deficiency seen in liver was a consequence of mtDNA depletion caused by ScaI RE cleaving both NZB and BALB haplotypes. Previous work has shown that cleaved mtDNA is very rapidly degraded.15 Given the rapidity and effectiveness of single cleavage site models, it is clear that a single cut is all that is required to initiate mtDNA degradation. This may have limited the extent of heteroplasmy shift seen in our model because as the proportion of NZB mtDNA drops, the relative abundance of ScaI sites on NZB versus BALB haplotype mtDNA also drops, decreasing the probability that further cleavages will occur within the NZB sub-population. The gradual reversion of the decrease in the proportion of NZB mtDNA in liver is likely related to (1) the transient expression of rAd[Mito-ScaI-HA] and (2) the natural non-random segregation towards an increasing proportion of NZB mtDNA in liver.20
Shifts in mtDNA heteroplasmy in skeletal muscle
Skeletal muscle, a post-mitotic tissue, is affected in many mitochondrial diseases32 and was therefore an attractive tissue for study in our system. Because rAd vectors transduce muscle inefficiently when injected systemically,33 intramuscular injections of rAd have been used extensively when targeting muscle.34 Existing examples include rAd encoding acid α-glucosidase, injected into the gastrocnemius of neonate mice for the treatment of glycogenosis type II,35 rAd-mediated gene transfer of a sarcoglycan used as a therapeutic strategy for a model of Duchenne muscular dystrophy,36 a similar approach to limb-girdle muscular dystrophy type 2D (LGMD 2D).37 Other rAd strategies have been applied for the treatment for Duchene muscular dystrophy.38 To avoid a neutralizing immune response, we performed intramuscular injections of rAd in mice less than 1 week old.15 Although expression of the transgenes was detected up to 4 weeks after injection, a shift in heteroplasmy was maintained up to 12 weeks after injection (the last time point analyzed). Based on the observations in liver, it is reasonable to assume that the COX defect in muscle was a consequence of mtDNA depletion. The long-term maintenance of the heteroplasmy shift in muscle is encouraging although mtDNA depletion remains a major concern. The increased SDH activity that accompanied the COX defect is indicative of increased mitochondrial proliferation, something that is also seen in muscle biopsies derived from patients with COX deficiency22 or carrying high mutant loads of mtDNA mutations.39 Importantly, COX-positive fibers adjacent to the regions of COX-negative fibers also showed a significant shift in mtDNA heteroplasmy. This implies that by controlling the expression of mitochondria-targeted RE to a level that promotes heteroplasmy, shift without a severe mtDNA depletion should be attainable.
Future perspectives
The critical goal of future work is to determine the conditions in which a mitochondria-targeted RE can be expressed in a differential multiple cleavage site model at levels sufficient to trigger a shift in mtDNA heteroplasmy with only a mild, transient mtDNA depletion. Inducible systems are being developed and optimized for animal use. Examples include the use of the heat-inducible mdr1 promoter40 or a multicistronic adenovirus vector with a streptogramin-inducible switch.41 Alternatively, controlled virus titer and frequency of injections can help adjust the therapeutic doses. The use of different viral vectors, which are optimized for specific tissues (for example AAV6 for skeletal muscle42) if highly regulated, should also be considered.
It is important to remember that small changes in heteroplasmy may be sufficient to restore OXPHOS function to non-pathological levels.4,5 A dramatic example of how small changes in heteroplasmy can affect the phenotype is illustrated by a study comparing the mtDNA heteroplasmy levels in single muscle fibers of patients harboring the 3243A→G mtDNA mutation. The mtDNA mutation load in COX-positive fibers could reach 90%, whereas COX-negative fibers had an average of 95% mutant mtDNA.43 Given the results presented here, an improved differential multiple cleavage site mitochondria-targeted RE system that avoids a severe mtDNA depletion could promote changes in mtDNA heteroplasmy with significant functional benefits.
Materials and methods
Animal handling
New Zealand Black (NZB)/BALB heteroplasmic female founders were produced as described.20 A pattern of segregation of the mtDNA haplotype with age has been described in tissues of these animals that is independent of the nuclear background. An increase in the proportion of the NZB haplotype occurs in liver and kidney and a decrease in blood and spleen. No significant changes in heteroplasmy are seen in skeletal or cardiac muscle (Figure 1c).
Mito-ScaI-HA rAd
The ScaI gene (from an Escherichia coli strain that carries the cloned ScaI gene from Streptomyces caespitosus, a kind gift from New England Biolabs) was cloned downstream of the human COX8A mitochondrial targeting sequence.44 An HA tag was added to the 3′ end. The final construct was cloned in a recombinant, E1-deleted, replication-deficient, adenovirus (Ad-track 5) encoding eGFP vector under control of CMV promoter. rAd [Mito-ScaI-HA] and control rAd[eGFP] adenovirus stocks were prepared by the Colorado State University Virus Core Facility (Ft Collins, CO, USA). The adenovirus titers were estimated by OD260: rAd[Mito-ScaI-HA]: 5.4 × 1012 particles/ml; rAd[eGFP]: 9.2 × 1012 particles/ml.
Adenovirus injections and sample preparation
For liver studies, anesthetized adult mice belonging to the young group (2.5- to 4-month-old) or old group (approximately 1-year-old) were injected in the right external jugular vein with 5 μl of rAd[Mito-ScaI-HA] or rAd[eGFP] (2.5 × 1010 particles) diluted in 30-50 μl of PBS with 30-gauge dental needle attached to a 100-μl gas-tight Hamilton syringe through a 30-gauge tygon tubing. Liver samples were obtained after anesthesia at day 0 and after perfusion of the animal with chilled PBS. For time-point studies, small liver biopsies were performed at day 0 and at different weeks after injection (2-14 weeks after virus administration). For pain relief, the analgesic buprenorphine (0.05-0.1 mg/kg) was administered to the mice during the following 48 h.
For muscle studies, 5-day-old mice were injected three consecutive times within about 2 min in the femoris muscle of the right leg with 1 × 1010 particles (2 μl) of rAd[Mito-ScaI-HA] or rAd[eGFP] with a 30-gauge dental needle attached to a 10-μl gas-tight Hamilton syringe via 18 cm of 30-gauge tygon tubing. When fluorescent latex microspheres were used (2 μm; Polysciences Inc., Warrington, PA, USA) as markers, they composed 0.25% of the volume of the injected virus solution. Left legs were not injected and were used as controls. After anesthesia (100 mg/kg ketamine and 10 mg/kg xylazine), mice were killed by cervical dislocation and the femoris muscles dissected.
Heteroplasmy analysis by ‘last-cycle hot’ PCR and RFLP
Total DNA from tissue samples was obtained by phenol-chloroform extraction or with the Nucleon BACC2 kit (Amersham Biosciences, Piscataway, NJ, USA). Total DNA from muscle samples microdissected by laser capture microscope (LCM) was extracted with the QIAamp DNA Micro scope Kit (Qiagen, Valencia, CA, USA) and amplified using the Expand long template PCR system (Roche, Indianapolis, IN, USA). ‘Last-cycle hot’ PCR45 was performed using mtDNA primers (5228-5250; 5665-5690), digested with ApaLI and subjected to electrophoreses in an 8% polyacrylamide gel. The radio-active signal was quantified using a Cyclone phosphor-imaging system (Perkin-Elmer, Waltham, MA, USA) as described.15
Southern blot analysis
Five microgram of total DNA was digested with SacI, which linearized mouse mtDNA by cleavage at position 9047. Digested samples were electrophoresed through 0.8% agarose gels and transferred to zeta-probe membranes (Bio-Rad, Hercules, CA, USA). mtDNA was detected by hybridization with a probe corresponding to positions 10-10 145 of the mouse mtDNA (accession number AJ512208). To quantify nuclear DNA, the blots were hybridized with a probe corresponding to positions (502-1515) of the mouse 18S rDNA (accession number BK000964). The radioactive signal was quantified using a Cyclone phosphor-imager system.
Western blots
Fifty microgram of protein from liver or cell homogenates was electrophoresed in a 4-20% Tris-HCl polyacrylamide gel (Bio-Rad) and transferred to a nitrocellulose membrane (Bio-Rad). Rat anti-HA antibody was obtained from Roche Biochemicals and a donkey anti-rat IgG IRDye 800-conjugated secondary antibody from Rockland (Gilbertsville, PA, USA). The Odyssey Infrared Imaging System (LI-COR) was used to scan western blots.
Histological and immunohistochemical studies
Mouse hepatocytes (~105 cells) in a six-well dish were infected with 5 × 107 particles of rAd[Mito-ScaI-HA] for 24 h. Hepatocytes were incubated for 30 min at 37°C with a mitochondrial dye Mitotracker red (200 nm) (Molecular Probes, Invitrogen, Carlsbad, CA, USA), and then fixed with 2% paraformaldehyde (PFA) in PBS for 20 min. After a brief treatment with methanol, a primary anti-HA antibody (Roche) (in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)) was left overnight at 4°C followed by 2 h incubation at room temperature with an Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, Invitrogen).
Liver biopsies (~50 mg) were collected at different times before or after injection and were snap-frozen in liquid nitrogen-cooled isopentane and stored at -80°C. Cryostat sections (20 μm) were cut and checked for eGFP expression using a fluorescence microscope. Positive samples were fixed in 4% PFA, treated with 0.5% Triton X-100 in PBS with 2% BSA for 30 min, then stained for HA as above.
Dissected femoris muscles were snap-frozen in liquid nitrogen-cooled isopentane and stored at -80°C. Twenty-micrometer cryostat sections were collected and analyzed in a fluorescence microscope for eGFP expression. When fluorescence microspheres were used to track transduced tissue, cryostat sections were collected and analyzed in a fluorescence microscope to localize the injection site. Positive samples were fixed in 4% PFA, treated with methanol for 5 min and then incubated with the primary anti-HA antibody (in 2% BSA in PBS) followed by 2 h with a secondary antibody conjugated to Alexa Fluor 594 (Molecular Probes, Invitrogen).
For immunohistochemical analysis, muscle sections were incubated overnight at 4°C with the monoclonal anti-SDHA primary antibody (Molecular Probes, Invitrogen) followed by 2 h at room temperature with an anti-MT-CO1-Alexafluor 488-conjugated monoclonal antibody (Molecular Probes, Invitrogen) diluted to a concentration of 2 μg/ml in 2% BSA in PBS.
For histological studies, muscle and liver tissue were frozen in isopentane cooled in liquid nitrogen. Frozen sections (8 μm thick) were stained for COX or COX/SDH activities as described.46
LCM microdissections
Muscle sections were collected on PEN-membrane slides (Leica) and stained for COX or COX/SDH activities as above. Slides that showed areas of COX deficiency were dehydrated as per the manufacturer’s protocol (Leica AS LMD, Allendale, NJ, USA). Areas from 10 000 to 30 000 μm2 were microdissected using standard laser settings and collected in 0.2-ml tubes.
Determination of enzyme activity of respiratory complexes
Liver homogenates were prepared in 10 mm 4-(2-hydroxy-ethyl)-1-piperazineethanesulfonic acid, 0.5 mm ethylene-diaminetetraacetic acid, 0.5 mm ethyleneglycoltetraacetic acid and 250 mm sucrose (pH 7.4) that contained a complete protease inhibitor cocktail (Roche Diagnostics), using a motor-driven Teflon-pestle homogenizer. Samples were centrifuged at 2000 g for 3 min. The supernatant was saved and re-centrifuged at 2000 g for 3 min. COX activity and citrate synthase activity were determined spectrophotometrically in homogenate preparations as described previously.47 Protein concentration was determined by the method of Bradford48 using BSA as a standard.
Supplementary Material
Acknowledgements
We are grateful to Dr Alexander Marcillo for assistance with surgical procedures. We are indebted to Brendan Battersby and Eric A Shoubridge for the NZB/BALB heteroplasmic mice and to New England Biolabs for the ScaI construct. We also thank the University of Miami Miller School of Medicine Imaging Core for their assistance. This work was supported by PHS grants EY10804 and NS041777. SB is supported by a supplement to PHS Grant EY10804.
References
- 1.Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders - past, present and future. Biochim Biophys Acta. 2004;1659:115–120. doi: 10.1016/j.bbabio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- 3.Solignac M, Monnerot M, Mounolou JC. Mitochondrial DNA heteroplasmy in Drosophila mauritiana. Proc Natl Acad Sci USA. 1983;80:6942–6946. doi: 10.1073/pnas.80.22.6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci USA. 1992;89:4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanna MG, Nelson IP, Morgan-Hughes JA, Harding AE. Impaired mitochondrial translation in human myoblasts har-bouring the mitochondrial DNA tRNA lysine 8344 A→G (MERRF) mutation: relationship to proportion of mutant mitochondrial DNA. J Neurol Sci. 1995;130:154–160. doi: 10.1016/0022-510x(95)00022-t. [DOI] [PubMed] [Google Scholar]
- 6.Taivassalo T, Fu K, Johns T, Arnold D, Karpati G, Shoubridge EA. Gene shifting: a novel therapy for mitochondrial myopathy. Hum Mol Genet. 1999;8:1047–1052. doi: 10.1093/hmg/8.6.1047. [DOI] [PubMed] [Google Scholar]
- 7.Shoubridge EA, Johns T, Karpati G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. Hum Mol Genet. 1997;6:2239–2242. doi: 10.1093/hmg/6.13.2239. [DOI] [PubMed] [Google Scholar]
- 8.Clark KM, Bindoff LA, Lightowlers RN, Andrews RM, Griffiths PG, Johnson MA, et al. Reversal of a mitochondrial DNA defect in human skeletal muscle. Nat Genet. 1997;16:222–224. doi: 10.1038/ng0797-222. [DOI] [PubMed] [Google Scholar]
- 9.Manfredi G, Gupta N, Vazquez-Memije ME, Sadlock JE, Spinazzola A, De Vivo DC, et al. Oligomycin induces a decrease in the cellular content of a pathogenic mutation in the human mitochondrial ATPase 6 gene. J Biol Chem. 1999;274:9386–9391. doi: 10.1074/jbc.274.14.9386. [DOI] [PubMed] [Google Scholar]
- 10.Taylor RW, Chinnery PF, Turnbull DM, Lightowlers RN. Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nat Genet. 1997;15:212–215. doi: 10.1038/ng0297-212. [DOI] [PubMed] [Google Scholar]
- 11.Chinnery PF, Taylor RW, Diekert K, Lill R, Turnbull DM, Lightowlers RN. Peptide nucleic acid delivery to human mitochondria. Gene Therapy. 1999;6:1919–1928. doi: 10.1038/sj.gt.3301061. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Schon EA, Wilichowski E, Vazquez-Memije ME, Davidson E, King MP. Rearrangements of human mitochondrial DNA (mtDNA): new insights into the regulation of mtDNA copy number and gene expression. Mol Biol Cell. 2000;11:1471–1485. doi: 10.1091/mbc.11.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cazzalini O, Lazze MC, Iamele L, Stivala LA, Bianchi L, Vaghi P, et al. Early effects of AZT on mitochondrial functions in the absence of mitochondrial DNA depletion in rat myotubes. Biochem Pharmacol. 2001;62:893–902. doi: 10.1016/s0006-2952(01)00713-4. [DOI] [PubMed] [Google Scholar]
- 14.Jazayeri M, Andreyev A, Will Y, Ward M, Anderson CM, Clevenger W. Inducible expression of a dominant negative DNA polymerase-gamma depletes mitochondrial DNA and produces a rho0 phenotype. J Biol Chem. 2003;278:9823–9830. doi: 10.1074/jbc.m211730200. [DOI] [PubMed] [Google Scholar]
- 15.Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci USA. 2005;102:14392–14397. doi: 10.1073/pnas.0502896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonu-clease. Hum Mol Genet. 2001;10:3093–3099. doi: 10.1093/hmg/10.26.3093. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka M, Borgeld HJ, Zhang J, Muramatsu S, Gong JS, Yoneda M, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci. 2002;9:534–541. doi: 10.1159/000064726. [DOI] [PubMed] [Google Scholar]
- 18.Huard J, Lochmuller H, Acsadi G, Jani A, Massie B, Karpati G. The route of administration is a major determinant of the transduction efficiency of rat tissues by adenoviral recombinants. Gene Therapy. 1995;2:107–115. [PubMed] [Google Scholar]
- 19.Sullivan DE, Dash S, Du H, Hiramatsu N, Aydin F, Kolls J, et al. Liver-directed gene transfer in non-human primates. Hum Gene Ther. 1997;8:1195–1206. doi: 10.1089/hum.1997.8.10-1195. [DOI] [PubMed] [Google Scholar]
- 20.Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 21.Moraes CT, Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, et al. mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991;48:492–501. [PMC free article] [PubMed] [Google Scholar]
- 22.Reichmann H. Enzyme activity analyses along ragged-red and normal single muscle fibres. Histochemistry. 1992;98:131–134. doi: 10.1007/BF00717004. [DOI] [PubMed] [Google Scholar]
- 23.D’Souza GG, Weissig V. Approaches to mitochondrial gene therapy. Curr Gene Ther. 2004;4:317–328. doi: 10.2174/1566523043346200. [DOI] [PubMed] [Google Scholar]
- 24.Bangari DS, Mittal SK. Current strategies and future directions for eluding adenoviral vector immunity. Curr Gene Ther. 2006;6:215–226. doi: 10.2174/156652306776359478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao H, Koehler DR, Hu J. Adenoviral vectors for gene replacement therapy. Viral Immunol. 2004;17:327–333. doi: 10.1089/vim.2004.17.327. [DOI] [PubMed] [Google Scholar]
- 26.Shayakhmetov DM, Li ZY, Ni S, Lieber A. Analysis of adenovirus sequestration in the liver, transduction of hepatic cells, and innate toxicity after injection of fiber-modified vectors. J Virol. 2004;78:5368–5381. doi: 10.1128/JVI.78.10.5368-5381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Everett RS, Hodges BL, Ding EY, Xu F, Serra D, Amalfitano A. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum Gene Ther. 2003;14:1715–1726. doi: 10.1089/104303403322611737. [DOI] [PubMed] [Google Scholar]
- 28.Nicklin SA, Wu E, Nemerow GR, Baker AH. The influence of adenovirus fiber structure and function on vector development for gene therapy. Mol Ther. 2005;12:384–393. doi: 10.1016/j.ymthe.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Xu ZL, Mizuguchi H, Sakurai F, Koizumi N, Hosono T, Kawabata K, et al. Approaches to improving the kinetics of adenovirus-delivered genes and gene products. Adv Drug Deliv Rev. 2005;57:781–802. doi: 10.1016/j.addr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Battersby BJ, Shoubridge EA. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet. 2001;10:2469–2479. doi: 10.1093/hmg/10.22.2469. [DOI] [PubMed] [Google Scholar]
- 31.Moreno-Loshuertos R, Acin-Perez R, Fernandez-Silva P, Movilla N, Perez-Martos A, Rodriguez de Cordoba S, et al. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–1268. doi: 10.1038/ng1897. [DOI] [PubMed] [Google Scholar]
- 32.Dimauro S. Mitochondrial myopathies. Curr Opin Rheumatol. 2006;18:636–641. doi: 10.1097/01.bor.0000245729.17759.f2. [DOI] [PubMed] [Google Scholar]
- 33.Menezes KM, Mok HS, Barry MA. Increased transduction of skeletal muscle cells by fibroblast growth factor-modified adenoviral vectors. Hum Gene Ther. 2006;17:314–320. doi: 10.1089/hum.2006.17.314. [DOI] [PubMed] [Google Scholar]
- 34.O’Hara AJ, Howell JM, Taplin RH, Fletcher S, Lloyd F, Kakulas B, et al. The spread of transgene expression at the site of gene construct injection. Muscle Nerve. 2001;24:488–495. doi: 10.1002/mus.1031. [DOI] [PubMed] [Google Scholar]
- 35.Martin-Touaux E, Puech JP, Chateau D, Emiliani C, Kremer EJ, Raben N, et al. Muscle as a putative producer of acid alpha-glucosidase for glycogenosis type II gene therapy. Hum Mol Genet. 2002;11:1637–1645. doi: 10.1093/hmg/11.14.1637. [DOI] [PubMed] [Google Scholar]
- 36.Dudley RW, Lu Y, Gilbert R, Matecki S, Nalbantoglu J, Petrof BJ, et al. Sustained improvement of muscle function one year after full-length dystrophin gene transfer into mdx mice by a gutted helper-dependent adenoviral vector. Hum Gene Ther. 2004;15:145–156. doi: 10.1089/104303404772679959. [DOI] [PubMed] [Google Scholar]
- 37.Allamand V, Donahue KM, Straub V, Davisson RL, Davidson BL, Campbell KP. Early adenovirus-mediated gene transfer effectively prevents muscular dystrophy in alpha-sarcoglycan-deficient mice. Gene Therapy. 2000;7:1385–1391. doi: 10.1038/sj.gt.3301247. [DOI] [PubMed] [Google Scholar]
- 38.Ragot T, Perricaudet M. Development of adenoviral vectors in gene therapy: application to gene transfer in muscles. C R Seances Soc Biol Fil. 1996;190:13–31. [PubMed] [Google Scholar]
- 39.Mitsuoka T, Kawarai T, Watanabe C, Katayama S, Nakamura S. Comparison of clinical pictures of mitochondrial encephalomyopathy with tRNA(Leu(UUR)) mutation in 3243 with that in 3254. No To Shinkei. 1998;50:1089–1092. [PubMed] [Google Scholar]
- 40.Walther W, Arlt F, Fichtner I, Aumann J, Stein U, Schlag PM. Heat-inducible in vivo gene therapy of colon carcinoma by human mdr1 promoter-regulated tumor necrosis factor-alpha expression. Mol Cancer Ther. 2007;6:236–243. doi: 10.1158/1535-7163.MCT-06-0070. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez-Nicolini V, Sanchez-Bustamante CD, Hartenbach S, Fussenegger M. Adenoviral vector platform for transduction of constitutive and regulated tricistronic or triple-transcript transgene expression in mammalian cells and microtissues. J Gene Med. 2006;8:1208–1222. doi: 10.1002/jgm.960. [DOI] [PubMed] [Google Scholar]
- 42.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3:449–454. doi: 10.1093/hmg/3.3.449. [DOI] [PubMed] [Google Scholar]
- 44.De Giorgi F, Ahmed Z, Bastianutto C, Brini M, Jouaville LS, Marsault R, et al. Targeting GFP to organelles. Methods Cell Biol. 1999;58:75–85. doi: 10.1016/s0091-679x(08)61949-4. [DOI] [PubMed] [Google Scholar]
- 45.Moraes CT, Ricci E, Petruzzella V, Shanske S, DiMauro S, Schon EA, et al. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nat Genet. 1992;1:359–367. doi: 10.1038/ng0892-359. [DOI] [PubMed] [Google Scholar]
- 46.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods. 2002;26:307–316. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 48.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.